Hakan R. Toka and Seth L. Alper
 INTRODUCTION
INTRODUCTIONCoordinated regulation of volume homeostasis and vascular tone maintains adequate tissue perfusion in response to varying environmental stimuli. This chapter discusses the pharmacologically relevant physiology of volume regulation, with emphasis on the hormonal pathways and renal mechanisms that modulate systemic volume. (Control of vascular tone is discussed in Chapter 22, Pharmacology of Vascular Tone.) Dysregulation of volume homeostasis can result in edema, the pathologic accumulation of fluid in the extravascular space. Pharmacologic modulation of volume is targeted at reducing volume excess; this is an effective treatment for hypertension and heart failure (HF), as well as for cirrhosis and the nephrotic syndrome. The two broad classes of pharmacologic agents used to modify volume status are modulators of neurohormonal regulators (such as angiotensin converting enzyme [ACE] inhibitors) and diuretics (agents that increase renal Na+ excretion). Drugs that modify volume regulation also have many other clinically important effects on the body, because these volume regulators act as diverse hormonal modulators in multiple physiologic pathways. Many of the clinical applications of these agents are discussed further in Chapter 26, Integrative Cardiovascular Pharmacology: Hypertension, Ischemic Heart Disease, and Heart Failure.
 PHYSIOLOGY OF VOLUME REGULATION
PHYSIOLOGY OF VOLUME REGULATION
An intricate set of mechanisms sense, signal, and modulate changes in plasma volume. Volume sensors are located throughout the vascular tree, including in the atria and in the kidneys. Many of the volume regulators activated by these sensors include systemic and autocrine hormones, while others involve neural circuits. The integrated result of these signaling mechanisms is to alter vascular tone and to regulate renal Na+ reabsorption and excretion. Vascular tone maintains end-organ tissue perfusion; changes in renal Na+ excretion alter total volume status.
 Mr. R, a 70-year-old male, is taken by ambulance to the emergency department at 1:00 AM after waking up with shortness of breath for the fourth night in a row. Each time, he “felt tight in the chest” and “couldn’t get a breath”; this discomfort was relieved somewhat by sitting up in bed. He also recalls previous episodes of shortness of breath while climbing stairs.
Mr. R, a 70-year-old male, is taken by ambulance to the emergency department at 1:00 AM after waking up with shortness of breath for the fourth night in a row. Each time, he “felt tight in the chest” and “couldn’t get a breath”; this discomfort was relieved somewhat by sitting up in bed. He also recalls previous episodes of shortness of breath while climbing stairs.
Physical exam reveals tachycardia (heart rate, 112/min), mild hypertension (blood pressure, 155/95 mm Hg), decreased oxygen saturation (90% on room air), increased respiratory rate (28/min), bilateral pulmonary crackles on inspiration, and 1–2+ edema of the feet. Serum troponin T level (a marker of cardiomyocyte injury) is normal, but serum creatinine (1.5 mg/dL) and blood urea nitrogen (BUN, 30 mg/dL) are mildly elevated. Urinalysis is normal. Electrocardiogram shows evidence of an old myocardial infarction (Q wave in leads II, III, and V4–V6). Echocardiography reveals diminished left ventricular ejection fraction (LVEF, 35%; the fraction of blood in the ventricle at the end of diastole that is ejected when the ventricle contracts) without ventricular dilatation.
Based on the clinical findings of decreased cardiac output, pulmonary congestion, and peripheral edema, Mr. R is diagnosed with acute heart failure. His increased creatinine and BUN also indicate an element of renal insufficiency. Pharmacologic therapy is started, including a coronary vasodilator, an antihypertensive calcium channel blocker, and a loop diuretic. After Mr. R’s condition stabilizes over the course of 3 days, the dose of the loop diuretic is decreased and then discontinued. Elective coronary angiography reveals significant stenosis of the left anterior descending coronary artery. Mr. R undergoes balloon angioplasty and stent placement and remains stable. Mr. R is discharged on a regimen that includes an ACE inhibitor and spironolactone.
Questions
1. What mechanisms led to Mr. R’s pulmonary congestion and pedal edema?
2. Why was Mr. R given a loop diuretic?
3. How do ACE inhibitors improve cardiovascular hemodynamics?
4. Why was Mr. R prescribed spironolactone?
Determinants of Intravascular Volume
Intravascular volume is a small proportion of total body water, but the amount of fluid in the vascular compartment critically determines the extent of tissue perfusion. Approximately 2/3 of total body water is intracellular, while 1/3 is extracellular. Of the extracellular fluid (ECF), approximately 3/4 resides in the interstitial space, while 1/4 of ECF is plasma.
