Jonathan Z. Li and Donald M. Coen
Inhibition of Viral Genome Replication—Polymerase Inhibitors |

Viral infections are among the leading causes of morbidity and mortality worldwide. Although much progress has been made on antiviral drug development, public health measures and prophylactic vaccines remain the primary means by which society controls the spread of viral infections. The persistence of the acquired immunodeficiency syndrome (AIDS) epidemic makes this painfully clear. Despite advances in anti-human immunodeficiency virus (HIV) drug therapies, AIDS continues to be a common cause of death, particularly in some African nations, where as many as one adult in four is infected with HIV. This enormous prevalence is largely attributable to failures in public health measures and the lack of an effective vaccine against HIV in a setting where anti-HIV drugs are too expensive and the healthcare delivery system is too fragmented.
Despite this bleak statistic, the array of drugs available to combat viruses has been instrumental in saving millions of lives each year and in improving the quality of life for countless others with viral illnesses. This chapter describes the physiology of viral replication and the steps in the viral life cycle that are targeted by current antiviral medications. Key concepts for the chapter include: (1) viruses replicate intracellularly and utilize host cell machinery; (2) despite this mode of replication, multiple targets have been exploited for antiviral drug therapy; and (3) most current antiviral drugs exploit differences between the structures and functions of viral and human proteins to achieve selectivity of antiviral action.
 The year is 1993. Mr. M, a 26-year-old man, complains to Dr. Rose, his primary care physician, of a sore throat, fever, and fatigue for the past several weeks. On physical examination, Dr. Rose notes bilateral cervical lymphadenopathy, consistent with the patient’s “flu-like symptoms.” Dr. Rose thinks it likely that Mr. M has an infection, possibly a simple “cold,” the “flu,” or strep throat. Because of Mr. M’s mononucleosis-like symptoms, Dr. Rose also includes human cytomegalovirus (HCMV), Epstein-Barr virus (EBV), toxoplasmosis, and HIV in her differential diagnosis. Laboratory antibody tests for Streptococcus, HCMV, EBV, toxoplasmosis, and HIV infection are negative. Mr. M is concerned about the possibility of HIV infection and the lack of truly effective therapies for AIDS, although he denies any unprotected sexual activity, intravenous (IV) drug use, or other potential exposure risks. Dr. Rose tells Mr. M that his symptoms will likely resolve with rest but that he should return for follow-up within 6 months. She explains to Mr. M that, if he has recently been infected with HIV, his body would not yet have produced sufficient antibodies to become evident on an anti-HIV antibody test.
The year is 1993. Mr. M, a 26-year-old man, complains to Dr. Rose, his primary care physician, of a sore throat, fever, and fatigue for the past several weeks. On physical examination, Dr. Rose notes bilateral cervical lymphadenopathy, consistent with the patient’s “flu-like symptoms.” Dr. Rose thinks it likely that Mr. M has an infection, possibly a simple “cold,” the “flu,” or strep throat. Because of Mr. M’s mononucleosis-like symptoms, Dr. Rose also includes human cytomegalovirus (HCMV), Epstein-Barr virus (EBV), toxoplasmosis, and HIV in her differential diagnosis. Laboratory antibody tests for Streptococcus, HCMV, EBV, toxoplasmosis, and HIV infection are negative. Mr. M is concerned about the possibility of HIV infection and the lack of truly effective therapies for AIDS, although he denies any unprotected sexual activity, intravenous (IV) drug use, or other potential exposure risks. Dr. Rose tells Mr. M that his symptoms will likely resolve with rest but that he should return for follow-up within 6 months. She explains to Mr. M that, if he has recently been infected with HIV, his body would not yet have produced sufficient antibodies to become evident on an anti-HIV antibody test.
