INTRODUCTION
The human heart is both a mechanical and an electrical organ. To perfuse the body adequately with blood, the mechanical and electrical components of the heart must work in precise concert with each other. The mechanical component pumps the blood; the electrical component controls the rhythm of the pump. When the mechanical component fails despite a normal rhythm, heart failure can result (see Chapter 26, Integrative Cardiovascular Pharmacology: Hypertension, Ischemic Heart Disease, and Heart Failure). When the electrical component goes awry (called an arrhythmia), cardiac myocytes fail to contract in synchrony, and effective pumping is compromised. Changes in the membrane potential of cardiac cells directly affect cardiac rhythm, and most antiarrhythmic drugs act by modulating the activity of ion channels in the plasma membrane. This chapter discusses the ionic basis of electric rhythm formation and conduction in the heart, the pathophysiology of electrical dysfunction, and the pharmacologic agents used to restore a normal cardiac rhythm.
 One winter morning, Dr. J, a 74-year-old professor, is lecturing on the treatment of cardiomyopathies to the second-year medical school class. He feels his heart beating irregularly and becomes nauseated. He is able to finish his lecture, but he continues to feel significantly short of breath throughout the morning. His persistent symptoms prompt him to walk down the street to the local emergency department.
One winter morning, Dr. J, a 74-year-old professor, is lecturing on the treatment of cardiomyopathies to the second-year medical school class. He feels his heart beating irregularly and becomes nauseated. He is able to finish his lecture, but he continues to feel significantly short of breath throughout the morning. His persistent symptoms prompt him to walk down the street to the local emergency department.
Physical examination reveals an irregular heartbeat ranging from 120 to 140 beats/min. Dr. J’s blood pressure is stable (132/76 mm Hg), and his oxygen saturation is 100% on room air. An electrocardiogram (ECG) confirms that Dr. J has atrial fibrillation, without any evidence of ischemia. Several intravenous boluses of diltiazem are administered, and his heart rate decreases to 80–100 beats/min, but his rhythm remains irregular. Further laboratory studies, an echocardiogram, and a chest x-ray do not reveal an underlying cause for Dr. J’s atrial fibrillation.
During observation over the next 12 hours, Dr. J remains in atrial fibrillation. Although his heart rate is under better control, he continues to experience palpitations. Under continuous ECG monitoring, a cardiologist administers an intravenous infusion of ibutilide. Twenty minutes after receiving the ibutilide, Dr. J’s ECG shows a return to normal sinus rhythm. At the time of discharge from the hospital, he is started on warfarin in order to reduce his risk of stroke.
Dr. J feels fine at first, but he develops recurrent palpitations within 3 weeks of his initial event. After discussion with his cardiologist, he elects to start amiodarone at a maintenance dose of 200 mg/day, in addition to continuing his warfarin. Dr. J tolerates the amiodarone well and reports no difficulty breathing. He remains symptom-free during the rest of his cardiology lectures.
Questions
1. Why were ibutilide and amiodarone effective in converting Dr. J’s heart rhythm to normal sinus rhythm?
2. Why should ibutilide be administered only under carefully monitored circumstances?
3. What adverse effects of amiodarone could develop at higher daily doses?
4. Why did diltiazem slow Dr. J’s heart rate without affecting his underlying heart rhythm, atrial fibrillation?
 ELECTRICAL PHYSIOLOGY OF THE HEART
ELECTRICAL PHYSIOLOGY OF THE HEART
Electrical activity in the heart, leading to rhythmic cardiac contraction, is a manifestation of the heart’s exquisite control of cell depolarization and impulse conduction. Once initiated, a cardiac action potential is a spontaneous event that proceeds based on the characteristic responses of ion channels to changes in membrane voltage. At the completion of a cycle, the spontaneous depolarization of pacemaker cells ensures that the process repeats without interruption.