Fluid exchange between plasma and interstitial compartments occurs as a result of changes in capillary permeability, oncotic pressure, and hydrostatic pressure. Capillary permeability is determined largely by the junctions between individual endothelial cells lining a vascular space. The capillary beds of some organs are more permeable than those of others and, as a result, allow larger intercompartmental fluid shifts. In the context of inflammation and other pathologic conditions (see below), increased capillary permeability allows proteins to shift, along with “oncotically obligated water,” between intravascular and perivascular compartments under the influence of the plasma oncotic pressure gradient. Oncotic pressure is determined by the molecular solute components of a fluid space that are differentially partitioned between adjacent compartments (such constituents are said to be osmotically active). Because albumin, globulins, and other large plasma proteins are normally confined to the plasma space, these oncotically active proteins serve to retain water in the vascular space. The hydrostatic pressure gradient across the capillary barrier between compartments is another force for water movement. An elevated intracapillary pressure favors increased transudation of fluid from plasma into the interstitial space.
The relationship between fluid filtration and capillary permeability, oncotic pressure, and hydrostatic pressure is represented by the following equation
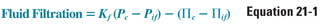
where Kf is the capillary permeability coefficient, Pc is capillary hydrostatic pressure, Pif is interstitial fluid hydrostatic pressure, Πc is capillary oncotic pressure, and Πif is interstitial fluid oncotic pressure. This equation emphasizes that transcapillary fluid movement is governed by intercompartmental gradients rather than by the absolute value of each compartmental pressure. Note that the hydrostatic and oncotic gradient terms have opposing vectors and therefore favor fluid movement in opposite directions. ΔPc normally favors transudation from the capillary lumen to the interstitium, whereas ΔΠc normally favors fluid retention within the capillary lumen.
The extent of fluid filtration that occurs along the length of the capillary differs for each tissue’s capillary bed and is determined by cellular and junctional permeability properties of tissue-specific capillary endothelial cells. In the example shown in Figure 21-1, liver capillaries filter fluid into the interstitium along their entire length. At the arterial end of the capillary bed, (Pc + Πif) exceeds (Pif + Πc), thus favoring plasma filtration from the capillary into the interstitial space. Pc gradually decreases along the length of the capillary, and the rate of fluid filtration into the interstitium decreases. At the venous end of the capillary, hydrostatic fluid filtration and oncotic fluid absorption are almost balanced. Liver sinusoids, which transfer fluid into the interstitial space during perfusion, return this fluid to the circulation via lymphatic flow. In capillary beds of other tissues, the integrated oncotic pressure gradient favoring fluid flow into the capillary balances the integrated hydrostatic pressure gradient, resulting in no net volume change between the vascular and interstitial spaces. Thus, the physiologic steady state of extracellular fluid represents a balance of driving forces between fluids of the intravascular and interstitial compartments. Pathologic alterations in the determinants of transcapillary fluid shifts, coupled with changes in renal Na+ handling, can result in the formation of edema, as discussed below.
Vascular volume sensors can be divided into low-pressure and high-pressure feedback systems. The low-pressure system consists of the atria and pulmonary vasculature. In response to decreased wall stress (e.g., caused by decreased intravascular volume), peripheral nervous system cells lining the atria and pulmonary vasculature transmit a signal to noradrenergic neurons in the medulla of the central nervous system (CNS). This signal is relayed to the hypothalamus, resulting in increased secretion by the posterior pituitary gland of antidiuretic hormone (ADH, also known as arginine vasopressin, AVP). ADH promotes vasoconstriction and antidiuresis (increased renal water reabsorption). Together with increased peripheral sympathetic tone, this maintains distal tissue perfusion. In response to increased wall stress (e.g., caused by increased intravascular volume), cells of the atria produce and secrete natriuretic peptide, which promotes vasodilation and natriuresis (increased renal Na+ excretion).
The high-pressure system consists of specialized baroreceptors in the aortic arch, carotid sinus, and juxtaglomerular apparatus. These sensors modulate hypothalamic control of ADH secretion and sympathetic outflow from the brainstem. In addition, sympathetic input stimulates the juxtaglomerular apparatus to secrete renin, a proteolytic enzyme that activates the renin-angiotensin-aldosterone system (see below).