Five years later, Mr. M returns to Dr. Rose’s office. He has not seen any physician in the interim and now presents with new symptoms. There are multiple open lesions on his lips and in his mouth, and he confides that he has similar lesions in his genital area. An ELISA test is positive for anti-HIV antibodies, and a viral load measurement shows high levels of HIV RNA in his blood. Mr. M’s CD4 count is 100 per mm3 (normal range, 800–1,200 per mm3). Dr. Rose immediately prescribes a drug regimen of zidovudine (AZT), lamivudine (3TC), and ritonavir, explaining to Mr. M that a combination of anti-HIV drugs is his best option for reducing the viral load and forestalling more serious disease. In addition, Dr. Rose prescribes oral valacyclovir, a prodrug of acyclovir, to treat Mr. M’s oral and genital herpes.
Over the next 3 years, Mr. M’s HIV viral load falls to undetectable levels and his condition improves. The herpes infections are also kept in check. Today, Mr. M appears in good health and he takes his medications diligently, which is easier now with a once-a-day pill containing efavirenz, emtricitabine, and tenofovir.
Questions
1. What is acyclovir’s mechanism of action?
2. Why does acyclovir not ordinarily cause significant toxicity in humans, while AZT does?
3. What are the mechanisms of action of the three anti-HIV drugs prescribed by Dr. Rose in 1998? In the once-a-day pill Mr. M is taking today?
4. Why is combination antiretroviral therapy required to effectively treat HIV infections?
5. What potential adverse effects could Mr. M experience from long-term treatment with ritonavir?
 PHYSIOLOGY OF VIRAL REPLICATION
PHYSIOLOGY OF VIRAL REPLICATION
Viruses replicate by co-opting the host cell’s metabolic machinery. Based on this fact, one might think that there would be fewer differences between viruses and their human hosts to exploit for drug development than between bacteria and humans. However, all viruses encode proteins that are substantially different from their human counterparts. Additionally, certain host proteins are more important for viral replication than they are for human health. In principle, antiviral drugs could target many of these proteins. In practice, however, relatively few viral proteins and even fewer host proteins have thus far served as useful targets for therapy. Nevertheless, it is a testament to the remarkable progress in antiviral drug development that the number of viral proteins that have been exploited for antiviral therapy is greater than the number of bacterial proteins that have been exploited for antibacterial therapy. However, most antiviral drugs are active against only one or a few viruses while most antibacterial drugs target multiple bacterial species. This difficulty arises because viruses are a heterogeneous group of infectious agents, whereas most bacteria share a common cell wall structure and distinct DNA replication, transcription, and translation machineries.
Viruses exist as small particles called virions. Virions consist of a nucleic acid genome packaged into a virus-encoded protein shell called a capsid. In some viruses, the capsid is surrounded by an envelope, a lipid bilayer membrane that contains virus-encoded envelope proteins. Viral genomes can consist of DNA or RNA and can be single- or double-stranded.
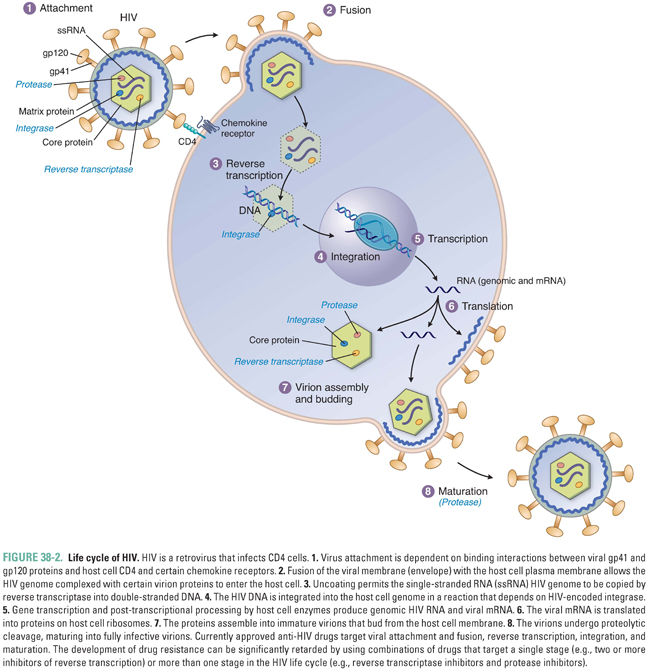
Almost all viruses have the same general life cycle for replication (Fig. 38-1) with some variations. Figure 38-2 illustrates the specific life cycle for HIV, which, as a retrovirus, contains RNA that is copied into DNA. At the start of infection, the virus attaches to the host cell. This attachment is mediated by proteins on the viral surface that bind specifically to a particular host membrane component. For example, the HIV viral envelope contains the glycoprotein gp120, a transmembrane protein that mediates binding and attachment of the virus to host cells expressing CD4 and chemokine receptors such as CCR5 or CXCR4 (Fig. 38-2). Next, the virion undergoes entry by crossing a host cell membrane into the cytoplasm. In the case of HIV, the process of entry depends on gp41, a viral envelope protein that fuses together the membranes of HIV and the target cell.
