INTRODUCTION
Blood carries oxygen and nutrients to tissues and takes metabolic waste products away from tissues. Humans have developed a well-regulated system of hemostasis to keep the blood fluid and clot-free in normal vessels and to form a localized plug rapidly in injured vessels. Thrombosis describes a pathologic state in which normal hemostatic processes are activated inappropriately. For example, a blood clot (thrombus) may form as the result of a relatively minor vessel injury and occlude a branch of the vascular tree. This chapter presents the normal physiology of hemostasis, the pathophysiology of thrombosis, and the pharmacology of drugs that can be used to prevent or reverse a thrombotic state. Drugs introduced in this chapter are used to treat a variety of cardiovascular diseases, such as deep vein thrombosis, stroke, and myocardial infarction.
 Mr. S, a 55-year-old man with a history of hypertension and cigarette smoking, is awakened in the middle of the night with substernal chest pressure, sweating, and shortness of breath. He calls 911 and is taken to the emergency department. An electrocardiogram shows deep T-wave inversions in leads V2 to V5. A cardiac biomarker panel shows a troponin T level of 3.2 μg/L (normal, <0.01 μg/L), consistent with myocardial infarction. He is treated with intravenous nitroglycerin, aspirin, unfractionated heparin, and eptifibatide, but his chest pain persists. He is taken to the cardiac catheterization laboratory, where he is found to have a 90% mid-LAD (left anterior descending artery) thrombus with sluggish distal flow. He undergoes successful angioplasty and stent placement. At the time of stent placement, an oral loading dose of clopidogrel is administered. The heparin is stopped, intravenous eptifibatide is continued for 18 more hours, and he is transferred to the telemetry ward. Six hours later, Mr. S is noted to have an expanding hematoma (an area of localized hemorrhage) in his right thigh below the arterial access site. The eptifibatide is stopped and pressure is applied to the access site, and the hematoma ceases to expand. He is discharged 2 days later with prescriptions that include clopidogrel and aspirin, which are administered to prevent thrombosis of the stent.
Mr. S, a 55-year-old man with a history of hypertension and cigarette smoking, is awakened in the middle of the night with substernal chest pressure, sweating, and shortness of breath. He calls 911 and is taken to the emergency department. An electrocardiogram shows deep T-wave inversions in leads V2 to V5. A cardiac biomarker panel shows a troponin T level of 3.2 μg/L (normal, <0.01 μg/L), consistent with myocardial infarction. He is treated with intravenous nitroglycerin, aspirin, unfractionated heparin, and eptifibatide, but his chest pain persists. He is taken to the cardiac catheterization laboratory, where he is found to have a 90% mid-LAD (left anterior descending artery) thrombus with sluggish distal flow. He undergoes successful angioplasty and stent placement. At the time of stent placement, an oral loading dose of clopidogrel is administered. The heparin is stopped, intravenous eptifibatide is continued for 18 more hours, and he is transferred to the telemetry ward. Six hours later, Mr. S is noted to have an expanding hematoma (an area of localized hemorrhage) in his right thigh below the arterial access site. The eptifibatide is stopped and pressure is applied to the access site, and the hematoma ceases to expand. He is discharged 2 days later with prescriptions that include clopidogrel and aspirin, which are administered to prevent thrombosis of the stent.
Questions
1. How did a blood clot arise in Mr. S’s coronary artery?
2. How do aspirin, heparin, clopidogrel, and eptifibatide act in the attempt to treat Mr. S’s blood clot and to prevent recurrent thrombus formation?
3. What accounts for the efficacy of eptifibatide (a platelet GPIIb–IIIa antagonist) in inhibiting platelet aggregation?
4. When the expanding hematoma was observed, could any measure other than stopping the eptifibatide have been used to reverse the effect of this agent?
5. If low-molecular-weight heparin had been used instead of unfractionated heparin, how would the monitoring of the patient’s coagulation status during the procedure have been affected?

An injured blood vessel must induce the formation of a blood clot to prevent blood loss and to allow healing. Clot formation must also remain localized to prevent widespread clotting within intact vessels. The formation of a localized clot at the site of vessel injury is accomplished in four temporally overlapping stages (Fig. 23-1). First, localized vasoconstriction occurs as a response to a reflex neurogenic mechanism and to the secretion of endothelium-derived vasoconstrictors such as endothelin. Immediately following vasoconstriction, primary hemostasis occurs. During this stage, platelets are activated and adhere to the exposed subendothelial matrix. Platelet activation involves both a change in shape of the platelet and the release of secretory granule contents from the platelet. The secreted granule substances recruit other platelets, causing more platelets to adhere to the subendothelial matrix and to aggregate with one another at the site of vascular injury. Primary hemostasis ultimately results in the formation of a primary hemostatic plug.
