Louise C. Ivers and Edward T. Ryan

More than one billion people worldwide are infected with parasites. Parasites of medical importance include protozoa (such as the organisms that cause malaria, toxoplasmosis, giardiasis, amebiasis, leishmaniasis, and trypanosomiasis) and helminths (“worms”). Worms that infect humans include cestodes (“flatworms” or “tapeworms,” such as the worm that causes taeniasis), nematodes (“roundworms,” which cause filariasis, strongyloidiasis, and ascariasis), and trematodes (“flukes,” such as the worms that cause schistosomiasis).
Ideally, antiparasitic drugs should be targeted to structures or biochemical pathways present or accessible only in parasites. Many antiparasitic drugs act by unknown or poorly defined mechanisms of action, however. This chapter focuses on a number of the better defined agents, including those active against Plasmodia species (which cause malaria), Entamoeba histolytica (which causes amebiasis), and Onchocerca volvulus (which causes onchocerciasis, a filarial infection referred to as river blindness). In each of these cases, antiparasitic agents interfere with metabolic requirements of the parasite: the dependence of malarial plasmodia on heme metabolism, the dependence of luminal parasites on specific fermentation pathways, and the dependence of helminths on neuromuscular activity. These three examples are not all-inclusive but rather emphasize opportunities to use or design pharmacologic agents to interrupt metabolic requirements specific to parasites.

Each year, approximately 200 million individuals in more than 90 countries develop malaria, and over a half million individuals die of malaria. Malaria is the most important parasitic disease of humans and one of the most important infections of humans. Human malaria is caused by one of five species of plasmodial parasites: Plasmodium falciparum, P. vivax, P. ovale, P. malariae, and P. knowlesi. The most serious type of malaria is that caused by P. falciparum.
 Binata, a 3-year-old girl living in a remote area of a central African country, is in good health when, one day, she begins to feel hot, has sweats and shaking chills, stops eating, and becomes intermittently listless and lethargic. Several days later, these symptoms climax in a seizure and coma, prompting Binata’s parents to rush her to the local health care clinic. In the clinic, the unconscious child’s neck is supple, but she is febrile to 103°F. Her lungs are clear to auscultation, and there is no rash. A smear of Binata’s peripheral blood discloses P. falciparum ring trophozoites in approximately 10% of her erythrocytes. Binata is given the only antimalarial medicines available at the clinic, chloroquine and pyrimethamine/sulfadoxine; however, the child does not improve, and she dies within 24 hours.
Binata, a 3-year-old girl living in a remote area of a central African country, is in good health when, one day, she begins to feel hot, has sweats and shaking chills, stops eating, and becomes intermittently listless and lethargic. Several days later, these symptoms climax in a seizure and coma, prompting Binata’s parents to rush her to the local health care clinic. In the clinic, the unconscious child’s neck is supple, but she is febrile to 103°F. Her lungs are clear to auscultation, and there is no rash. A smear of Binata’s peripheral blood discloses P. falciparum ring trophozoites in approximately 10% of her erythrocytes. Binata is given the only antimalarial medicines available at the clinic, chloroquine and pyrimethamine/sulfadoxine; however, the child does not improve, and she dies within 24 hours.
Questions
1. Why did Binata die?
2. Why did Binata not improve after receiving antimalarial drugs?
3. How often does a child die of malaria?
 Mr. G is a 36-year-old married software engineer who was born and raised in India. He comes to the United States and is completely well for 6 months. He then begins to experience episodes of fever, headache, and body aches. One week later, he goes to his physician, who examines a smear of Mr. G’s blood, diagnoses malaria, and prescribes chloroquine for treatment. Therapy with chloroquine resolves his symptoms completely. However, Mr. G notes recurrence of fevers and the other symptoms 3 months later and returns to his doctor’s office.
Mr. G is a 36-year-old married software engineer who was born and raised in India. He comes to the United States and is completely well for 6 months. He then begins to experience episodes of fever, headache, and body aches. One week later, he goes to his physician, who examines a smear of Mr. G’s blood, diagnoses malaria, and prescribes chloroquine for treatment. Therapy with chloroquine resolves his symptoms completely. However, Mr. G notes recurrence of fevers and the other symptoms 3 months later and returns to his doctor’s office.