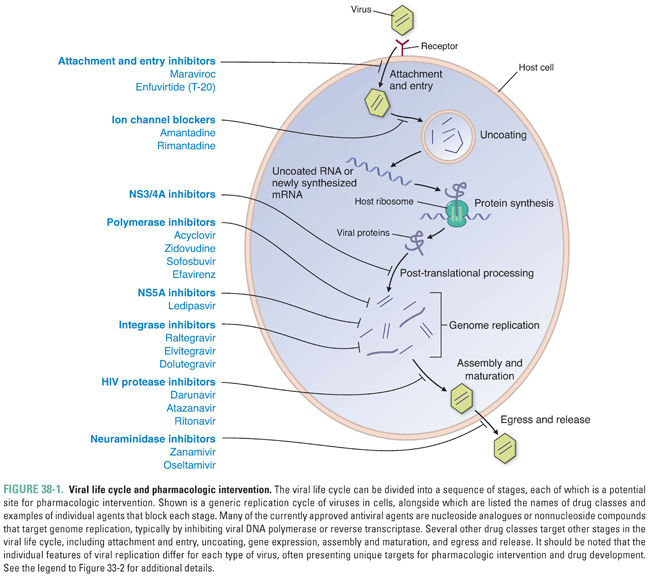
The virion then loses enough of its capsid proteins—the stage of uncoating—that its nucleic acid becomes available for gene expression. (For retroviruses, uncoating does not lead directly to gene expression; instead, it allows reverse transcription of the viral RNA genome into DNA to occur—as described below, this is a step in genome replication.) Viral gene expression entails transcription of the viral genome into mRNA, translation of mRNA into protein on cellular ribosomes, and a variety of processing events including splicing of mRNA precursors and proteolytic cleavage of viral polyproteins into their individual protein units. For many viruses, gene expression begins with transcription. For certain RNA viruses, such as hepatitis C virus (HCV), the first step in gene expression is translation of the viral RNA. Many viruses encode proteins that execute or abet certain of these steps in gene expression, and these proteins can serve as drug targets.
Genome replication is the next stage of the cycle. This stage requires a supply of ribonucleoside triphosphates for RNA viruses and deoxyribonucleoside triphosphates for DNA viruses. For DNA viruses, the generation of these deoxyribonucleoside triphosphates occurs via two pathways: the salvage pathway, which employs the pharmacologically relevant enzyme thymidine kinase, and the de novo pathway, which includes the enzyme thymidylate kinase. Nucleoside triphosphates are incorporated into new viral genomes by a viral or cellular polymerase (see Chapter 39, Pharmacology of Cancer: Genome Synthesis, Stability, and Maintenance, for more detail on nucleotide metabolism). In the case of herpes simplex viruses 1 and 2 (which will be collectively referred to as HSV), the generation of deoxyribonucleoside triphosphates includes phosphorylation of nucleosides via the salvage pathway by a viral thymidine kinase; a viral DNA polymerase then adds deoxyribonucleoside triphosphates to the growing DNA genome. Exploitation of this two-step process has led to the development of some of the most effective and safe antivirals currently available, because differences between human and viral kinases and polymerases allow drugs to take advantage of two different steps in a single pathway. For many viruses, genome replication requires other kinds of proteins. One example is the HCV NS5A protein.