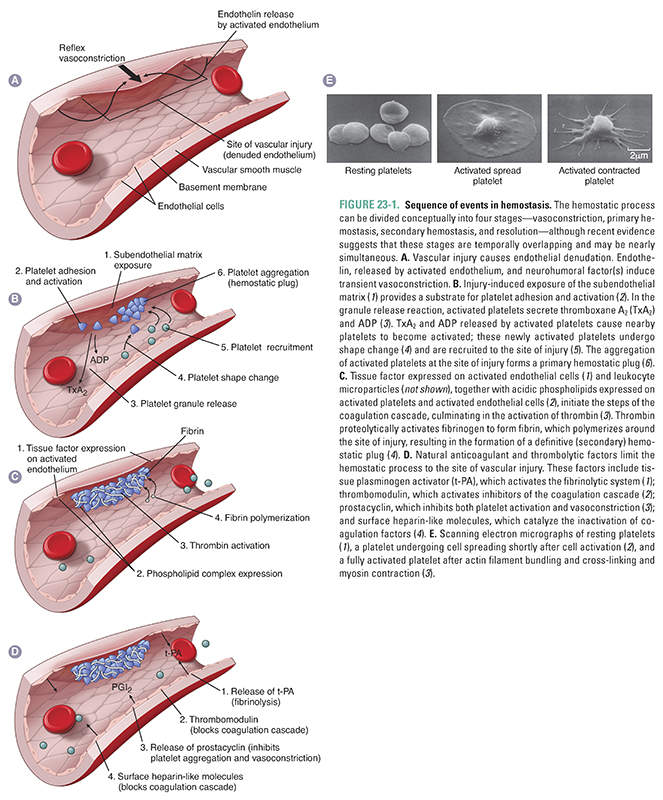
The goal of the final two stages of hemostasis is to form a stable, permanent plug. During secondary hemostasis, also known as the coagulation cascade, the activated endothelium and other nearby cells (see below) express a membrane-bound procoagulant factor called tissue factor, which complexes with coagulation factor VII to initiate the coagulation cascade. The end result of this cascade is the activation of thrombin, a critical enzyme. Thrombin serves two pivotal functions in hemostasis: (1) it converts soluble fibrinogen to an insoluble fibrin polymer that forms the matrix of the clot and (2) it induces more platelet recruitment and activation. Evidence indicates that fibrin clot formation (secondary hemostasis) overlaps temporally with platelet plug formation (primary hemostasis), and that each process reinforces the other. During the final stage, platelet aggregation and fibrin polymerization lead to the formation of a stable, permanent plug. In addition, antithrombotic mechanisms restrict the permanent plug to the site of vessel injury, ensuring that the permanent plug does not inappropriately extend to occlude the vascular tree.
Transient arteriolar vasoconstriction occurs immediately after vascular injury. This vasoconstriction is mediated by a poorly understood reflex neurogenic mechanism. Local endothelial secretion of endothelin, a potent vasoconstrictor, potentiates the reflex vasoconstriction. Because the vasoconstriction is transient, bleeding would resume if primary hemostasis were not activated.
The goal of primary hemostasis is to form a platelet plug that rapidly stabilizes vascular injury. Platelets play a pivotal role in primary hemostasis. Platelets are cell fragments that arise by budding from megakaryocytes in the bone marrow; these small, membrane-bound discs contain cytoplasm but lack nuclei. Glycoprotein receptors in the platelet plasma membrane are the primary mediators by which platelets are activated. Primary hemostasis involves the transformation of platelets into a hemostatic plug through three reactions: (1) adhesion, (2) the granule release reaction, and (3) aggregation and consolidation.
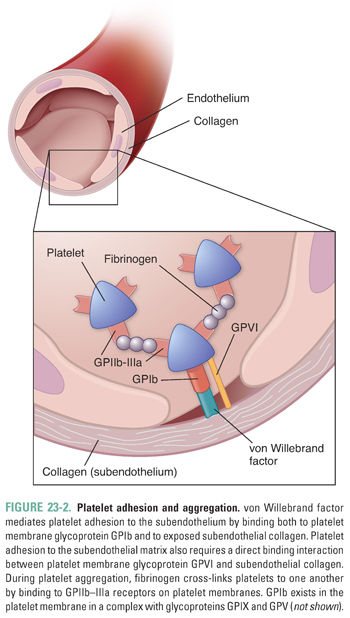
In the first reaction, platelets adhere to subendothelial collagen that is exposed after vascular injury (Fig. 23-2). This adhesion is mediated initially by two molecular interactions. First, von Willebrand factor (vWF), a large multimeric protein that is secreted by both activated platelets and the injured endothelium, binds both to surface receptors (especially glycoprotein Ib [GPIb]) on the platelet membrane and to the exposed collagen. This “bridging” action mediates adhesion of platelets to the collagen. Second, platelet glycoprotein VI (GPVI) interacts directly with collagen in the exposed vessel wall. Both the GPIb:vWF:collagen interaction and the GPVI:collagen interaction are required for initiation of primary hemostasis.
