David M. Slovik and Ehrin J. Armstrong
Calcitonin, Glucocorticoids, Thyroid Hormone, and Gonadal Steroids |
Treatment of Secondary Hyperparathyroidism in Chronic Kidney Disease |

The 206 bones of the human skeleton are far from the lifeless structures they are commonly imagined to be. Bones are remodeled continuously and are involved in many functions besides structural support and protection of internal organs, including hematopoiesis and mineral storage. The focus of this chapter is on the critical role of bone in mineral homeostasis, the process and regulation of bone remodeling, the diseases that can result when the delicate balances of mineral homeostasis and bone remodeling are perturbed, and the pharmacologic therapies employed to treat these conditions. A key concept regarding the pharmacologic agents discussed in this chapter is the distinction between bone antiresorptive agents, which slow bone loss, and bone anabolic agents, which have the potential to increase overall bone mass.
 RS is a 60-year-old Caucasian female living in the northeast who comes to her physician with the recent onset of low back pain that began when she inadvertently stepped into a pothole. She is otherwise healthy and has no history of prior fractures.
RS is a 60-year-old Caucasian female living in the northeast who comes to her physician with the recent onset of low back pain that began when she inadvertently stepped into a pothole. She is otherwise healthy and has no history of prior fractures.
Her menstrual periods ceased when she was 54 years old. She had little in the way of postmenopausal symptoms and never took hormone replacement therapy. Menarche was at age 11. She has one child who was born when RS was 38 years old. Her mother died at age 55 with breast cancer, and her sister, age 58, was recently diagnosed with breast cancer. She is lactose intolerant and avoids dairy. Additionally, she does not take calcium or vitamin D supplements and she has very little sunlight exposure with her daily activity. There is no known family history of osteoporosis. Her father and maternal aunt died in their 60s with coronary artery disease.
Her physical examination is unremarkable except for point tenderness over lumbar vertebra L1. Her weight is 135 lb, and she is 64 inches tall but believes she has lost some height over the last year. Laboratory studies are all within normal limits except for a low 25-OH vitamin D level. Lateral x-ray of the spine shows a compression fracture of L1 and generalized osteopenia. Measurement of bone mineral density (BMD) at the spine and hip reveals values that are 2.6 standard deviations below the healthy peak female value for both sites. Her physician diagnoses postmenopausal osteoporosis and a recent compression fracture of L1. RS asks her physician to discuss with her the available therapeutic options and is particularly interested in the potential risks and benefits of each option.
Questions
1. What medical conditions should be investigated to rule out reversible causes of RS’s osteoporosis?
2. Why is RS at particularly high risk for osteoporosis?
3. Given her family history, RS has an increased risk for breast cancer and cardiovascular disease. How does this alter the choice of therapeutic agents that could be prescribed?
4. What are the therapeutic options available for RS? What are the advantages and disadvantages of each option?
5. Should RS take calcium and vitamin D in addition to another therapeutic agent?
 PHYSIOLOGY OF BONE MINERAL HOMEOSTASIS
PHYSIOLOGY OF BONE MINERAL HOMEOSTASIS
Specialized cells called osteoblasts and osteoclasts continually remodel the human skeleton in response to mechanical forces and endocrine and paracrine factors. Two of the endocrine factors—parathyroid hormone and vitamin D—control bone metabolism for the purpose of maintaining extracellular calcium homeostasis. Other hormones, such as glucocorticoids, thyroid hormone, gonadal steroids, and fibroblast growth factor 23 (FGF-23), also have important effects on bone integrity. This section reviews the cellular and molecular mechanisms that mediate bone formation and bone resorption and the mechanisms by which hormones (especially parathyroid hormone and vitamin D) maintain plasma calcium levels within a narrow concentration range.