Viral proteins that are synthesized intracellularly assemble with viral genomes within the host cell in a process known as assembly. For a number of viruses, assembly is followed by a process known as viral maturation, which is essential for newly formed virions to become infectious. This process typically involves cleavage of viral polyproteins by proteases. For some viruses, maturation occurs within the host cell; for others, such as HIV, it occurs outside the host cell. Viruses egress from the cell either by cell lysis or by budding through the cell membrane. For influenza viruses, the newly formed virions require an additional step of release from the extracellular surface of the host cell membrane.
In summary, nearly all viruses replicate via the following stages: attachment, entry, uncoating, gene expression, genome replication, assembly, and egress. Some viruses have additional stages such as maturation and release. The stages of retrovirus infection occur in a different order from those of most other viruses, and retroviruses have additional steps and stages in their life cycle. For example, genome replication of HIV includes the additional step of integration, in which the viral genome is incorporated into the host genome (Fig. 38-2). Specific host and/or viral proteins are involved in each of these stages. Differences between viral and host proteins at any of these stages can be targeted for antiviral therapy.
Different viruses have vastly different arrays of genes. Some, such as hepatitis B virus (HBV), have compact genomes that encode only coat proteins and a few proteins used mainly in gene expression and genome replication. Others, such as herpesviruses, encode scores of proteins that perform many different functions. The viral proteins that most frequently have served as targets for antiviral drugs are enzymes involved in genome replication, although multiple other proteins acting at different stages in the viral life cycle serve as targets, too.
 PHARMACOLOGIC CLASSES AND AGENTS
PHARMACOLOGIC CLASSES AND AGENTS
This section of the chapter reviews the mechanisms of antiviral drugs that target different stages of the viral life cycle. Understanding the mechanisms of antiviral drugs has relied strongly on studies of viruses that are resistant to these drugs. Resistance to an antiviral drug usually implies that the drug acts, at least in part, by interfering directly with a virus-specific process rather than by incapacitating a host cell process. In most cases, then, mutations that confer resistance to an antiviral drug affect the target(s) of that drug and suggest that the drug acts selectively against that target. Thus, mapping drug-resistance mutations to viral genes, along with demonstrating that the drug interacts with the product of that viral gene in a way that explains drug action, has been a major method by which the drug mechanisms described below have been elucidated.
Inhibition of Viral Attachment and Entry
All viruses must infect cells to replicate. Therefore, inhibiting the initial stages of viral attachment and entry provides a conceptual “preventive” measure against infection. Drugs that act at these stages do not need to enter cells, which can be an advantage. Two anti-HIV drugs, maraviroc and enfuvirtide (T-20), act at these stages. Both drugs have unusual properties for antiviral agents: maraviroc targets a host protein rather than a viral protein, and enfuvirtide is a peptide.
Maraviroc targets the chemokine receptor CCR5, which is a host plasma membrane protein. The development of maraviroc stemmed from clinical studies of individuals who had been exposed repeatedly to HIV, yet did not develop AIDS. It was found that some of these individuals have a deletion in the CCR5 gene. Absence of the CCR5 gene product prevents infection by the strains of HIV that are most frequently transmitted between individuals. The deletion also causes increased risk of clinical manifestations from West Nile disease but otherwise appears to have little negative impact on human health. Drug companies performed screens for compounds that could prevent binding of chemokines to CCR5 and chemically modified the leading candidate compounds to optimize their pharmacodynamic and pharmacokinetic properties. (Such “target-based screens” had earlier achieved success in the development of anti-HIV nonnucleoside reverse transcriptase inhibitors; see Box 38-1.) Maraviroc, the end result of this process, blocks infection of HIV strains that use CCR5 for attachment and entry (Fig. 38-3). However, maraviroc is not active against HIV strains that use the CXCR4 receptor. It is approved for use in combination with other anti-HIV drugs in patients who have undetectable levels of CXCR4-using virus (which requires a genotypic or phenotypic diagnostic test of viral tropism).