Platelet Granule Release Reaction
Adherent platelets undergo a process of activation (Fig. 23-3) during which the cells’ granule contents are released. The release reaction is initiated by agonist binding to cell surface receptors, which activates intracellular protein phosphorylation cascades and ultimately causes release of granule contents. Specifically, stimulation by adenosine diphosphate (ADP), epinephrine, and collagen leads to activation of platelet membrane phospholipase A2 (PLA2). PLA2 cleaves membrane phospholipids and liberates arachidonic acid, which is converted into a cyclic endoperoxide by platelet cyclooxygenase. Thromboxane synthase subsequently converts the cyclic endoperoxide into thromboxane A2 (TxA2). TxA2, via a G protein-coupled receptor, causes vasoconstriction at the site of vascular injury by inducing a decrease in cAMP levels within vascular smooth muscle cells. TxA2 also stimulates the granule release reaction within platelets, thereby propagating the cascade of platelet activation and vasoconstriction.
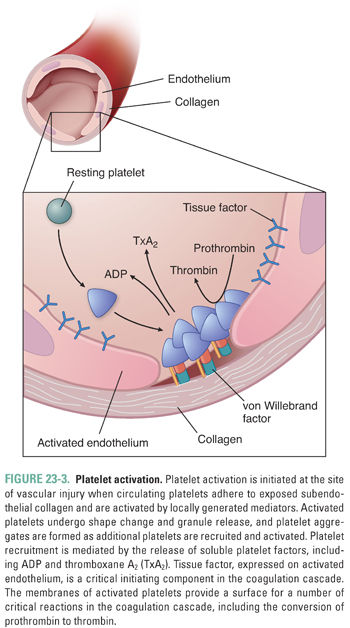
During the release reaction, large amounts of ADP, Ca2+, adenosine triphosphate (ATP), serotonin, vWF, and platelet factor 4 are actively secreted from platelet granules. ADP is particularly important in mediating platelet aggregation, causing platelets to become “sticky” and adhere to one another (see below). Although strong agonists (such as thrombin and collagen) can trigger granule secretion even when aggregation is prevented, ADP can trigger granule secretion only in the presence of platelet aggregation. Presumably, this difference is due to the set of intracellular effectors that are coupled to the various agonist receptors. Release of Ca2+ ions is also important for the coagulation cascade, as discussed below.
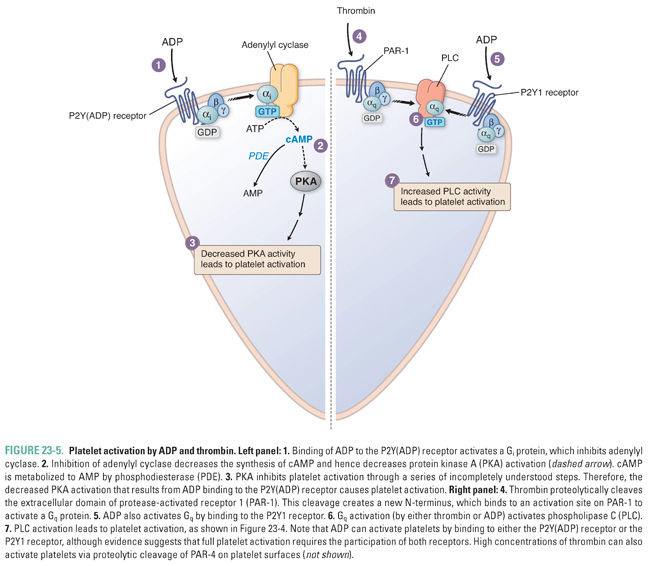
Although platelet activation can be initiated via exposure of subendothelial collagen, a separate and parallel process of platelet activation occurs without disruption of the endothelium and without the involvement of von Willebrand factor. This second pathway of platelet activation is initiated by tissue factor, a lipoprotein expressed by activated endothelial cells, activated leukocytes, and microparticles derived from activated leukocytes (see below). As in the coagulation cascade, tissue factor forms a complex with factor VIIa, and the tissue factor–factor VIIa complex activates factor IX. Factor IX activation leads to a proteolytic cascade that results in the generation of thrombin (factor IIa), a multifunctional enzyme that plays a critical role in the coagulation cascade (see below). In the tissue factor-initiated pathway of platelet activation, thrombin cleaves protease-activated receptor 1 (PAR-1) and protease-activated receptor 4 (PAR-4) on the platelet surface and thereby causes the platelets to release ADP, serotonin, and TxA2. By activating other nearby platelets, these agonists amplify the signal for thrombus formation.