Together, the low-pressure and high-pressure feedback systems integrate neurohumoral volume signals to maintain volume homeostasis in the face of volume perturbations. The neurohormonal response to a change in volume status is controlled by four main systems: the renin-angiotensin-aldosterone system (RAAS), natriuretic peptides, ADH, and renal sympathetic nerves. The RAAS, ADH, and renal sympathetic nerves are active in situations of intravascular volume depletion, while natriuretic peptides are released in response to intravascular volume overload.
Renin-Angiotensin-Aldosterone System
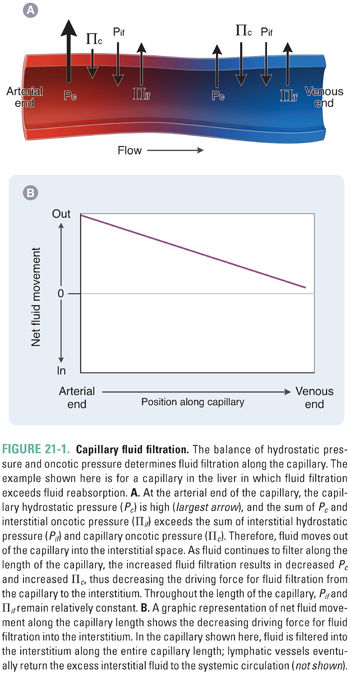
Renin is an aspartyl protease that activates the RAAS by cleavage of the circulating prohormone angiotensinogen to generate angiotensin I. Renin is produced and secreted by the juxtaglomerular apparatus (JGA), a specialized set of granule-containing smooth muscle cells derived from the afferent arteriole that line the afferent and efferent arterioles of the renal glomerulus. The JGA is also adjacent to the macula densa, a nephron segment between the end of the thick ascending limb of the loop of Henle and the distal convoluted tubule, containing specialized tubular epithelial cells capable of sensing distal chloride (and/or sodium) delivery. The JGA in the renal cortex represents the major structural component of the RAAS and one of the most important regulatory sites of renal volume conservation and blood pressure maintenance. The ultimate result of renin secretion is vasoconstriction and Na+ retention, actions that maintain tissue perfusion and increase extracellular fluid volume (Fig. 21-2).
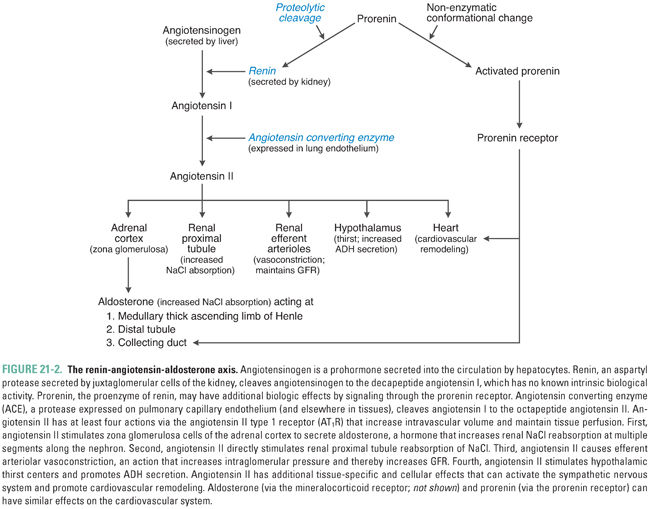
At least three mechanisms are thought to control juxtaglomerular cell renin release (Fig. 21-3). First, a direct pressure-sensing mechanism of the afferent arteriole, equivalent to an intrarenal baroreceptor, responds to changes in renal perfusion pressure (arteriolar wall tension) to increase juxtaglomerular cell release of renin. The detailed molecular mechanism of this sensory transduction is unknown in humans; in rodents, it involves autocrine prostaglandin and purinergic signaling. Second, sympathetic innervation of juxtaglomerular cells promotes renin release via β1-adrenoceptor stimulation. Third, the autoregulatory mechanism known as tubuloglomerular feedback senses distal nephron delivery of chloride (and/or sodium) to modulate renin release. Nephron anatomy is organized such that the distal end of the cortical thick ascending limb (TAL) of each nephron is closely apposed to the juxtaglomerular mesangium of the same nephron. This spatial proximity allows rapid integrative regulation of afferent arteriolar diameter and glomerular mesangial contractility by distal nephron electrolyte concentration and/or salt load. Macula densa cells of the cortical thick ascending limb respond to increased luminal NaCl delivery by increasing extracellular adenosine in the juxtaglomerular interstitium, thereby activating A1 receptors on the juxtaglomerular mesangial cells to decrease renin release. Conversely, decreased luminal NaCl delivery activates a mesangial prostaglandin signaling cascade that culminates in increased renin release. Macula densa cells sense luminal NaCl delivery by monitoring both luminal NaCl concentration and luminal fluid flow rate as sensed by shear stress. NaCl delivery may be sensed directly by receptors in the apical sensory monocilia of macula densa cells; fluid flow may be sensed by direct bending of the monocilia. Molecular components in the extraciliary apical membrane likely also contribute to these signal transduction processes.