Questions
4. What is a likely explanation for the return of Mr. G’s fever?
5. How can Mr. G’s treatment be modified so that his illness will not return?
Physiology of Malarial Plasmodia
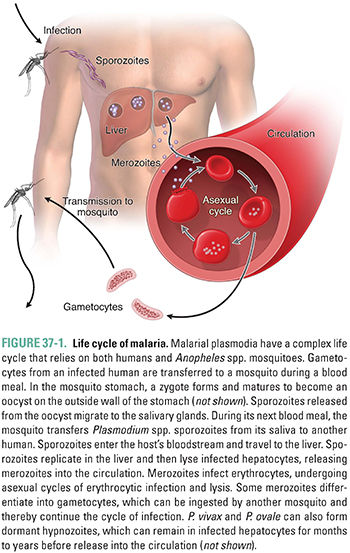
The life cycle of malaria involves a parasite, a mosquito vector, and a human host (Fig. 37-1). An Anopheles spp. mosquito can ingest sexual forms of malarial parasites (gametocytes) when taking a blood meal from an infected human. After fusion of the male and female gametocytes and maturation of the zygote within the mosquito, sporozoites are released from an oocyst. The sporozoites, which migrate to the mosquito’s salivary glands, can be inoculated into the blood of another human host during a subsequent blood meal. In the human, sporozoites leave the blood and multiply in the liver, forming tissue schizonts. This exoerythrocytic hepatic stage is asymptomatic. In a typical P. falciparum infection, 1–12 weeks after the infective bite, the liver cells release parasites into the bloodstream as merozoites. A single sporozoite can produce more than 30,000 merozoites. Merozoites invade erythrocytes, multiply asexually, and form blood schizonts. This is the erythrocytic stage. Infected erythrocytes eventually rupture, releasing another generation of merozoites that continue the erythrocytic cycle. Rare merozoites also mature into gametocytes. Ingestion of these circulating gametocytes by an appropriate mosquito completes the life cycle. The clinical symptoms of malaria, most distinctively fever, are caused by the intravascular lysis of erythrocytes and subsequent release of merozoites into the blood. The fevers that Binata and Mr. G experienced were associated with these hemolytic episodes. Binata, unfortunately, developed cerebral malaria due to P. falciparum.
P. falciparum-infected erythrocytes express “knobs” on their surface that are composed of both host and parasite proteins. Parasite proteins include PfEMP-1, a protein family composed of approximately 100–150 gene products that mediate attachment of infected erythrocytes to cellular receptors—including CD36, ICAM-1, ELAM-1, and chondroitin sulfate—on endothelial surfaces in the human host. This intravascular binding during a malarial episode occurs only during P. falciparum infection and contributes to intravascular “sludging” of erythrocytes. Endothelial attachment lessens the amount of time during which infected erythrocytes circulate systemically, thereby decreasing the likelihood that infected erythrocytes will be cleared via splenic sequestration. Sludging also accounts, in large part, for the pathophysiology of malaria caused by P. falciparum. Sludging can affect any organ, including the brain, lungs, and kidneys; damage to these organs leads to tissue hypoxia, focal necrosis, and hemorrhage. In Binata’s case, the brain was involved (so-called cerebral malaria).
Untreated, cerebral malaria is almost uniformly fatal, and, even with optimal treatment, cerebral malaria has a case fatality rate exceeding 20%. Binata was treated with two drugs that have historically been quite important in treating individuals with malaria but which, unfortunately, are now ineffective in most places in the world because of widespread drug-resistant P. falciparum. Largely because of their low cost and availability, these drugs (chloroquine and a fixed combination of pyrimethamine and sulfadoxine) were widely used in many developing areas of the world to treat older children and adults with partial immunity to malaria, but these agents have little clinical use in treating nonimmune individuals such as Binata. Due to the increasing ineffectiveness of these older agents, it is now recommended that individuals in sub-Saharan Africa with malaria be treated with an artemisinin derivative in combination with a second agent (see below).
Unfortunately, Binata’s story has been all too common. On average, worldwide, a child dies of malaria every 75 seconds; of these deaths, more than 90% occur in sub-Saharan Africa, more than 90% occur in children under 5 years of age, and more than 95% are caused by P. falciparum infection. No pharmacologic agent has yet been developed that interferes with the recently elucidated role of PfEMP-1 in the endothelial attachment of malarially infected erythrocytes.
In Mr. G’s case, a peripheral blood smear showed P. vivax parasites inside his erythrocytes. Because P. falciparum and P. malariae infections involve only one cycle of hepatic cell invasion, drugs that eliminate these species from erythrocytes are usually sufficient to clear the infection. Unfortunately, P. vivax and P. ovale also have dormant hepatic forms (hypnozoites) that release merozoites over months to 1–2 years. Therefore, individuals infected with P. vivax or P. ovale should be treated with agents that are effective against not only blood-stage plasmodia but also liver-stage parasites (see below). Because chloroquine does not eliminate hepatic forms of P. vivax and P. ovale, Mr. G’s P. vivax infection recurred.