BOX 38-1 Development of Nonnucleoside Reverse Transcriptase Inhibitors and CCR5 Antagonist |
The nonnucleoside reverse transcriptase inhibitors (NNRTIs) were discovered by using target-based, high-throughput screening methods. The gene encoding HIV RT was overexpressed in E. coli, and large amounts of RT were purified and used in an RT assay that could be easily automated. Using this assay, many thousands of compounds were screened for the ability to inhibit RT. Candidate compounds were then tested for specificity in a counter-screen by checking their ability to inhibit an unrelated polymerase. The compounds that emerged were chemically modified to improve their stability, pharmacokinetics, and toxicity profile. This process eventually yielded NNRTIs that are highly specific, inhibiting HIV-1 RT at low concentrations while not inhibiting even the RT of the closely related virus HIV-2. The CCR5 antagonist maraviroc was also developed using a target-based, high-throughput screen. In this case, the assay was designed to discover lead compounds that prevented the binding of endogenous ligands (chemokines) to CCR5. As with the development of the NNRTIs, the lead compounds were then tested for specificity against CCR5 and chemically modified to optimize their potency, antiviral activity, pharmacokinetics, and toxicity profile. The end result was maraviroc, a selective CCR5 antagonist that is used in combination antiretroviral treatment of adults infected with CCR5-tropic HIV-1. |

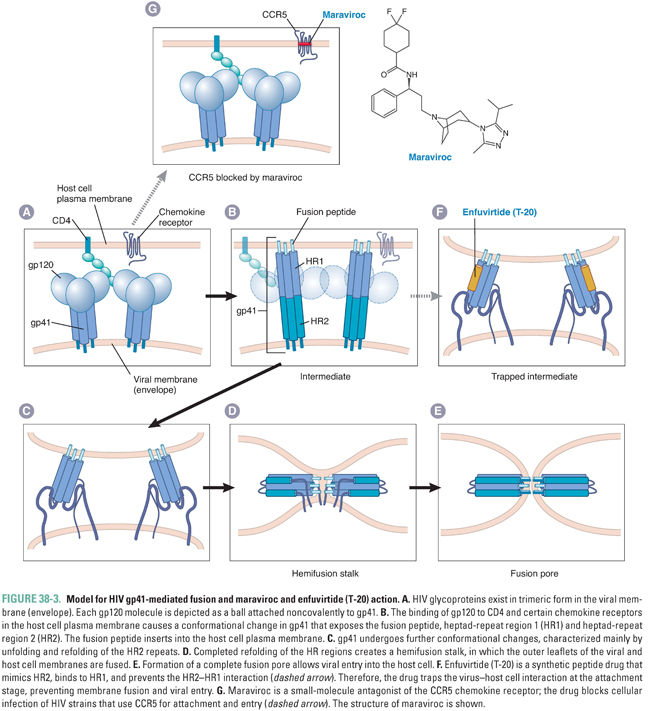
Enfuvirtide, also known as T-20, is a peptide that is structurally similar to a segment of gp41, the HIV protein that mediates membrane fusion. The proposed mechanism for gp41-mediated membrane fusion and T-20 action is illustrated in Figure 38-3. In the native virion, gp41 is held in a conformation that prevents it from fusing membranes or binding T-20. Attachment of HIV to its cellular receptors triggers a conformational change in gp41 that exposes a segment that can insert into membranes (fusion peptide), a heptad repeat region (HR1), and a second heptad repeat region mimicked by T-20 (HR2). The gp41 then refolds, so that the HR2 segments bind directly to the HR1 segments. This refolding brings the virion envelope and the cell membrane into close proximity, allowing membrane fusion to occur (by mechanisms that remain incompletely understood). When T-20 is present, however, the drug binds to the exposed HR1 segments and prevents the refolding process, thereby preventing fusion of the HIV envelope with the host cell membrane.