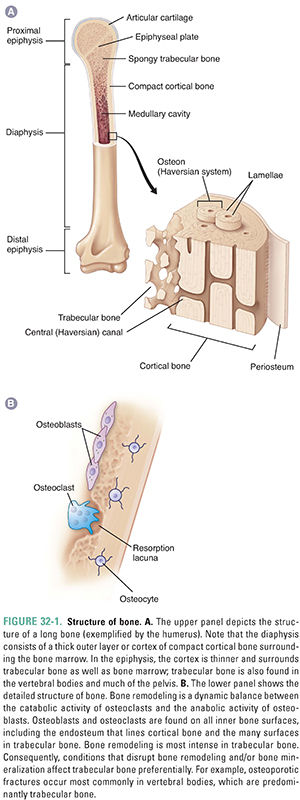
Bone consists of 25% organic and 75% inorganic components. The organic component includes the cells (osteoblasts, osteoclasts, osteocytes, bone lining cells, bone stromal cells) and osteoid (a matrix consisting primarily of type I collagen fibers and several low-abundance proteins). The inorganic component consists of crystalline calcium phosphate salts, primarily hydroxyapatite. The chemical formula of hydroxyapatite is (Ca)5(PO4)3OH. Ninety-nine percent of the calcium in the body is stored in the skeleton, mostly as hydroxyapatite. Figure 32-1 illustrates the structure of a long bone.
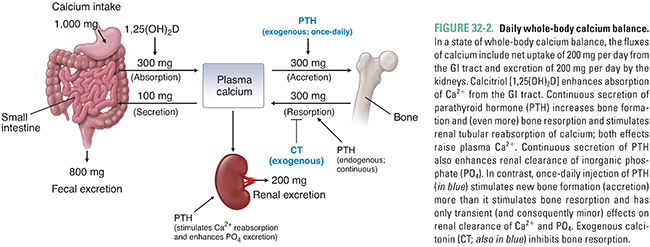
Calcium is absorbed in the small intestine by two mechanisms: facilitated transport, which occurs throughout the small intestine, and calcitriol-dependent active transport, which occurs mainly in the duodenum. In people ingesting 1,000 mg of dietary calcium per day, approximately 300 mg/day is normally absorbed by the intestines (Fig. 32-2). At lower calcium intakes, the efficiency of intestinal calcium absorption is higher, and at higher calcium intakes, the efficiency of absorption is lower. These adjustments contribute importantly to calcium homeostasis, and intestinal calcium absorption can increase to as much as 600 mg/day in the presence of high levels of calcitriol (the active form of vitamin D), as discussed below. The absorption of calcium from the intestine is normally balanced by calcium losses through renal excretion (about 200 mg/day) and salivary and biliary secretion (about 100 mg/day; Fig. 32-2). In contrast to calcium absorption, intestinal absorption of inorganic phosphate is not homeostatically regulated and is typically about two-thirds of the ingested phosphate irrespective of dietary intake.
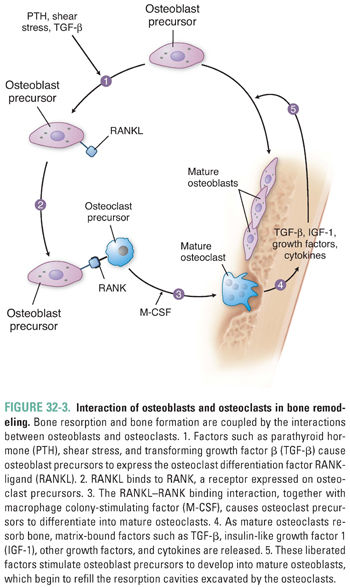
Osteoclasts are the cells responsible for bone resorption. Osteoblasts are the cells responsible for bone formation. Regulation of these two cell types by mechanical, endocrine, and paracrine factors determines the balance between bone formation and bone resorption (see below).
The two signaling proteins RANK ligand (RANKL) and macrophage colony-stimulating factor (M-CSF) are together both necessary and sufficient for the maturation of osteoclasts. RANKL, a member of the tumor necrosis factor (TNF) superfamily, is synthesized by osteoblasts and by osteoblast precursors, which express RANKL on their cell membranes. RANKL binds to RANK, a receptor expressed on osteoclasts and osteoclast precursor cells in the bone marrow. This binding interaction promotes the differentiation of osteoclast precursors into mature osteoclasts (Fig. 32-3). Alternatively, RANKL binds with high affinity to osteoprotegerin (OPG), a soluble extracellular protein synthesized and secreted by osteoblasts. OPG is called a decoy receptor because it prevents RANKL from interacting with RANK. Inherited deficiency of RANKL or RANK causes a form of osteopetrosis (defined as defective bone resorption and increased bone mass), while inherited deficiency of OPG causes increased bone resorption and osteoporosis.