Prorenin, the renin proenzyme, was for many years considered an inactive precursor of renin lacking any intrinsic function. It now seems, however, that prorenin can be activated either by proteolytic cleavage to renin or by nonenzymatic conformational change induced by acid pH or elevated temperature. Renin activation typically occurs before its release from juxtaglomerular cells. Proconvertase I and cathepsin B are among the several enzymes that have been proposed to mediate proteolytic activation of renin; the potential roles of plasma and tissue kallikrein in renin cleavage have been disputed.
Recent studies have suggested that prorenin has cleavage-independent functions mediated through its binding to the (pro)renin receptor (PRR). Reversible binding of prorenin to PRR in tissues promotes a nonenzymatic conformational change that allows the uncleaved prorenin to act as an angiotensinogen convertase, thereby converting angiotensinogen to angiotensin I (Ang I; see below). In addition, binding of prorenin to PRR triggers downstream activation of the MAP kinase ERK1/2 signaling pathway, leading to up-regulation of profibrotic and cyclooxygenase-2 genes and to regulation of the vacuolar H+ ATPase in the collecting duct (see below). Interestingly, circulating plasma levels of prorenin are higher than those of renin, particularly in the setting of diabetic kidney disease. Transgenic mice overexpressing PRR develop hypertension and glomerulosclerosis, whereas genetic engineering of PRR loss-of-function leads to embryonic lethality, suggesting that essential cellular functions of PRR are not yet understood.
Secreted renin (produced by cleavage of prorenin) acts as a protease to cleave the first 10 amino acids of the circulating prohormone angiotensinogen, generating angiotensin I (Ang I). Ang I (Ang 1-10) is then cleaved to the active octapeptide angiotensin II (Ang II, AT II, or Ang 1-8) by the carboxypeptidase angiotensin converting enzyme (ACE) located on the endothelial cell surface. Although ACE is expressed primarily in the pulmonary vascular endothelium and coronary circulation, ACE activity regulates local production of AT II in all vascular beds. Indeed, an incompletely understood “local” renin-angiotensin system is also expressed in the vasculature, producing these substances as autocrine factors independently of the kidney and liver. ACE has a broad proteolytic substrate specificity that includes many other substrates such as neuropeptides and kinins (e.g., bradykinin) that are venodilatory autacoids released in response to inflammation. For this reason, ACE is also known as kininase II. Kininase activity has important pharmacologic consequences, as discussed below.
The recently identified ACE homolog ACE2 is highly expressed in the kidney, where it is localized predominantly in tubular epithelial cells and less prominently in glomeruli and the renal vasculature. ACE2 degrades both Ang I and Ang II to the heptapeptide Ang 1-7. In contrast to the vasoconstrictor and pro-proliferative peptide Ang II, Ang 1-7 is considered an antiproliferative vasodilator that counteracts the cardiovascular and baroreflex actions of Ang II. Ang 1-7 binds to the G protein-coupled receptor MAS rather than to the angiotensin II (AT II) receptor (see below). ACE2 activity is altered in diabetic kidney disease and hypertensive renal disease and in experimental models of kidney injury. Dissociation between tubular and glomerular ACE2 expression can occur in diabetic kidney disease, in which ACE2 expression is increased in tubular epithelial cells but decreased in glomeruli. In addition to Ang I (Ang 1-10), Ang II (Ang 1-8), and Ang 1-7, angiotensin III (Ang 2-8) and angiotensin IV (Ang 3-8) have been identified. Ang 2-8 is a less potent vasopressor than Ang II but retains 100% of its aldosterone-stimulating activity. The vasopressor activity of Ang 3-8 resembles that of Ang 2-8.