Platelet Aggregation and Consolidation
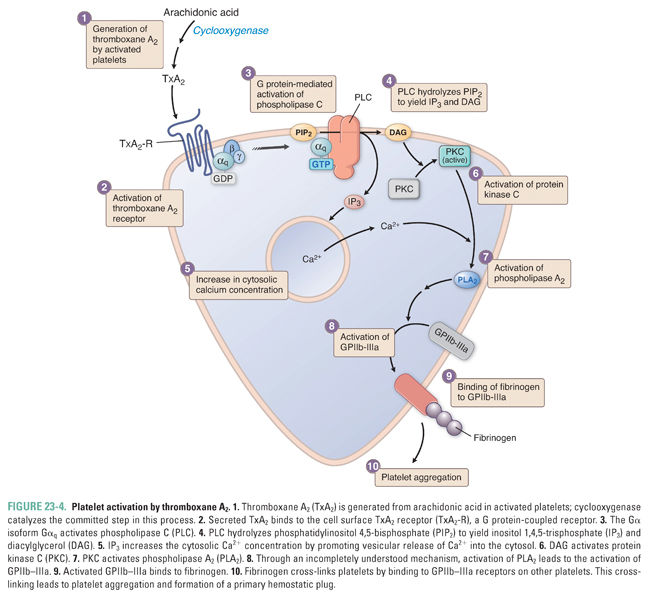
TxA2, ADP, and fibrous collagen are all potent mediators of platelet aggregation. TxA2 promotes platelet aggregation through stimulation of G protein-coupled TxA2 receptors in the platelet membrane (Fig. 23-4). Binding of TxA2 to platelet TxA2 receptors leads to activation of phospholipase C (PLC), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PI[4,5]P2) to yield inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 raises the cytosolic Ca2+ concentration, and DAG activates protein kinase C (PKC), which in turn promotes the activation of PLA2. Through an incompletely understood mechanism, PLA2 activation induces the expression of functional GPIIb–IIIa, the membrane integrin that mediates platelet aggregation.
ADP triggers platelet activation by binding to G protein-coupled ADP receptors on the platelet surface (Fig. 23-5). The two subtypes of G protein-coupled platelet ADP receptors are termed P2Y1 receptors and P2Y(ADP) receptors. P2Y1, a Gq-coupled receptor, releases intracellular calcium stores through activation of phospholipase C. P2Y(ADP), a Gi-coupled receptor, inhibits adenylyl cyclase. The P2Y(ADP) receptor is the target of the antiplatelet agents ticlopidine, clopidogrel, prasugrel, and ticagrelor (see below). Activation of ADP receptors mediates platelet shape change and expression of functional GPIIb–IIIa.
Fibrous collagen activates platelets by binding directly to platelet glycoprotein VI (GPVI). Activation of GPVI by collagen initiates signaling cascades that promote the granule release reaction and that induce conformational changes in cell surface integrins (especially GPIIb–IIIa and α2β1) that promote the direct or indirect binding of these integrins to collagen. These additional binding interactions further strengthen the adhesion of activated platelets to the subendothelial matrix.
Platelets aggregate with one another through a bridging molecule, fibrinogen, which has multiple binding sites for functional GPIIb–IIIa (Fig. 23-2). Just as the vWF:GPIb interaction is important for platelet adhesion to exposed subendothelial collagen, the fibrinogen:GPIIb–IIIa interaction is critical for platelet aggregation. Platelet aggregation ultimately leads to the formation of a reversible clot, or a primary hemostatic plug.
Activation of the coagulation cascade proceeds nearly simultaneously with the formation of the primary hemostatic plug, as described below. Activation of the coagulation cascade leads to the generation of fibrin, initially at the periphery of the primary hemostatic plug. Platelet pseudopods attach to the fibrin strands at the periphery of the plug and contract (Fig. 23-1E). Platelet contraction yields a compact, solid, irreversible clot, or a secondary hemostatic plug.