To repair its strength over time and to respond adaptively to mechanical stresses, human bone is continually resorbed and reformed. This process is called remodeling. Partly because of its large surface area on which remodeling can take place, 25% of trabecular bone is remodeled each year in adults. In contrast, only 3% of cortical bone is remodeled each year. This difference is important because pathologic conditions that disturb bone remodeling preferentially affect bones with a high content of trabecular bone, such as the vertebral bodies.
Remodeling is carried out by the coordinated activity of millions of cellular units—basic multicellular units (BMU)— consisting of osteoblasts and osteoclasts. The process of resorption begins when physical and/or chemical signals (discussed below) recruit osteoclasts to form a tight ring-like seal with the bone surface and extend villus-like projections toward the surface within this ring. These villi secrete lactic acid, carbonic acid, and citric acid and use carbonic anhydrase to generate protons and a H+-ATPase to pump the protons onto the bone surface. (Individuals and experimental animals deficient in this carbonic anhydrase have osteopetrosis.) The tight seal creates a closed, ring-shaped microenvironment beneath the osteoclast, within which the secretion of organic acids and protons consumes hydroxide at the bone surface and dissolves hydroxyapatite. The dissolution of hydroxyapatite can be expressed as follows:
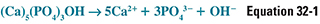
According to Le Chatelier’s principle, OH− consumption drives this reaction to the right. This is an important mechanism exploited by osteoclasts to resorb the mineral component of bone.
Demineralization of the bone matrix exposes it to proteolysis by cathepsin K, collagenases, and other proteases that are concomitantly secreted by the villi. Although this proteolysis totally degrades much of the exposed bone matrix, some of the type I collagen peptide chains escape into the circulation after partial proteolysis. The blood or urine level of such type I collagen metabolites (e.g., NTX, CTX) is an index of bone turnover, reflecting breakdown of type I collagen and total body bone resorption. Because of its large hydroxyapatite-covered surface area, bone normally adsorbs various nonskeletal proteins and peptides from its environment, including IGF-I and TGF-β. Demineralization exposes these adsorbed growth factors to the proteolytic enzymes secreted by osteoclast villi, but some escape proteolysis and affect the cellular activity of neighboring osteoclasts, osteoblasts, and osteocytes.
After about 3 weeks of such bone resorption, cytokines and growth factors liberated from the matrix, together with hormonal and other factors (see below), begin to stimulate local accumulation of osteoblasts via proliferation, differentiation, and reduced apoptosis (programmed cell death). These osteoblasts replace the osteoclasts in the resorption cavity (lacuna) and begin to refill the cavity with concentric layers, or lamellae, of unmineralized organic matrix (osteoid) (Fig. 32-3). As osteoblasts fill the cavity with new osteoid, they also secrete alkaline phosphatase, which hydrolyzes phosphate esters including pyrophosphate (a potent inhibitor of bone mineralization). The hydrolysis of pyrophosphate also increases the local concentration of inorganic phosphate. Together, the alkaline phosphatase-catalyzed hydrolysis of pyrophosphate and the liberation of inorganic phosphate promote the crystallization of calcium phosphate salts and mineralization of the bone matrix.
As osteoblasts continue to lay down matrix, some eventually become completely surrounded by it and are then called osteocytes (Fig. 32-1). Osteocytes are the most numerous bone cell type, comprising 90–95% of the cells in bone. Osteocytes respond to changes in mechanical strain and help control the balance between bone formation and resorption via their secretion of sclerostin (a protein that inhibits bone formation) and other factors. Genetic mutations that delete or inactivate sclerostin increase bone formation without a corresponding increase in bone resorption. Such uncoupling leads to a marked increase in bone mass and bone strength in humans and experimental animals. A monoclonal antibody that inactivates sclerostin, and thereby increases bone mass and bone strength throughout the skeleton, is being evaluated as a potential treatment for osteoporosis. Mature osteocytes normally alter their secretion of sclerostin in bone regions subjected to mechanical loads and thereby play a critical role in skeletal adaptations to gravity and other mechanical loads by localizing bone remodeling responses to such loads.