Ang II (AT II) binding to the AT II receptor subtype 1 (G protein-coupled AT1 receptor, AT1R) produces at least four stimulatory physiologic responses: (1) stimulation of aldosterone secretion by zona glomerulosa cells of the adrenal glands, (2) increased reabsorption of NaCl from the proximal tubule and other nephron segments, (3) arteriolar vasoconstriction, and (4) central stimulation of thirst and ADH secretion. All four of these actions increase intravascular volume and therefore help to maintain perfusion pressure: aldosterone secretion increases distal tubule Na+ reabsorption; proximal tubule NaCl reabsorption increases the fraction of filtered Na+ that is reabsorbed; arteriolar vasoconstriction maintains blood pressure; stimulation of thirst increases free water absorbed into the vasculature; and secretion of ADH increases collecting duct free water absorption. Ang II also negatively regulates renin secretion through binding to AT1R on juxtaglomerular cells. Ang II has many other AT1R-mediated tissue- and cell type-specific effects, leading to sympathetic nervous system activation, generation of reactive oxygen species (ROS), and cell growth. Conversion of Ang I to Ang II by tissue-specific ACE (chymase) of cardiac myocytes can promote cardiac remodeling, hypertrophy, and fibrosis. Similar cardiovascular effects are also potentiated through aldosterone activation of extrarenal mineralocorticoid receptors.
The actions of Ang II are best understood in vascular smooth muscle cells, where AT1R activates phospholipase C, leading to release of Ca2+ from intracellular stores, activation of protein kinase C, and vasoconstriction. Inhibition of AT1R decreases vascular smooth muscle cell contractility and thereby decreases systemic vascular resistance and blood pressure (see the following discussion). The related G protein-coupled AT II receptor AT2R has a vasodilator role, in part by increasing nitric oxide production. AT2R is highly expressed in fetal kidney and intestine, whereas high expression in adults is restricted to myometrium, with lower levels in adrenals and oviducts. AT2R action has recently been implicated in some types of refractory pain syndromes.
Natriuretic peptides are hormones released by atria, ventricles, and vascular endothelium in response to volume overload. The classical natriuretic peptides are A-type, B-type, and C-type natriuretic peptides. A-type natriuretic peptide (ANP) is released primarily by the atria, while B-type natriuretic peptide (BNP) is released mainly by the ventricles. C-type natriuretic peptide (CNP) is released by vascular endothelial cells. The natriuretic peptide uroguanylin (UGN) is released by enterocytes in response to dietary ingestion of salt.
Vascular natriuretic peptides are released in response to increased intravascular volume, an effect that may be signaled by increased stretch of natriuretic peptide-secreting cells. Circulating natriuretic peptides bind to one of three receptors, termed NPR-A, NPR-B, and NPR-C. NPR-A and NPR-B are transmembrane proteins with cytoplasmic guanylyl cyclase domains (see Chapter 1, Drug–Receptor Interactions); activation of these receptors increases intracellular cGMP levels. NPR-C lacks an intracellular guanylyl cyclase domain and may serve as a “decoy” or “buffer” receptor to reduce the level of circulating natriuretic peptides available to bind to the two signaling receptors. Both ANP and BNP bind with high affinity to NPR-A, while only CNP binds to NPR-B. All three natriuretic peptides bind to NPR-C (Fig. 21-4A). Deletion of the ANP gene (Nppa) in mice causes salt-sensitive hypertension. Common human allelic gene variants in the ANP (NPPA) and BNP (NPPB) genes have been associated with higher levels of ANP and BNP, higher blood pressure, and risk of hypertension. In mice, UGN binds and activates transmembrane guanylyl cyclase C in both enterocytes (promoting enteric Cl− secretion) and renal proximal tubule cells (reducing renal Na+ and Cl− reabsorption). UGN also promotes natriuresis in the renal collecting duct by additional, less well characterized mechanisms.
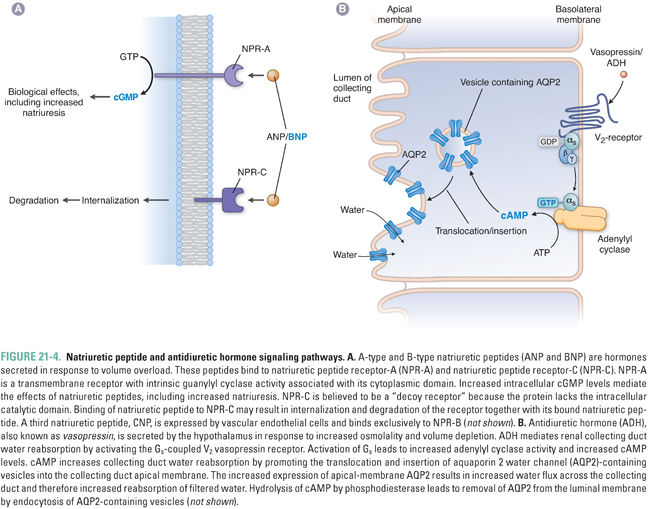
Natriuretic peptides affect the cardiovascular system, the kidney, and the central nervous system. Integration of natriuretic peptide-derived signals serves to decrease volume overload and its sequelae. ANP relaxes vascular smooth muscle by increasing intracellular cGMP, which causes dephosphorylation of myosin light chain and subsequent vasorelaxation (see Chapter 22). ANP also increases capillary endothelial permeability, which reduces blood pressure by favoring fluid filtration from the plasma into the interstitium (see Equation 21-1).