Secondary Hemostasis: The Coagulation Cascade
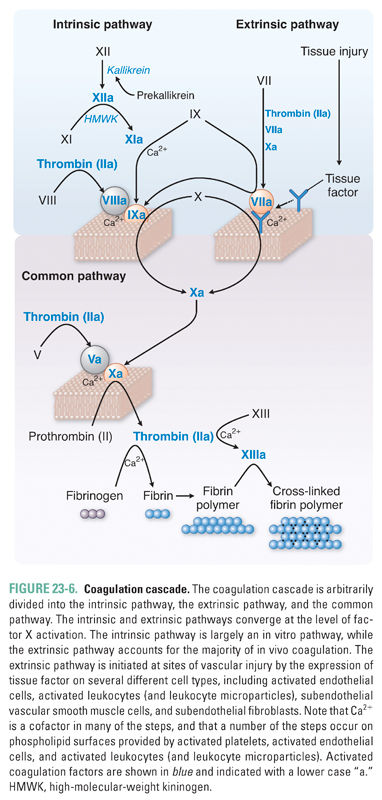
Secondary hemostasis is also termed the coagulation cascade. The goal of this cascade is to form a stable fibrin clot at the site of vascular injury. Details of the coagulation cascade are presented schematically in Figure 23-6. Several general principles should be noted.
First, the coagulation cascade is a sequence of enzymatic events. Most plasma coagulation factors circulate as inactive proenzymes, which are synthesized by the liver. These proenzymes are proteolytically cleaved, and thereby activated, by the activated factors that precede them in the cascade. The activation reaction is catalytic and not stoichiometric. For example, one “unit” of activated factor X can potentially generate 40 “units” of thrombin. This robust amplification process rapidly generates large amounts of fibrin at a site of vascular injury.
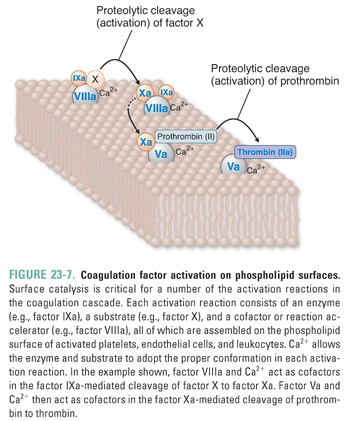
Second, the major activation reactions in the cascade occur at sites where a phospholipid-based protein–protein complex has formed (Fig. 23-7). This complex is composed of a membrane surface (provided by activated platelets, activated endothelial cells, and possibly activated leukocyte microparticles [see below]), an enzyme (an activated coagulation factor), a substrate (the proenzyme form of the downstream coagulation factor), and a cofactor. The presence of negatively charged phospholipids, especially phosphatidylserine, is critical for assembly of the complex. Phosphatidylserine, which is normally sequestered in the inner leaflet of the plasma membrane, translocates to the outer leaflet of the membrane in response to agonist stimulation of platelets, endothelial cells, or leukocytes. Calcium is required for the enzyme, substrate, and cofactor to adopt the proper conformation for the proteolytic cleavage of a coagulation factor proenzyme to its activated form.
Third, the coagulation cascade has been divided traditionally into the intrinsic and extrinsic pathways (Fig. 23-6). This division is a result of in vitro testing and is essentially arbitrary. The intrinsic pathway is activated in vitro by factor XII (Hageman factor), while the extrinsic pathway is initiated in vivo by tissue factor, activated endothelial cells, subendothelial smooth muscle cells, and subendothelial fibroblasts at the site of vascular injury. Although these two pathways converge at the activation of factor X, there is also much interconnection between the two pathways. Because factor VII (activated by the extrinsic pathway) can proteolytically activate factor IX (a key factor in the intrinsic pathway), the extrinsic pathway is regarded as the primary pathway for the initiation of coagulation in vivo.
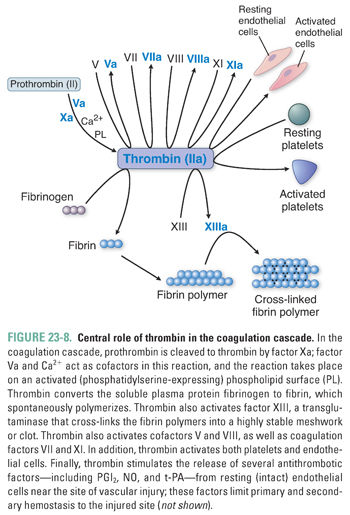
Fourth, both the intrinsic and extrinsic coagulation pathways lead to the activation of factor X. In an important reaction that requires factor V, activated factor X proteolytically cleaves prothrombin (factor II) to thrombin (factor IIa) (Fig. 23-8). Thrombin acts in the coagulation cascade in four important ways: (1) it converts the soluble plasma protein fibrinogen into fibrin, which then forms long, insoluble polymer fibers; (2) it activates factor XIII, which cross-links the fibrin polymers into a highly stable meshwork or clot; (3) it amplifies the clotting cascade by catalyzing the feedback activation of factors VIII and V; and (4) it strongly activates platelets, causing granule release, platelet aggregation, and platelet-derived microparticle generation. In addition to its procoagulant properties, thrombin acts to modulate the coagulation response. Thrombin activates protease-activated receptors (PARs) on the intact vascular endothelial cells adjacent to the area of vascular injury and stimulates these cells to release the platelet inhibitors prostacyclin (PGI2) and nitric oxide (NO), the profibrinolytic protein tissue plasminogen activator (t-PA), and the endogenous t-PA modulator plasminogen activator inhibitor 1 (PAI-1) (see below).