Pacemaker and Nonpacemaker Cells
The heart contains cardiac myocytes that can spontaneously initiate action potentials and myocytes that cannot. Cells possessing the ability to initiate spontaneous action potentials are termed pacemaker cells. All pacemaker cells possess automaticity, the ability to depolarize above a threshold voltage in a rhythmic fashion. Automaticity results in the generation of spontaneous action potentials. Pacemaker cells are found in the sinoatrial node (SA node), the atrioventricular node (AV node), and the ventricular conducting system (bundle of His, bundle branches, and Purkinje fibers). Together, the pacemaker cells constitute the specialized conducting system that governs the electrical activity of the heart. The second type of cardiac cells, the nonpacemaker cells, includes the atrial and ventricular myocytes. The nonpacemaker cells contract in response to depolarization and are responsible for the majority of cardiac contraction. In pathologic conditions, these nonpacemaker cells can acquire automaticity and thereby also act as pacemaker cells.
Ions are not distributed equally across cell membranes. Transporters (pumps) drive K+ into cells while pumping Na+ and Ca2+ out, giving rise to electrical and chemical gradients across the membrane. These gradients ultimately determine the membrane potential of a cardiac cell. The Nernst equilibrium potential for each ion (ENa = +70 mV, EK = −94 mV, and ECa = +150 mV) depends on the relative concentrations of ions inside and outside the cell. The difference between an ion’s Nernst potential and the cell’s membrane potential determines the driving force for ions into or out of the cell. Refer to Chapter 8, Principles of Cellular Excitability and Electrochemical Transmission, for a detailed discussion of the Nernst equilibrium potential.
When an ion-selective channel opens, the membrane potential approaches the equilibrium potential for that ion. For example, opening a K+-selective channel drives the membrane potential toward EK (−94 mV). When a Na+-selective channel opens, the membrane potential is driven toward ENa (+70 mV), and opening a Ca2+-selective channel drives the membrane potential toward ECa (+150 mV). Note that the reversal potential for a nonselective ion channel (e.g., a channel that passes all cations nonselectively) is 0 mV. The final membrane potential depends on the number of channels of each type, their conductances (i.e., the ability of each channel to pass ions), and the duration for which each channel remains open. The resting membrane of the cardiac myocyte is relatively permeable to K+ (because some types of K+-selective channels are open) but not to Na+ or Ca2+; hence, the resting membrane potential is close to the equilibrium potential for K+. (The actual cardiac myocyte membrane potential is a bit more positive than the equilibrium potential for K+, due to the contribution of other ion channels to the resting membrane potential.)
Changing the membrane potential requires the movement of relatively few ions across the membrane. Therefore, despite the opening and closing of ion channels, the ionic concentration gradients across the membrane remain relatively stable, and the Nernst potential for each ion remains relatively constant.
Cardiac action potentials are strikingly longer than those of nerve or skeletal muscle, lasting for almost half a second. Prolonged cardiac action potentials provide the sustained depolarization and contraction needed to empty the heart’s chambers. Sinoatrial (SA) nodal cells pace the heart at normal resting heart rates between 60 and 100 beats/min, while ventricular muscle cells orchestrate the contraction that ejects blood from the heart (Fig. 24-1).
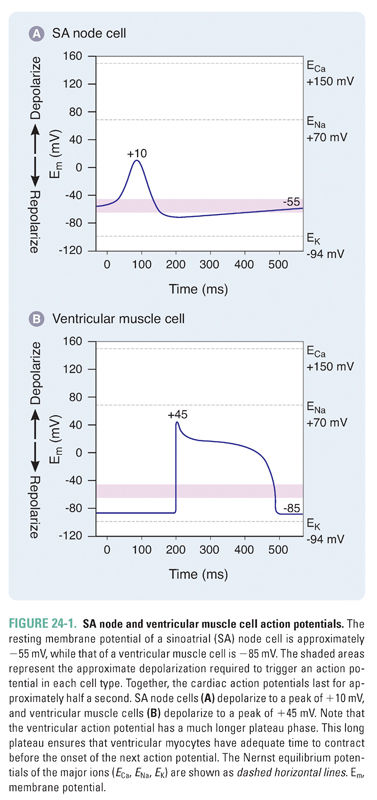
SA nodal cells fire spontaneously in a cycle defined by three phases, referred to as phase 4, phase 0, and phase 3 (Fig. 24-2 and Table 24-1). Phase 4 consists of a slow, spontaneous depolarization that is caused by an inward pacemaker current (If –encoded by HCN). This spontaneous depolarization accounts for the automaticity of the SA node. The channels that carry the If current are activated during the repolarization phase of the previous action potential. The If channels are relatively nonselective cation channels. Phase 0 consists of a more rapid depolarization mediated by highly selective voltage-gated Ca2+ channels that, upon opening, drive the membrane potential toward ECa (+150 mV). In phase 3, the Ca2+ channels slowly close and K+-selective channels open, resulting in membrane repolarization. Once the membrane potential repolarizes to approximately −60 mV, the opening of If channels is triggered and the cycle begins again.