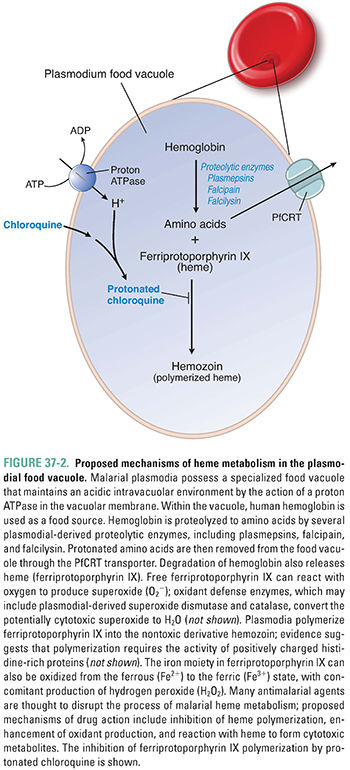
Plasmodia have a limited capacity for de novo amino acid synthesis; instead, they rely on amino acids released from ingested host hemoglobin molecules. Within red blood cells, plasmodia degrade hemoglobin in a digestive vacuole, which is an elaborate lysosome with an acidic pH (Fig. 37-2). Hemoglobin is sequentially degraded to its constituent amino acids by plasmodial aspartic proteases (plasmepsins), a cysteine protease (falcipain), and a metalloprotease (falcilysin). Degradation of hemoglobin releases protonated basic amino acids and a toxic heme metabolite, ferriprotoporphyrin IX. Ferriprotoporphyrin IX is detoxified by polymerization to crystalline hemozoin. If ferriprotoporphyrin IX does not polymerize, it causes lysosomal membrane damage and toxicity to the malarial parasite. Quinoline antimalarials (see below) are believed to act by inhibiting heme polymerization, thereby creating an environment that is toxic to intraerythrocytic plasmodia.
Malarial plasmodia also possess mitochondria with a tiny genome (approximately 6 kb) that encodes only three cytochromes (large protein complexes involved in electron transport and oxidative phosphorylation). These cytochromes, together with a number of mitochondrial-targeted proteins derived from the plasmodial nuclear genome, make up a rudimentary electron transport chain similar in organization to that found in mammals (Fig. 37-3). In this electron transport chain, integral proteins of the mitochondrial inner membrane are reduced and then oxidized as they transport electrons from one intermediate protein to another. The energy liberated by electron transport is used to drive proton pumping across the mitochondrial membrane, and the energy stored in the proton gradient drives ATP synthesis. In this electron transport chain, oxygen is the final electron acceptor, resulting in the reduction of oxygen to water.
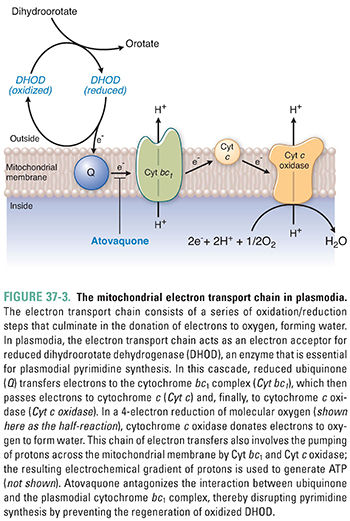
Plasmodia derive most of their ATP directly from glycolysis and probably do not use mitochondrial electron transport as a significant source of energy. However, plasmodia do rely on electron transport for the oxidation of key enzymes involved in nucleotide synthesis. For example, dihydroorotate dehydrogenase (DHOD), the enzyme that mediates an early step in pyrimidine synthesis (see Chapter 39, Pharmacology of Cancer: Genome Synthesis, Stability, and Maintenance), catalyzes the oxidation of dihydroorotate to orotate. As part of this reaction, DHOD is reduced, and the enzyme must be reoxidized before it can continue with another cycle of catalysis. Ubiquinone, an integral membrane protein located near the beginning of the electron transport chain, accepts electrons from reduced DHOD, thus regenerating the oxidized form of DHOD necessary for pyrimidine synthesis. Because plasmodia depend on de novo pyrimidine synthesis for DNA replication, interrupting the ability of ubiquinone to oxidize DHOD can disrupt plasmodial DNA replication (see below).
Pharmacology of Antimalarial Agents
The currently available antimalarial agents target four physiologic pathways in plasmodia: heme metabolism (chloroquine, quinine, mefloquine, and artemisinin), electron transport (primaquine and atovaquone), protein translation (doxycycline, tetracycline, and clindamycin), and folate metabolism (sulfadoxine-pyrimethamine and proguanil). The following section discusses the pharmacologic agents that target these pathways.