Thrombin binds to protease-activated receptors (PARs), which are G protein-coupled receptors expressed in the plasma membrane of platelets, vascular endothelial cells, monocytes, vascular smooth muscle cells, and fibroblasts. Activation of PARs involves proteolytic cleavage of an extracellular domain of the receptor by thrombin. The new NH2-terminal-tethered ligand binds intramolecularly to a discrete site within the receptor and initiates intracellular signaling. Activation of PAR-1 results in G protein-mediated activation of PLC (Fig. 23-5) and inhibition of adenylyl cyclase. Four different PARs have been identified (PARs 1–4). PAR-1 and PAR-4 are expressed on platelets; PARs 1–4 are expressed on vascular endothelial cells; and some or all of the PARs are expressed on other cell types including monocytes and vascular smooth muscle cells. Of the two protease-activated receptors expressed on platelets, PAR-1 has a higher affinity for thrombin, and PAR-1 is the target of the new antiplatelet agent vorapaxar (see below).
Evidence from intravital (in vivo) microscopy experiments suggests that microparticles also have an important role in coupling platelet plug formation (primary hemostasis) to fibrin clot formation (secondary hemostasis). Microparticles are vesicular structures derived from leukocytes, monocytes, platelets, endothelial cells, and smooth muscle cells; they display proteins of the cells from which they were derived. For example, a subpopulation of microparticles is released from monocytes that are activated in the context of tissue injury and inflammation. These microparticles express both tissue factor and P-selectin glycoprotein ligand-1 (PSGL-1). In turn, PSGL-1 on the microparticles binds to the P-selectin adhesion receptor expressed on activated platelets. By recruiting tissue factor-bearing microparticles throughout the developing platelet plug (primary hemostasis), thrombin generation and fibrin clot formation (secondary hemostasis) could be greatly accelerated within the plug itself. Indeed, both vessel-wall tissue factor (expressed by activated endothelial cells, subendothelial fibroblasts, and smooth muscle cells) and microparticle tissue factor are important for the formation of a stable clot.
Hemostasis is exquisitely regulated for two major reasons. First, hemostasis must be restricted to the local site of vascular injury. That is, activation of platelets and coagulation factors in the plasma should occur only at the site of endothelial damage, tissue factor expression, and procoagulant phospholipid exposure. Second, the size of the primary and secondary hemostatic plugs must be restricted so that the vascular lumen remains patent. After vascular injury, intact endothelium in the immediate vicinity of the injury becomes “activated.” This activated endothelium presents a set of procoagulant factors that promote hemostasis at the site of injury and anticoagulant factors that restrict propagation of the clot beyond the site of injury. The procoagulant factors, such as tissue factor and phosphatidylserine, tend to be membrane-bound and localized to the site of injury—these factors provide a surface on which the coagulation cascade can proceed. In contrast, the anticoagulant factors are generally secreted by the endothelium and are soluble in the blood. Thus, the activated endothelium maintains a balance of procoagulant and anticoagulant factors to limit hemostasis to the site of vascular injury.
After vascular injury, the endothelium surrounding the injured area participates in five separate mechanisms that limit the initiation and propagation of the hemostatic process to the immediate vicinity of the injury. These mechanisms involve prostacyclin (PGI2), antithrombin III, proteins C and S, tissue factor pathway inhibitor (TFPI), and tissue-type plasminogen activator (t-PA).
Prostacyclin (PGI2) is an eicosanoid (i.e., a metabolite of arachidonic acid) that is synthesized and secreted by the endothelium. By acting through Gs protein-coupled platelet-surface PGI2 receptors, this metabolite increases cAMP levels within platelets and thereby inhibits platelet aggregation and platelet granule release. PGI2 also has potent vasodilatory effects; this mediator induces vascular smooth muscle relaxation by increasing cAMP levels within the vascular smooth muscle cells. (Note that these mechanisms are physiologically antagonistic to those of TxA2, which induces platelet activation and vasoconstriction.) Therefore, PGI2 both prevents platelets from adhering to the intact endothelium that surrounds the site of vascular injury and maintains vascular patency around the site of injury.