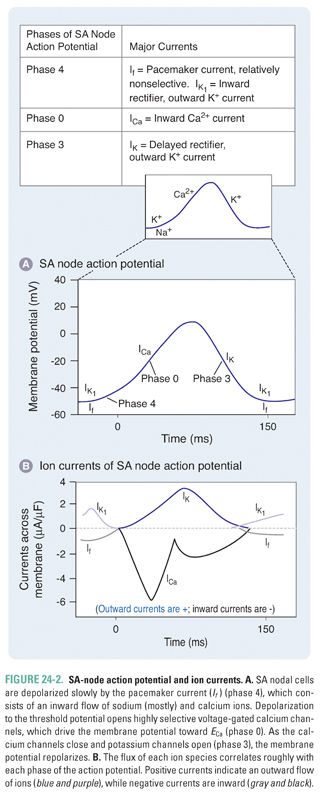
Although the If (inward pacemaker) current is responsible for the slow spontaneous depolarization in phase 4 of the SA-node action potential, the kinetics of this depolarization are modulated by voltage-gated Na+ channels that are also expressed in the SA node. There are gradients of expression of If channels and of the more selective voltage-gated Na+ and Ca2+ channels within the SA node, such that cells at the border of the node express relatively more voltage-gated Na+ channels and cells in the center of the node express relatively more If and voltage-gated Ca2+ channels. The expression of voltage-gated Na+ channels in the SA node is partly responsible for the effect of certain antiarrhythmics on the automaticity of SA nodal cells (see below).
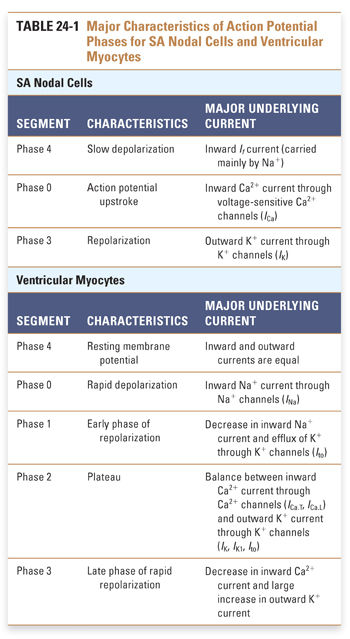
Unlike SA nodal cells, ventricular myocytes do not depolarize spontaneously under physiologic conditions. As a result, the membrane potential of the resting ventricular myocyte remains near EK until the cell is stimulated by a wave of depolarization that is initiated by nearby pacemaker cells. The five phases of the ventricular myocyte action potential result from an intricately woven cascade of channel openings and closings; the phases are numbered from 0 to 4 (Fig. 24-3 and Table 24-1).
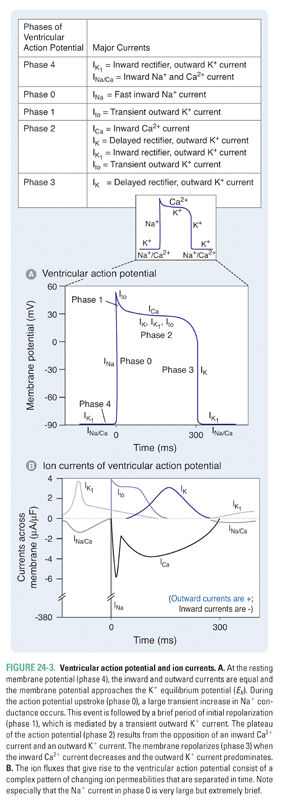
In phase 0, an action potential upstroke of very rapid depolarization is caused by a transient increase in inward Na+ current through voltage-gated Na+ channels. (Note that currents in phase 0 of the SA nodal and ventricular myocyte action potentials are carried by different ions—Ca2+ and Na+, respectively.) The opening of Na+ channels leads to a rapid influx of Na+ (INa), which accounts for the depolarization and drives the membrane potential toward ENa (+70 mV). Although large, the increase in Na+ conductance during phase 0 lasts for only 1–2 milliseconds because the Na+ channels inactivate as a function of time and voltage. Inactivation of the fast Na+ channels causes a dramatic decrease in the inward Na+ current. The time it takes for Na+ channels to recover from their voltage-dependent and time-dependent inactivation determines the refractory period of the myocyte. The refractory period is the time during which another action potential cannot fire. This serves as a protective mechanism to ensure that the heart has sufficient time to eject blood from its chambers. The refractory period lasts from the initiation of the action potential upstroke until the repolarization phase. INa is the major determinant of the velocity of impulse conduction throughout the ventricle.