Hormonal Control of Calcium and Phosphate
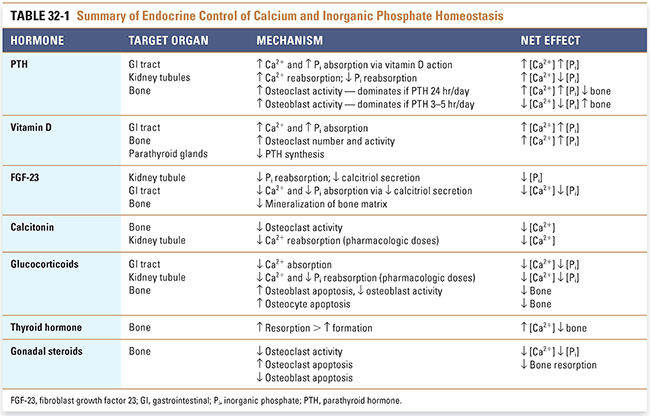
Calcium is essential for many important physiologic processes, such as neurotransmitter release, muscle contraction, and blood coagulation, and deviations in extracellular calcium levels can have serious consequences. Therefore, the plasma calcium level is tightly regulated. Inorganic phosphate concentrations must also be regulated, in part because changes in plasma inorganic phosphate concentrations affect plasma calcium levels (see below). Three main hormones—parathyroid hormone (PTH), vitamin D, and FGF-23—mediate calcium and phosphate homeostasis. In addition, calcitonin, glucocorticoids, thyroid hormone, and gonadal steroids have lesser effects on calcium and phosphate homeostasis. Table 32-1 summarizes the mechanisms and effects of these hormones on calcium and phosphate homeostasis.
The most important endocrine regulator of calcium homeostasis is parathyroid hormone, an 84-amino acid peptide hormone secreted by the parathyroid glands. The secretion of PTH is finely regulated in response to plasma calcium levels. Calcium-sensing receptors reside on the plasma membrane of chief cells in the parathyroid gland; when bound by extracellular calcium ions, these G protein-coupled receptors mediate increases in the level of intracellular free calcium, which, in turn, decreases secretion of preformed PTH. By this mechanism, high plasma calcium levels suppress PTH secretion, while low plasma calcium levels stimulate PTH secretion. (Note: In many other secretory tissues, an increase in intracellular calcium enhances secretion. Thus, the parathyroid chief cell is unusual in its response to changes in intracellular calcium.)
PTH acts on three organs to raise the plasma calcium concentration: it acts directly on kidney and bone and indirectly on the gastrointestinal (GI) tract (Fig. 32-4). The most rapid physiologic effects of PTH are to increase reabsorption of calcium and decrease reabsorption of inorganic phosphate by the kidney tubules. These actions decrease renal clearance of calcium while increasing renal clearance of inorganic phosphate. In this manner, PTH raises plasma calcium levels and decreases plasma inorganic phosphate concentrations.
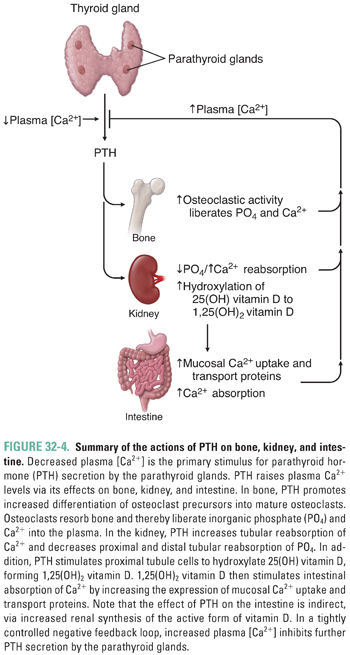
Another important, although slower, effect of PTH results from its direct actions on bone cells. Physiologic levels of PTH stimulate cell surface PTH receptors on osteoblasts, causing these cells to increase their expression of the osteoclast differentiation factor RANKL (Fig. 32-3) and decrease their expression of its antagonist OPG. The resulting increase in osteoclastic activity increases bone resorption and thereby increases the release of calcium and inorganic phosphate into the circulation. PTH also induces bone marrow stromal cells to secrete cytokines such as IL-6, and these cytokines ultimately stimulate osteoclast proliferation and bone resorption.
Finally, PTH raises plasma calcium levels by an indirect effect on the intestine. PTH stimulates the kidney not only to increase calcium reabsorption and decrease phosphate reabsorption, as described above, but also to increase the enzymatic conversion of 25-hydroxy vitamin D to 1,25-dihydroxy vitamin D (calcitriol). This hydroxylation takes place in cells of the proximal renal tubules. Calcitriol, in turn, increases small intestinal absorption of calcium and (to a lesser extent) inorganic phosphate (discussed below).