Enfuvirtide is approved for use in combination with other anti-HIV drugs in patients whose HIV infection has not been controlled by first-line anti-HIV medications. Enfuvirtide is rarely used in clinical practice as it must be administered by twice-daily subcutaneous injections. Local injection site reactions are common and have been associated with bacterial pneumonia and a hypersensitivity reaction. Resistance mutations appear to emerge rapidly in patients who have detectable viremia despite enfuvirtide treatment.
The adamantanes amantadine and rimantadine (structures in Fig. 38-4) are inhibitors of viral uncoating that are active exclusively against influenza A virus (and not against influenza B or C viruses). Influenza viruses (in particular influenza A viruses) cause 50 million illnesses each year in the United States and tens to hundreds of thousands of hospitalizations. Current vaccines generally are not completely efficacious and are useful for only a year at a time due to the rapid evolution of new influenza strains. Thus, there has long been a considerable need for anti-influenza drugs.
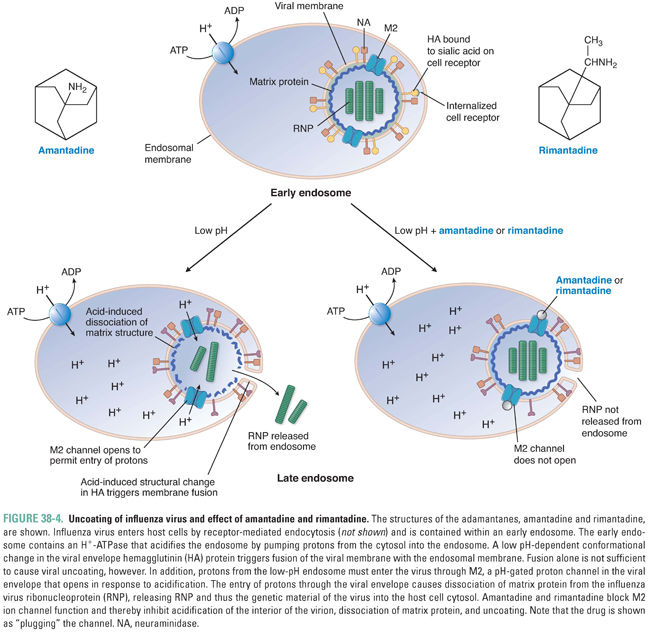
A well-supported model for the mechanism of action of the adamantanes is diagrammed in Figure 38-4. Influenza virions enter cells via receptor-mediated endocytosis and are internalized into endosomes (see Chapter 1, Drug–Receptor Interactions). As endosomes acidify because of the action of an endosomal proton pump, two events occur. First, the conformation of the viral envelope protein hemagglutinin changes drastically. This conformational change permits fusion of the influenza virus envelope with the endosome membrane (see the above discussion of HIV-mediated membrane fusion). By itself, this action could liberate viral ribonucleoprotein (including the virion’s RNA genome), but that would not be sufficient to permit its transcription: a second pH-dependent event within the virion is also required. This entails the influx of protons through a proton channel called M2 in the viral envelope, which causes dissociation of the virion matrix protein from the rest of the ribonucleoprotein. Amantadine and rimantadine inhibit the influx of protons through M2. As hydrophobic molecules with a positive charge at one end, these drugs resemble blockers of cellular ion channels (see Chapters 12, Local Anesthetic Pharmacology, and 24, Pharmacology of Cardiac Rhythm), and indeed the adamantanes appear to simply “plug” (physically occlude) the channel.