The threshold-dependent activation of INa quickly depolarizes the membrane. The upstroke terminates before reaching ENa, however, and is followed by an early phase of rapid repolarization to about +20 mV. This phase 1 repolarization is a consequence of two events: (1) the rapid voltage-dependent inactivation of INa and (2) the activation of transient K+ currents (transient outward; Ito).
Phase 2, the plateau phase of the ventricular action potential, is unique to cardiac cell electrophysiology. The plateau is maintained by a finely tuned balance between an inward Ca2+ current through two types of Ca2+ channels (ICa.T, ICa.L) and an outward K+ current through several types of K+ channels (IK, IK1, Ito). Remarkably, only a few hundred channels per cell are used to maintain this fine balance. Because only a small number of channels are open, the total membrane conductance is low. The high membrane resistance during the plateau phase insulates the cardiac cells electrically, allowing rapid propagation of the action potential with little current dissipation.
During the plateau phase, two distinct Ca2+ currents—the transient Ca2+ current, ICa.T, and the long-lasting Ca2+ current, ICa.L—mediate the influx of Ca2+ needed to initiate cardiac myocyte contraction. T-type Ca2+ channels inactivate with time and are insensitive to block by dihydropyridines such as nifedipine. Current through the L-type Ca2+ channels (ICa.L) provides the dominant Ca2+ current in virtually all cardiac cells. ICa.L is activated at −30 mV and inactivates slowly (hundreds of milliseconds). It is sensitive to block by dihydropyridines (nifedipine), benzothiazepines (diltiazem), and phenylalkylamines (verapamil), as discussed below. L-type Ca2+ channels carry inward current throughout the plateau phase; because Ca2+ stimulates the contraction of cardiac myocytes, these channels are crucial for coupling membrane excitability to myocardial contraction.
Opposing the inward Ca2+ currents are outward currents through the K+ channels that are activated during the plateau phase. As the time-dependent inward Ca2+ currents inactivate, the outward K+ currents (mostly IK) rapidly drive the membrane potential toward EK, thus repolarizing the cell in phase 3. However, these channels are unable to drive the membrane potential all the way to EK because they deactivate at −40 mV. In phase 4, the resting membrane potential is reestablished by the activation of time-independent K+ currents (IK1), which drive the membrane potential close to the K+ equilibrium potential.
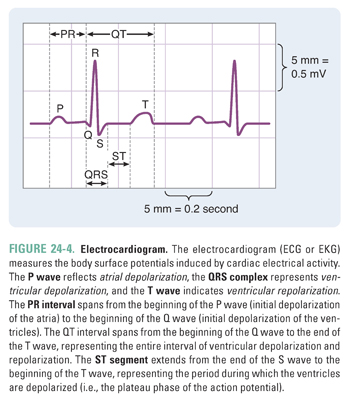
In clinical practice, the overall electrical activity of the heart is measured rather than the ionic changes that occur at a single-cell level. This overall activity is reported in the electrocardiogram, or ECG (Box 24-1 and Fig. 24-4).