Although the release of skeletal calcium and inorganic phosphate could be considered catabolic, PTH simultaneously stimulates new bone formation by promoting differentiation of osteoblast precursors to mature osteoblasts and by enhancing osteoblast survival. Interaction of PTH with its receptor on mature osteoblasts stimulates Gαs, which increases adenylyl cyclase activity, which, in turn, increases intracellular cAMP. The PTH-induced increase in cAMP has an anti-apoptotic effect on osteoblasts. In addition, the increase in cAMP promotes osteoblast release of IGF-1, which induces osteoblast precursor cells in the bone marrow to differentiate into mature osteoblasts (Fig. 32-3).
The balance between PTH’s catabolic and anabolic effects on bone depends on the length of time extracellular PTH remains in contact with PTH receptors on osteoblasts. Specifically, intermittent, brief (1- to 3-hour) elevations in extracellular PTH increase bone formation more than bone resorption and cause a net increase in bone mass. Consequently, intermittent PTH administration by once-daily injection or by some other drug delivery technology increases bone matrix production, bone mass, bone mineral density, and bone strength (see below). In contrast, continuous elevation of extracellular PTH increases bone resorption more than bone formation and thereby causes net bone loss in patients with primary or secondary hyperparathyroidism.
Despite its name, vitamin D3 is produced in the skin and is not required in the diet if sun exposure is generous. Because it is produced endogenously and travels in the blood to effect responses in distant target tissues, vitamin D3 is more correctly considered a hormone. The term vitamin D applies to two related compounds, cholecalciferol and ergocalciferol. Cholecalciferol, or vitamin D3, is generated nonenzymatically in the skin when 7-dehydrocholesterol absorbs a photon of short ultraviolet light (UV-B; Fig. 32-5). Ergocalciferol, or vitamin D2, is produced when ergosterol in plants absorbs such a photon. Vitamins D2 and D3 are each added to dairy products and some other foods; each is available as a dietary supplement; and each is available (in much higher doses) as a prescription drug. Vitamins D2 and D3 have equal biological activities, and “vitamin D” in subsequent paragraphs refers to both the D2 and D3 forms of the hormone.
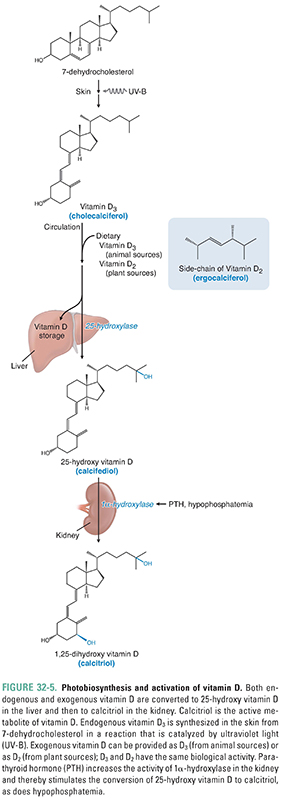
Whether from an endogenous (skin) or an exogenous (dietary) source, vitamin D travels to the liver, where it is either stored or converted to calcifediol [25-hydroxy vitamin D, or 25(OH)D] by the first of two enzymatic hydroxylation steps. The second enzymatic hydroxylation converts calcifediol to the final, active form of vitamin D called calcitriol [1α,25-dihydroxy vitamin D, or 1,25(OH)2D]. This second hydroxylation takes place in many tissues, particularly in the proximal tubule of the kidney (where it is PTH-dependent), but does not take place in the intestines because they lack the 1α-hydroxylase enzyme required for the second hydroxylation. Calcitriol’s primary effect on calcium balance is in the small intestine, where it increases the absorption of dietary calcium. Calcitriol enhances Ca2+ absorption by acting on nuclear receptors in the enterocyte to up-regulate the expression of genes coding for multiple brush border proteins. Calcitriol also promotes the transcellular transport of Ca2+ through the enterocyte by inducing the expression of (1) a calcium uptake pump on the luminal surface of the enterocyte; (2) calbindin, an intracellular Ca2+-binding protein; and (3) an ATP-dependent Ca2+ pump that extrudes Ca2+ from the enterocyte into the surrounding capillaries. Because enterocytes do not express the 1α-hydroxylase enzyme needed to form 1,25(OH)2D from 25(OH)D, their absorption of calcium is regulated by blood levels of 1,25(OH)2D, which in turn depend on renal tubular function and blood levels of PTH.