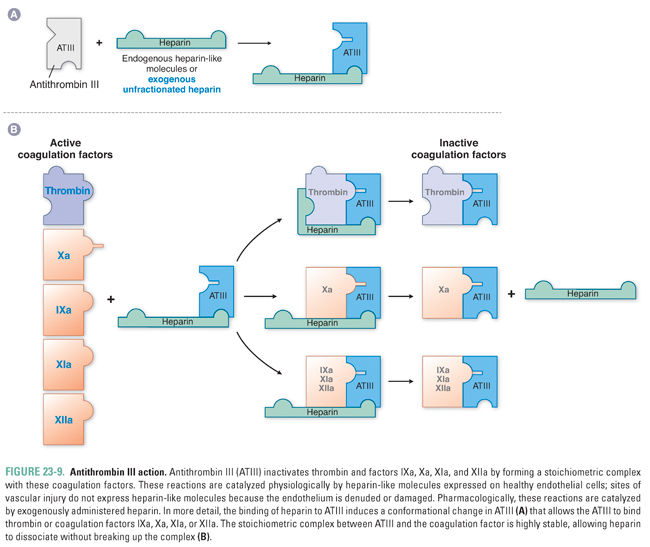
Antithrombin III inactivates thrombin and other coagulation factors (IXa, Xa, XIa, and XIIa, where “a” denotes an “activated” factor) by forming a stoichiometric complex with the coagulation factor (Fig. 23-9). These interactions are enhanced by a heparin-like molecule that is expressed at the surface of intact endothelial cells, ensuring that this mechanism is operative at all locations in the vascular tree except where endothelium is denuded at the site of vascular injury. (These endothelial cell surface proteoglycans are referred to as heparin-like because they are the physiologic equivalent of the pharmacologic agent heparin, discussed below.) Heparin-like molecules on the endothelial cells bind to and activate antithrombin III, which is then primed to complex with (and thereby inactivate) the activated coagulation factors.
Protein C and protein S are vitamin K-dependent proteins that slow the coagulation cascade by inactivating coagulation factors Va and VIIIa. Protein C and protein S are part of a feedback control mechanism, in which excess thrombin generation leads to activation of protein C, which, in turn, helps to prevent the enlarging fibrin clot from occluding the vascular lumen. Specifically, the endothelial cell surface protein thrombomodulin is a receptor for both thrombin and protein C in the blood. Thrombomodulin binds these proteins in such a way that thrombomodulin-bound thrombin cleaves protein C to activated protein C (also known as protein Ca). In a reaction that requires the cofactor protein S, activated protein C then inhibits clotting by cleaving (and thereby inactivating) factors Va and VIIIa.
Tissue factor pathway inhibitor (TFPI), as its name indicates, limits the action of tissue factor (TF). The coagulation cascade is initiated when factor VIIa complexes with TF at the site of vascular injury (Fig. 23-6). The resulting VIIa:TF complex catalyzes the activation of factors IX and X. After limited quantities of factors IXa and Xa are generated, the VIIa:TF complex is feedback inhibited by TFPI in a two-step reaction. First, TFPI binds to factor Xa and neutralizes its activity in a Ca2+-independent reaction. Subsequently, the TFPI:Xa complex interacts with the VIIa:TF complex via a second domain on TFPI, so that a quaternary Xa:TFPI:VIIa:TF complex is formed. The molecular “knots” of the TFPI molecule hold the quaternary complex tightly together and thereby inactivate the VIIa:TF complex. In this manner, TFPI prevents excessive TF-mediated activation of factors IX and X.
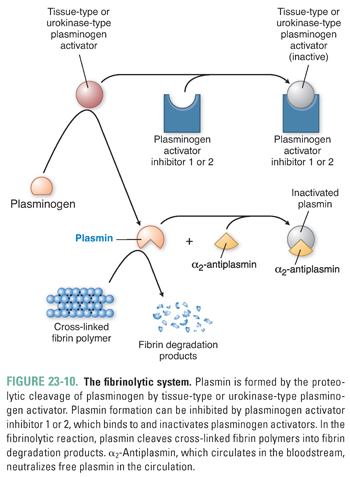
Plasmin exerts its anticoagulant effect by proteolytically cleaving fibrin into fibrin degradation products. Because plasmin has powerful antithrombotic effects, the formation of plasmin has intrigued researchers for many years, and a number of pharmacologic agents have been developed to target the plasmin formation pathway (Fig. 23-10). Plasmin is generated by the proteolytic cleavage of plasminogen, a plasma protein that is synthesized in the liver. The proteolytic cleavage is catalyzed by tissue plasminogen activator (t-PA), which is synthesized and secreted by the endothelium. Plasmin activity is carefully modulated by three regulatory mechanisms in order to restrict plasmin action to the site of clot formation. First, t-PA is most effective when it is bound to a fibrin meshwork. Second, t-PA activity can be inhibited by plasminogen activator inhibitor (PAI). When local concentrations of thrombin and inflammatory cytokines (such as IL-1 and TNF-α) are high, endothelial cells increase the release of PAI, preventing t-PA from activating plasmin. This ensures that a stable fibrin clot forms at the site of vascular injury. Third, α2-antiplasmin is a plasma protein that neutralizes free plasmin in the circulation and thereby prevents systemic degradation of plasma fibrinogen. Plasma fibrinogen is important for platelet aggregation in primary hemostasis (see above), and it is also the precursor for the fibrin polymer that is required to form a stable clot.