Clinically, antimalarials can be classified into agents used for prophylaxis (to prevent malaria in individuals residing in or traveling through a malaria zone), agents used for treating individuals with acute blood-stage malaria, and agents used to eliminate hypnozoite liver-stage malarial infections. Generally, agents used for prophylaxis must be well tolerated and easy to administer.
For many centuries, agents that disrupt intraerythrocytic malarial parasites have been the foundation of antimalarial treatment regimens. Most of these compounds are congeners of quinoline and, as a result, are all believed to possess similar mechanisms of action. Artemisinin, discussed at the end of this section, is also thought to act by inhibiting heme metabolism, although its structure is different from that of the quinolines.
Chloroquine
For the past 2,000 years, humans have used the roots of Dichroa febrifuga or the leaves of hydrangea in the treatment of individuals with malaria. More recently, the bark of the cinchona tree was found to be a more effective remedy. In all these plants, a quinoline compound is the pharmacologically active antiplasmodial agent. Chloroquine, a 4-aminoquinoline, was introduced in 1935 for use in the treatment of malaria. Chloroquine is a weak base that, in its neutral form, freely diffuses across the membrane of the parasite’s food vacuole. Once inside the acidic environment of the vacuole, chloroquine is rapidly protonated, making it unable to diffuse out of the vacuole. As a result, protonated chloroquine accumulates to high concentrations inside the parasite’s food vacuole, where it binds to ferriprotoporphyrin IX and inhibits the polymerization of this heme metabolite. Accumulation of unpolymerized ferriprotoporphyrin IX leads to oxidative membrane damage and is toxic to the parasite. Chloroquine thus poisons the parasite by preventing the detoxification of a toxic product of hemoglobin catabolism (Fig. 37-2).
Chloroquine is concentrated by as much as 100-fold in parasitized erythrocytes compared to uninfected erythrocytes. In addition, the concentration of chloroquine required to alkalinize lysosomes of mammalian cells is much higher than that needed to raise the pH in malarial food vacuoles. Therefore, chloroquine is relatively nontoxic to humans, although the drug commonly causes pruritus in darkly pigmented individuals, and it can exacerbate psoriasis and porphyria. Taken in supratherapeutic doses, however, chloroquine can cause vomiting, retinopathy, hypotension, confusion, and death. In fact, chloroquine is used globally in suicides each year (largely because it is inexpensive, available, and toxic at high doses), and accidental ingestion by children can be fatal.
When initially introduced, chloroquine was a first-line drug used against all types of malaria; however, it is now ineffective against most strains of P. falciparum in Africa, Asia, and South America (Fig. 37-4). Hypotheses regarding the mechanisms responsible for chloroquine resistance are based on the finding that chloroquine-resistant plasmodia accumulate less chloroquine inside food vacuoles than chloroquine-sensitive plasmodia do. In the food vacuole, protonated amino acids are generated by the parasite as it degrades hemoglobin. These protonated amino acids exit the lysosome by means of a transmembrane protein called PfCRT, encoded by pfcrt on P. falciparum chromosome 7. A number of mutations in PfCRT have been associated with chloroquine resistance; for example, a substitution of threonine for lysine at position 76 (K76T) is highly correlated with chloroquine resistance. This mutated PfCRT probably pumps protonated chloroquine out of the food vacuole. This altered pump action could also be detrimental to the parasite, perhaps because of altered amino acid export and/or changes in vacuole pH. Many P. falciparum strains with mutations in pfcrt carry a second mutation in the gene pfmdr1 encoding Pgh1, a food vacuole membrane protein involved in pH regulation. It is speculated that this second mutation provides a “corrective” action that allows chloroquine-resistant P. falciparum to continue growth in the presence of a pfcrt mutation.
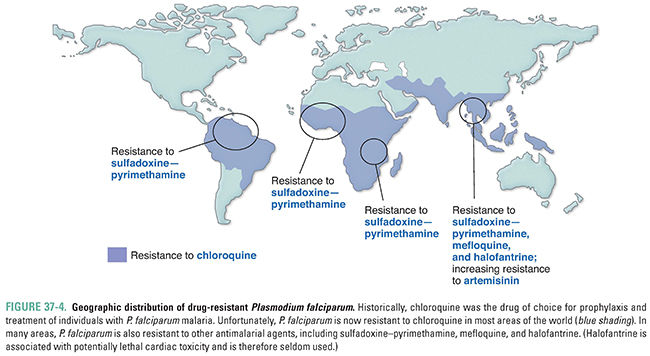
Strains of P. vivax with decreased susceptibility to chloroquine are now reported with increasing frequency in areas of Papua New Guinea, Indonesia, and other focal areas of Oceania and Latin America, although the exact mechanism of decreased susceptibility to chloroquine in these strains has not been fully established. Despite concerns regarding increasing resistance, chloroquine remains a drug of choice for treating most individuals with malaria caused by P. vivax, P. ovale, P. malariae, and P. knowlesi and by chloroquine-sensitive strains of P. falciparum. It can also be used prophylactically to prevent malaria caused by sensitive strains of plasmodia.