Amantadine can cause light-headedness and difficulty concentrating; these adverse effects are likely due to its effects on host ion channels. Indeed, the unintended effects of amantadine on host channels likely account for this drug’s other therapeutic use—the treatment of Parkinson’s disease (see Chapter 14, Pharmacology of Dopaminergic Neurotransmission). Rimantadine is an analogue of amantadine that has a similar antiviral mechanism and fewer adverse effects compared to amantadine, especially the neurological effects that can be problematic in the elderly. However, resistance to adamantanes develops rapidly, and resistant viruses retain nearly complete replication capacity (fitness) and pathogenicity. Indeed, adamantanes are no longer recommended for use in the United States due to the high rates of drug resistance, and they have been supplanted by neuraminidase inhibitors (see “Inhibition of Viral Release”).
Inhibition of Viral Gene Expression
HCV causes serious liver disease and more deaths in the United States than does HIV. After entry of HCV into the cell and uncoating in endosomes, the first step in HCV gene expression is translation of the viral genome. The translation product is a so-called polyprotein that encompasses proteins from multiple viral genes, and the next, crucial step in HCV gene expression is cleavage of the polyprotein into individual proteins (Fig. 38-5A). Certain of these individual proteins then replicate HCV RNA, producing new genomes to be translated and cleaved. Other viral proteins assemble RNA-containing viral particles that are then released from the cell. HCV encodes a protease, called NS3/4A, which is essential for several of the cleavages of the polyprotein. Additionally, this enzyme may play a role in counteracting host innate immune responses, particularly those elicited by interferon alpha.
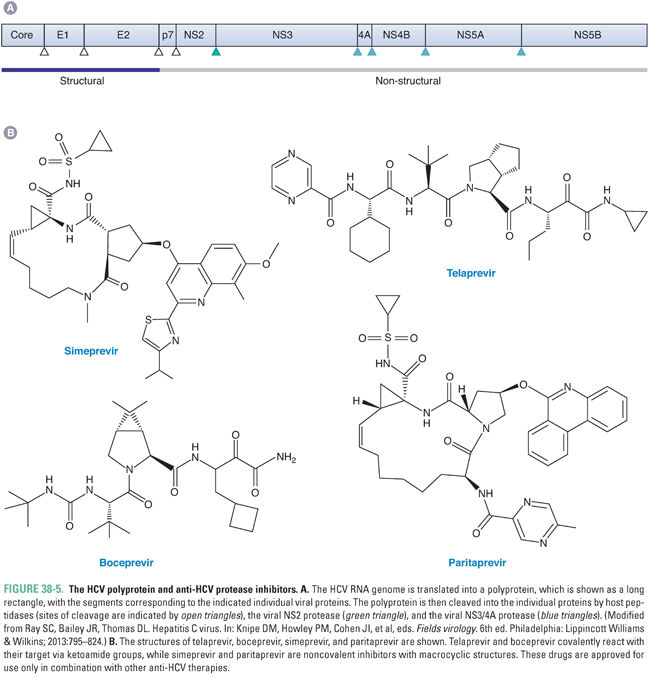
Given the success of drugs targeting the HIV protease, which is required for the maturation step in its virus’ life cycle (see “Inhibition of Viral Maturation”), there was considerable interest in developing drugs that target the HCV NS3/4A protease. In 2011, two such drugs, telaprevir (which is no longer marketed) and boceprevir (Fig. 38-5B), were the first agents that act directly against HCV (direct-acting antivirals, DAAs) to be approved by the US Food and Drug Administration (FDA). In 2013 and 2014, respectively, the NS3/4A inhibitors simeprevir and paritaprevir (Fig. 38-5B), which can be administered once a day, were approved. All four drugs were developed by an iterative approach similar to that used to discover inhibitors of the HIV protease (see discussion below and Fig. 38-11), although, among other differences, telaprevir and boceprevir covalently react with their target via ketoamide groups rather than binding tightly via noncovalent interactions like the anti-HIV drugs. Simeprevir and paritaprevir are noncovalent inhibitors with macrocyclic structures that increase the affinity of binding by decreasing entropic effects. All four drugs inhibit HCV NS3/4A protease with higher potency than they do human proteases.