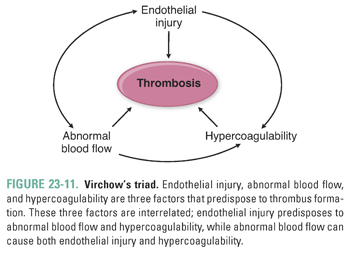
Thrombosis is the pathologic extension of hemostasis. In thrombosis, coagulation reactions are inappropriately regulated so that a clot uncontrollably enlarges and occludes the lumen of a blood vessel. The pathologic clot is now termed a thrombus. Three major factors predispose to thrombus formation—endothelial injury, abnormal blood flow, and hypercoagulability. These three factors influence one another and are collectively known as Virchow’s triad (Fig. 23-11).
Endothelial injury is the dominant influence on thrombus formation in the heart and the arterial circulation. There are many possible causes of endothelial injury, including changes in shear stress associated with hypertension or turbulent flow, hyperlipidemia, elevated blood glucose in diabetes mellitus, traumatic vascular injury, and some infections. (Recall that Mr. S developed coronary artery thrombosis, which was probably attributable to endothelial injury secondary to hypertension and cigarette smoking.)
Endothelial injury predisposes the vascular lumen to thrombus formation through three mechanisms. First, platelet activators, such as exposed subendothelial collagen, promote platelet adhesion to the injured site. Second, exposure of tissue factor on injured endothelium initiates the coagulation cascade. Third, natural antithrombotics, such as t-PA and PGI2, become depleted at the site of vascular injury because these mechanisms rely on the functioning of an intact endothelial cell layer.
Abnormal blood flow refers to a state of turbulence or stasis rather than laminar flow. Atherosclerotic plaques commonly predispose to turbulent blood flow in the vicinity of the plaque. Bifurcations of blood vessels can also create areas of turbulent flow. Turbulent blood flow causes endothelial injury, forms countercurrents, and creates local pockets of stasis. Local stasis can also result from formation of an aneurysm (a focal outpouching of a vessel or a cardiac chamber) and from myocardial infarction. In the latter condition, a region of noncontractile (infarcted) myocardium serves as a favored site for stasis. Cardiac arrhythmias, such as atrial fibrillation, can also generate areas of local stasis. Stasis is also the major cause for the formation of venous thrombi, which typically occur in the deep veins of the leg.
Disruption of normal blood flow by turbulence or stasis promotes thrombosis by three major mechanisms. First, the absence of laminar blood flow allows platelets to come into close proximity to the vessel wall. Second, stasis inhibits the flow of fresh blood into the vascular bed, so that activated coagulation factors in the region are not removed or diluted. Third, abnormal blood flow promotes endothelial cell activation, which leads to a prothrombotic state.
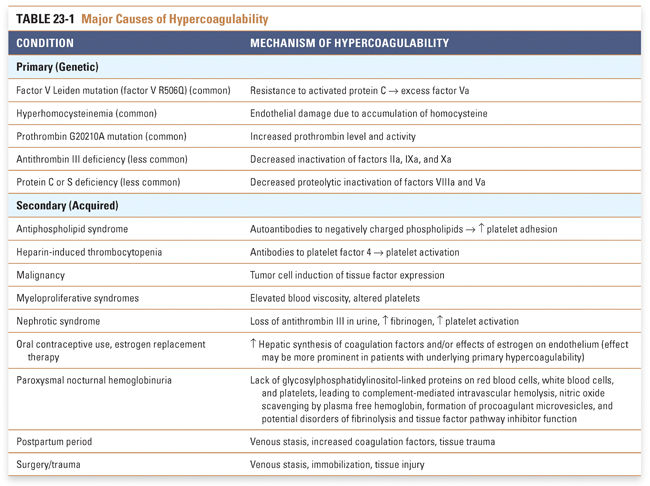
Hypercoagulability is generally less important than endothelial injury and abnormal blood flow in predisposing to thrombosis, but this condition can be an important factor in some patients. Hypercoagulability refers to an abnormally heightened coagulation response to vascular injury, resulting from either (1) primary (genetic) disorders or (2) secondary (acquired) disorders (see Table 23-1). (Hypocoagulable states, or hemorrhagic disorders, can also result from primary or secondary causes; see Box 23-1 for an example.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