In the kidney, natriuretic peptides promote both increased glomerular filtration rate (GFR) and natriuresis. GFR is increased because of constriction of the efferent arteriole and dilation of the afferent arteriole, resulting in higher intraglomerular pressure and therefore increased plasma filtration. The natriuretic effects on the kidney result from antagonism of ADH action in the collecting ducts and antagonism of Na+ reabsorption in multiple nephron segments.
The central effects of natriuretic peptides are less well understood, but they include decreased perception of thirst (and therefore decreased fluid intake), decreased release of antidiuretic hormone, and decreased sympathetic tone. The signaling mechanisms mediating these actions are uncertain, but may be via CNP, as this natriuretic peptide is expressed at high levels in the brain.
Although many of the effects of natriuretic peptides remain incompletely understood, these hormones appear to play an important role in regulating the pathophysiology of volume excess. Much interest has recently focused on the relationship between natriuretic peptides and heart failure. In particular, BNP and N-terminal proBNP (NT-proBNP) have emerged as promising markers for heart failure diagnosis, prognosis, and treatment. The physiology and pharmacology of natriuretic peptides and their receptors remain subjects for active investigation.
Antidiuretic hormone (ADH, arginine vasopressin, or vasopressin) is a nonapeptide hormone secreted by the posterior pituitary gland in response to increased plasma osmolality or severe hypovolemia. ADH constricts the peripheral vasculature and promotes water reabsorption in the renal collecting duct. Its actions are mediated by two distinct G protein-coupled receptors. The V1 receptor, present predominantly in vascular smooth muscle cells, stimulates vasoconstriction through a Gq–mediated mechanism. The V2 receptor, expressed in collecting duct principal cells, stimulates water reabsorption by a Gs-mediated mechanism (Fig. 21-4B). This Gs signal increases cytosolic cAMP, which leads to activation of protein kinase A (PKA). PKA phosphorylates the water channel aquaporin 2 and activates transport and fusion of aquaporin 2-containing vesicles into the apical membrane of the principal cell. Increased aquaporin 2 expression at the apical membrane promotes increased water reabsorption. Regulation of renal water reabsorption in the collecting duct modulates urine and plasma osmolality and serves as a reserve mechanism for increasing intravascular volume in situations of severe dehydration.
Renal sympathetic nerves innervate both afferent and efferent arterioles. In response to a decrease in intravascular volume, the renal sympathetic nerves decrease GFR by stimulating constriction of the afferent arteriole to a greater degree than the efferent arteriole. The decreased GFR resulting from preferential constriction of the afferent arteriole ultimately leads to decreased natriuresis. Renal sympathetic nerves also increase renin production by stimulation of β1-adrenergic receptors on juxtaglomerular mesangial cells and increase proximal tubule NaCl reabsorption. Since transplanted kidneys function normally in the initial absence of sympathetic nerve input, renal innervation is not required for clinically normal kidney function.
Renal Control of Na+ Excretion
Over the course of 24 hours, the kidneys filter approximately 180 L of fluid. To increase or decrease body fluid volume, the kidneys must increase or decrease renal Na+ reabsorption from the large daily volume of glomerular filtrate. For this reason, the neurohormonal mechanisms controlling extracellular volume status have important actions on the kidney. An understanding of the renal control of Na+ excretion is crucial to understanding the role of the kidney in regulation of body fluid volume.