BOX 24-1 The Electrocardiogram |
The electrocardiogram (ECG or EKG) is used to infer changes in cardiac impulses by recording electrical potentials at various locations on the surface of the body. An ECG recording reflects changes in the excitation of the myocardium. A basic understanding of the ECG is useful for discussions of the clinical applications of the various antiarrhythmic agents. A normal electrocardiogram contains three electrical waveforms: the P wave, the QRS complex, and the T wave (Fig. 24-4). The P wave represents atrial depolarization; the QRS complex represents ventricular depolarization; and the T wave represents ventricular repolarization. The ECG does not show atrial repolarization explicitly because the atrial repolarization is “drowned out” by the QRS complex. The ECG also contains two intervals and one segment: the PR interval, the QT interval, and the ST segment. The PR interval spans from the beginning of the P wave (initial depolarization of the atria) to the beginning of the Q wave (initial depolarization of the ventricles). Hence, the length of the PR interval varies with conduction velocity through the AV node. For example, if a patient has an electrical block in the AV node, then the conduction velocity through the AV node decreases and the PR interval increases. The QT interval spans from the beginning of the Q wave to the end of the T wave, representing the entire sequence of ventricular depolarization and repolarization. The ST segment extends from the end of the S wave to the beginning of the T wave; this segment, which represents the period during which the ventricles are depolarized, corresponds to the plateau phase of the ventricular action potential. |

The specialized conduction system of the heart consists of the SA node, AV node, bundle of His, and Purkinje system. These different populations of cells have different intrinsic rates of firing. Three factors determine the firing rate. First, as the rate of spontaneous depolarization in phase 4 increases, the rate of firing increases because the threshold potential (the minimum potential necessary to trigger an action potential) is reached more quickly at the end of phase 4. Second, if the threshold potential becomes more negative, the rate of firing increases because the threshold potential is reached more quickly at the end of phase 4. Third, if the maximum diastolic potential (the resting membrane potential) becomes more positive, the rate of firing increases because less time is needed to repolarize the membrane fully at the end of phase 3.
Because the various populations of pacemaker cells possess different intrinsic rates of firing, the pacemaker population with the fastest firing rate sets the heart rate. The SA node possesses the fastest intrinsic firing rate (60–100 times per minute) and is the native pacemaker of the heart. The cells of the atrioventricular (AV) node and bundle of His fire intrinsically between 50 and 60 times per minute, and the cells of the Purkinje system have the slowest intrinsic firing rate at 30–40 times per minute. The cells of the AV node, bundle of His, and Purkinje system are termed latent pacemakers because their intrinsic rhythm is overridden by the faster SA-node automaticity. In a mechanism termed overdrive suppression, the SA node suppresses the intrinsic rhythm of the other pacemaker populations and entrains them to fire at the SA nodal firing rate.
 PATHOPHYSIOLOGY OF ELECTRICAL DYSFUNCTION
PATHOPHYSIOLOGY OF ELECTRICAL DYSFUNCTION
Causes of electrical dysfunction in the heart can be divided into defects in impulse formation and defects in impulse conduction. In the former case, SA-node automaticity is interrupted or altered, leading to missed beats or ectopic beats, respectively. In the latter case, impulse conduction is altered (e.g., in the case of reentrant rhythms), and sustained arrhythmias can result.
Defects in Impulse Formation (SA Node)
As the native pacemaker of the heart, the SA node has a pivotal role in normal impulse formation. Electrical events that alter SA nodal function or disturb overdrive suppression can lead to impaired impulse formation. Two mechanisms commonly associated with defective impulse formation are altered automaticity and triggered activity.
Some mechanisms that alter automaticity of the SA node are physiologic. In particular, the autonomic nervous system often modulates automaticity of the SA node as part of a physiologic response. In sympathetic stimulation during exercise, an increased concentration of catecholamines leads to greater β1-adrenergic receptor activation. Activation of β1 receptors causes the opening of a greater number of pacemaker channels (If channels); a larger pacemaker current is then conducted through these channels; and faster phase 4 depolarization results. Sympathetic stimulation also causes the opening of a greater number of Ca2+ channels and thereby shifts the threshold to more negative potentials. Both of these mechanisms increase heart rate. The parasympathetic vagus nerve affects the SA node by a number of mechanisms that oppose the sympathetic regulation of heart rate. Vagus nerve release of acetylcholine initiates an intracellular signaling cascade that (1) reduces the pacemaker current by decreasing pacemaker channel opening, (2) shifts the threshold to more positive potentials by reducing Ca2+ channel opening, and (3) makes the maximum diastolic potential (analogous to the resting membrane potential in these spontaneously firing cells) more negative by increasing K+ channel opening. The SA node, atria, and AV node are highly innervated and are thus more sensitive than the ventricular conducting system to the effects of vagal stimulation.