Quinine and Quinidine
Quinine is an alkaloid that consists of a quinoline ring linked by a secondary carbinol to a quinuclidine ring. Its optical isomer, quinidine, has identical pharmacologic actions. Because of quinine’s structural similarity to other antimalarial quinolines, quinine is thought to attack plasmodia by the mechanism described above. Quinine has also been shown to intercalate into DNA through hydrogen bonding, thus inhibiting DNA strand separation, transcription, and translation. The overall effect is a decrease in the growth and replication of the erythrocytic form of plasmodia. Quinine and quinidine have been used to treat individuals with acute blood-stage malaria but are not used prophylactically. Use of quinine can cause cinchonism, a syndrome that includes tinnitus, deafness, headaches, nausea, vomiting, and visual disturbances. Quinine and quinidine can also prolong the cardiac QT interval (see Chapter 24, Pharmacology of Cardiac Rhythm).
Mefloquine
Mefloquine is a quinoline compound that is structurally related to other antimalarial agents. Unlike quinine, mefloquine does not bind to DNA. Its exact mechanism of action is unknown, although mefloquine appears to disrupt polymerization of heme in intraerythrocytic malarial parasites. Mefloquine has a number of adverse effects, including nausea, cardiac conduction abnormalities (including bradycardia, prolongation of the QT interval, and arrhythmia), and neuropsychiatric effects, including vivid dreams/nightmares, insomnia, anxiety, depression, hallucinations, seizures, and, rarely, psychosis. The mechanism(s) responsible for these adverse effects is unknown. In 2013, the US Food and Drug Administration (FDA) issued a black box warning related to these neurologic and psychiatric adverse effects. Mefloquine can be used both therapeutically and prophylactically. Strains of P. falciparum resistant to both chloroquine and mefloquine have been reported in areas of Southeast Asia.
Artemisinin
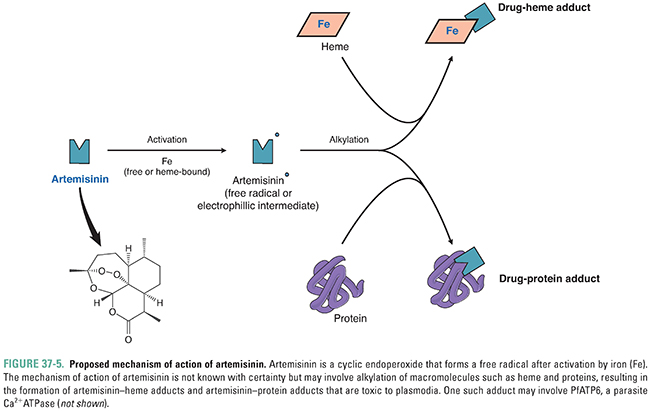
Artemisinin, from the wormwood plant Artemisia annua, has been used in China (where it is known as qinghao) for centuries in the treatment of individuals with fever. Artemisinin derivatives have now become the first-line drug for treating individuals with falciparum malaria. The compound is both a sesquiterpene lactone and a cyclic endoperoxide. When activated by free or heme-bound iron, it forms a carbon-centered free-radical compound (Fig. 37-5). This free radical has the ability to alkylate many proteins as well as heme. The mechanism of specificity of the drug for plasmodia-infected erythrocytes is unknown—potential sources of specificity include artemisinin’s requirement for heme for free radical formation and artemisinin’s preferential accumulation in plasmodia. Drug action may relate to free radical production in the food vacuole of the parasite and subsequent inhibition of PfATP6, the parasite Ca2+ ATPase that is the ortholog of the mammalian SERCA calcium pump (see Chapter 25, Pharmacology of Cardiac Contractility). Administration of artemisinin and its derivatives (artesunate, artemether, dihydroartemisinin) is associated with a rapid decrease in the level of malaria parasites in the blood of an infected individual and rapid resolution of symptoms in patients with blood-stage malaria. Unlike many of the other antimalarials, artemisinins affect blood-stage gametocytes and thus can decrease transmission of malaria from an infected human. Artemisinin is not effective as a prophylactic agent against malaria.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