The renal glomerulus produces an ultrafiltrate of plasma that flows through and is processed by renal tubules of the nephron, the functional unit of the kidney (Fig. 21-5). The postglomerular nephron is responsible for solute and water reabsorption from the filtrate, as well as for excretion of metabolic waste products and xenobiotics, including drugs. The renal tubular epithelial cells of the postglomerular nephron enclose a lengthy tubular lumen, the “urinary space,” which leads to the ureters, urinary bladder, and urethra. The initial glomerular ultrafiltrate contains solutes of low molecular weight at concentrations similar to those in the plasma. As the ultrafiltrate passes through the nephron, substrate-specific transporters and channels in the luminal (apical) membrane of polarized renal tubular epithelial cells sequentially alter the solute concentrations of the tubular fluid. The function of these transporters and channels is, in turn, influenced by changes in solute concentrations in the cells themselves, as regulated in part by channels and transporters on the contraluminal (basolateral) side of the cells. Systemic volume regulation by the kidney is accomplished by tubular solute reabsorption through integrated action of ion channels and ion transporters in the apical and basolateral membranes of tubular epithelial cells and by the accompanying reabsorption of water.
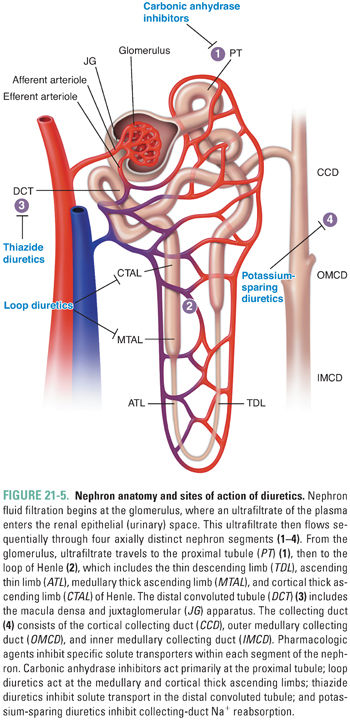
The postglomerular nephron exhibits remarkable heterogeneity along its length. Four segments of the nephron are especially relevant to the pharmacology of systemic volume regulation (Fig. 21-5). These are the proximal tubule, the thick ascending limb (TAL) of the loop of Henle, the distal convoluted tubule (DCT), and the cortical collecting duct (CCD). In each tubular segment, a complex but tightly choreographed group of segment-specific ion transporters and channels collaborate in the reabsorption of NaCl from the lumen across the cellular monolayer of tubular epithelium into the interstitial space. NaCl reabsorption is key for systemic water retention. Solute and water transport across each segment requires coordination of transporter function in the luminal and basolateral membranes. In addition, paracellular transport of ions across the tight junctions between cells requires regulated communication between adjacent cells of the tubular epithelium. Integration of the transcellular and paracellular components of transepithelial transport requires integration of signals transmitted by sensors of extracellular and intracellular ion concentrations and of intracellular, local extracellular, and systemic volume. Alteration of ion transport by drugs in any nephron segment can induce compensatory regulation locally and in more distally located nephron segments.
The proximal tubule (PT) is the first reabsorptive site in the nephron. It is responsible for approximately two-thirds of sodium reabsorption, 85–90% of bicarbonate reabsorption, and approximately 60% of chloride reabsorption (Fig. 21-6). Specific sodium-coupled symporters in the proximal tubule apical membrane drive renal reabsorption of all glucose, amino acids, phosphate, and sulfate from the glomerular filtrate. The proximal tubule also mediates secretion and reabsorption of weak organic acids and weak organic bases; these are coupled to processes of sodium or proton symport or antiport or to anion exchange mechanisms. Among these weak acids and bases are many of the drugs used to regulate systemic volume (see below).
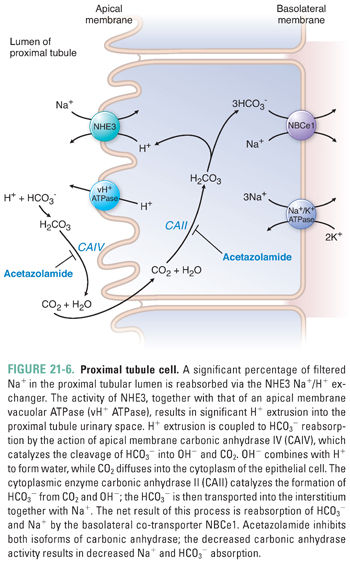
Bicarbonate reabsorption requires the coordinated action of apical and basolateral ion transporters together with apical and intracellular enzymatic activities (Fig. 21-6). At the luminal surface of the proximal tubule, filtered bicarbonate encounters active proton secretion across the proximal tubule brush-border microvilli. Two-thirds of the proton efflux is in exchange for influx of Na+, largely via the NHE3 Na+/H+ exchanger. The remaining third of proton efflux is mediated by the vacuolar H+ ATPase (vH+ ATPase).