In pathologic conditions, automaticity can be altered when latent pacemaker cells take over the SA node’s role as the pacemaker of the heart. When the SA nodal firing rate becomes pathologically slow or when conduction of the SA impulse is impaired, an escape beat may occur as a latent pacemaker initiates an impulse. A series of escape beats, known as an escape rhythm, may result from prolonged SA nodal dysfunction. On the other hand, an ectopic beat occurs when latent pacemaker cells develop an intrinsic rate of firing that is faster than the SA nodal rate, in some cases despite the presence of a normally functioning SA node. A series of ectopic beats, termed an ectopic rhythm, can result from ischemia, electrolyte abnormalities, or heightened sympathetic tone.
Direct tissue damage (such as can occur after a myocardial infarction) also results in altered automaticity. Tissue injury can cause structural disruption of the cell membrane. Disrupted membranes are unable to maintain ion gradients, which are critical for maintaining appropriate membrane potentials. If the resting membrane potential becomes sufficiently positive (more positive than −60 mV), nonpacemaker cells may begin to depolarize spontaneously. Another mechanism by which tissue damage leads to altered automaticity is through the loss of gap junction connectivity. Direct electrical connectivity is important for the effective delivery of overdrive suppression from the SA node to the rest of the cardiac myocytes. When connectivity is disrupted due to tissue injury, overdrive suppression is not efficiently relayed, and the unsuppressed cells can initiate their own rhythm. This abnormal rhythm can lead to cardiac arrhythmia.
Afterdepolarizations occur when a normal action potential triggers extra abnormal depolarizations. That is, the first (normal) action potential triggers additional oscillations of membrane potential, which may lead to arrhythmia. There are two types of afterdepolarizations—early afterdepolarizations and delayed afterdepolarizations.
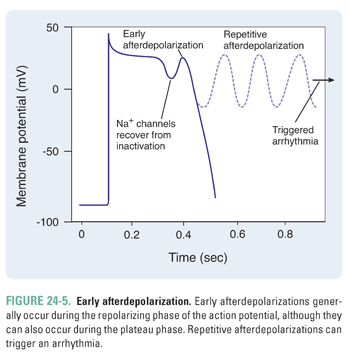
If the afterdepolarization occurs during the inciting action potential, it is termed an early afterdepolarization (Fig. 24-5). Conditions that prolong the action potential (e.g., drugs that prolong the QT interval, such as procainamide and ibutilide) tend to trigger early afterdepolarizations. Specifically, an early afterdepolarization can occur during the plateau phase (phase 2) or the rapid repolarization phase (phase 3). During the plateau phase, because most of the Na+ channels are inactivated, an inward Ca2+ current is responsible for the early afterdepolarization. On the other hand, during the rapid repolarization phase, partially recovered Na+ channels can conduct an inward Na+ current that contributes to the early afterdepolarization. If an early afterdepolarization is sustained, it can lead to a type of ventricular arrhythmia termed torsades de pointes. Torsades de pointes, French for “twisting of the points,” is characterized by QRS complexes of varying amplitudes as they “twist” along the baseline; this rhythm is a medical emergency that can lead to death if not treated immediately.
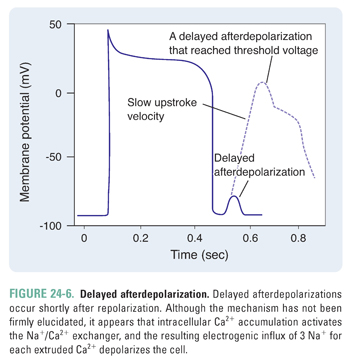
In contrast to early afterdepolarizations, delayed afterdepolarizations occur shortly after the completion of repolarization (Fig. 24-6). The mechanism of delayed afterdepolarizations is not well understood; it has been proposed that high intracellular Ca2+ concentrations (such as in digoxin toxicity) lead to an inward Na+ current, which, in turn, triggers the delayed afterdepolarization.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