None of the NS3/4A inhibitors are approved for use as monotherapies. Telaprevir, boceprevir, and simeprevir are approved for use in combination with interferon alpha (modified by pegylation to permit less frequent dosing) and ribavirin (see below for further information on these two agents), which was the previous standard of care for HCV therapy. The addition of the protease inhibitors results in a substantially higher rate of “sustained virological response,” which is tantamount to cure of HCV infection. However, these interferon-based combinations entail 6 months or more of treatment and are limited by the development of resistance in some patients, by the requirement for injection of interferon alpha, and by major adverse effects of interferon alpha and ribavirin. Relative to telaprevir and boceprevir, simeprevir and paritaprevir represent an advance, in that they can be administered once a day, have a more favorable adverse effect profile, and are also approved for use in combination with particular DAAs that inhibit HCV genome replication (see below)—simeprevir with sofosbuvir; paritaprevir with ombitasvir and dasabuvir—in a regimen that requires no interferon alpha and, in many cases, only 3 months of treatment. However, each of the protease inhibitors is approved for only certain genotypes of HCV, and resistance can arise. Indeed, it is strongly recommended that testing for resistance occur prior to use of simeprevir. Paritaprevir is formulated together with the anti-HIV protease inhibitor ritonavir (see below), which increases paritaprevir serum levels by blocking its hepatic metabolism and is not recommended for use in patients with decompensated liver disease. Depending on the HCV subtype, the DAA combination containing paritaprevir may be co-administered with ribavirin, which has its own toxicities. Newer NS3/4A protease inhibitors are under development.
Inhibition of Viral Genome Replication—Polymerase Inhibitors
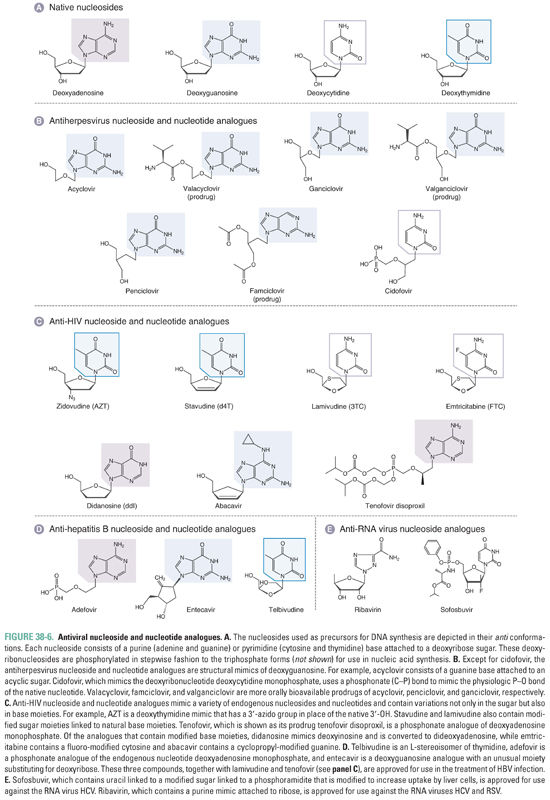
The vast majority of drugs that inhibit viral genome replication inhibit a polymerase. Most viruses encode their own polymerases. These enzymes are required to replicate the viral genomes and differ in various ways from human polymerases, making them excellent targets for antiviral drugs. Viruses expressing polymerases that have been successfully targeted to yield drugs approved by the FDA include certain human herpesviruses, HIV, HBV, and HCV. Most of these drugs are so-called nucleoside analogues (Fig. 38-6). Several, as discussed below, are nonnucleoside inhibitors of a polymerase. The latter do not structurally resemble physiologic nucleosides.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