The HCO3− permeability of the luminal membrane of the proximal tubular cell is low. However, the outer leaflet of the luminal membrane harbors the glycosylphosphatidylinositol-linked exoenzyme carbonic anhydrase IV (CAIV). CAIV converts luminal HCO3− to CO2 and OH−. The OH− is rapidly hydrated to water by the abundance of local protons, and the CO2 freely diffuses into the cytoplasm of the proximal tubular epithelial cell. The intracellular CO2 is rapidly rehydrated to HCO3− by cytoplasmic carbonic anhydrase II (CAII); this reaction consumes the intracellular OH− accumulated as a result of the H+-extruding activities of apical NHE3 and vH+ ATPase. The HCO3− produced by the CAII reaction is then co-transported with Na+ across the basolateral membrane of the epithelial cell, accounting for the net reabsorption of sodium and bicarbonate. The Na+/HCO3− co-transporter NBCe1 mediates electrogenic basolateral efflux of three HCO3− ions with each co-transported Na+ ion. Basolateral K+ channels maintain an inside-negative membrane potential to enhance the driving force for net efflux of two negative charges per NBCe1 transport cycle. Emerging evidence also suggests the presence of several types of transmembrane ecto-carbonic anhydrases in the basolateral membrane that help dissipate the local accumulation of bicarbonate within the interstitial space between the epithelial cells and peritubular capillaries.
Solute absorption in the proximal tubule is iso-osmotic—water accompanies reabsorbed ions to maintain osmotic balance. In the past, water flow was assumed to be largely paracellular. However, data from mice genetically modified to lack the aquaporin water channel AQP1 (and from rare cases of humans lacking AQP1) demonstrate that most water reabsorption across the proximal tubule—and, beyond that, across the thin descending limb of Henle—is transcellular. Aquaporins are central to transepithelial water permeability in all water-permeable nephron segments. Thus, the transition from the water-permeable thin descending limb of Henle to the water-impermeable ascending thin limb is paralleled by decreased AQP1 expression.
Proximal tubule Na+ reabsorption is a key component of the intrarenal RAAS. Ang II is filtered into the tubular fluid and activates AT1 receptors expressed on the apical membrane of renal tubular epithelial cells. Proximal tubular lumen concentrations of Ang I and Ang II have been found to be higher than their corresponding plasma concentrations, leading to the discovery that proximal tubular epithelial cells express angiotensinogen that is converted to Ang I and Ang II through intrarenal (pro)renin and ACE activity. Elevated Ang II levels sustain or up-regulate tubular epithelial cell AT1R but downregulate vascular AT1R. The critical importance of kidney AT1R has been demonstrated by using proximal tubule-specific AT1R knockout mice (with intact expression of AT1R in all other tissues). Infusion of Ang II into these mice failed to increase blood pressure to the same degree as in control mice. The reduced positive Na+ balance observed in these mice was consistent with facilitated natriuresis as a mechanism for resistance to hypertension.
Thick Ascending Limb of the Loop of Henle
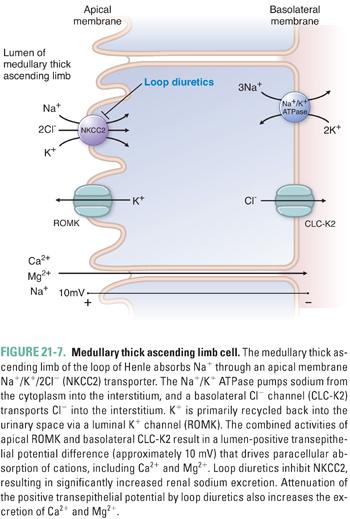
The tubular fluid emerging from the ascending thin limb is hypertonic and has an elevated NaCl concentration. The three nephron segments into which this fluid flows, the thick ascending limb (TAL), the distal convoluted tubule (DCT), and the connecting tubule or segment (CNT), together constitute “the diluting segment.” The apical membrane of the thick ascending limb of Henle is devoid of aquaporins, as is the apical membrane of the rest of the diluting segment; therefore, these nephron segments reabsorb NaCl and urea without accompanying water (Fig. 21-7), thus diluting the solutes of the tubular fluid. Reabsorption of NaCl and urea across the TAL provides the interstitial solute that generates and maintains the corticomedullary osmotic gradient of the kidney, allowing operation of the “countercurrent multiplier” that can concentrate the urine of humans to 1,200 mOsM and that of desert rodents to 4,000 mOsM.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