Calcitriol has important effects on other target organs, including the parathyroid glands, bone, kidneys, and immune system. Calcitriol binds to nuclear receptors in parathyroid cells and thereby inhibits PTH synthesis and release. In bone, calcitriol increases osteoclast number and activity, resulting in increased bone resorption. High blood levels of calcitriol, and lower levels of certain calcitriol analogues, increase bone formation. In the distal tubule of the kidney, calcitriol increases the reabsorption of both calcium and phosphate. In the immune system, calcitriol production by macrophages may act as a local suppressant of adaptive immune cells; this observation has led to the use of calcitriol and calcitriol analogues in the treatment of psoriasis.
Fibroblast Growth Factor 23 and Phosphatonins
It has long been known that renal clearance of inorganic phosphate is increased by high-phosphate diets and decreased by low-phosphate diets in both normal and hypoparathyroid humans and animals. Recent research has begun to elucidate the mechanisms that regulate this homeostatic response. One responsible factor is fibroblast growth factor 23 (FGF-23), a 251-amino acid protein. Injection of FGF-23 rapidly alters the activity of the renal tubular sodium-phosphate co-transporters NaPi-2a and NaPi-2c and thereby increases renal clearance of inorganic phosphate. FGF-23 also suppresses renal tubular 25-hydroxy vitamin D 1α-hydroxylase and induces renal tubular 25-hydroxy vitamin D 24-hydroxylase, thereby decreasing renal secretion of 1,25(OH)2D. The resulting decrease in blood 1,25(OH)2D reduces active transport of calcium and phosphate across the intestines and also increases parathyroid hormone secretion both directly and indirectly. Proteins with similar effects on renal tubular clearance of phosphate and/or secretion of 1,25(OH)2D include secreted frizzled-related protein (sFRP-4), FGF-7, and matrix extracellular phosphoglycoprotein [MEPE, which does not suppress renal 1,25(OH)2D secretion]. These proteins, together with FGF-23, are collectively called phosphatonins.
FGF-23 is expressed by many cells, including osteocytes. Animal models and humans with increased osteocyte expression of FGF-23 exhibit increased renal clearance of phosphate, hypophosphatemia, low or inappropriately normal blood levels of 1,25(OH)2D, and defective mineralization of bone matrix. These findings suggest that osteocytes are a particularly important source of circulating FGF-23. Mutations that generate proteolysis-resistant FGF-23 produce a nearly identical syndrome in mice and humans (e.g., human autosomal dominant hypophosphatemic rickets, ADHR). A more common hereditary form of FGF-23/phosphatonin excess in humans is X-linked hypophosphatemic rickets (XLH), caused by mutations in the endopeptidase PHEX. How PHEX mutations elevate blood FGF-23 and/or phosphatonin levels remains controversial.
Nonhereditary causes of hypophosphatemia with increased renal clearance of phosphate, low or inappropriately normal blood levels of 1,25(OH)2D, and defective mineralization of bone matrix include repeated intravenous administration of saccharated iron (which elevates blood levels of FGF-23) and rare benign phosphatonin-secreting mesenchymal tumors that elevate blood levels of FGF-23 and/or other phosphatonins (oncogenic osteomalacia). Cessation of saccharated iron therapy or ablation of the mesenchymal tumor rapidly reverses these two syndromes.
Patients undergoing chronic dialysis for chronic renal insufficiency have markedly elevated blood levels of FGF-23. The levels of FGF-23 are independently related to mortality, cardiac hypertrophy, and indices of endothelial dysfunction, but it is unclear whether lowering or neutralizing blood FGF-23 in such patients prolongs their lives.
Conditions associated with decreased levels of FGF-23 provide further support for the importance of this hormone in calcium and phosphate homeostasis. Experimental deletion of the FGF-23 gene in mice causes hyperphosphatemia and high blood levels of 1,25(OH)2D, with resulting calcitriol intoxication (hypercalcemic renal failure), extraskeletal calcium phosphate deposits (ectopic calcification, labeled tumoral calcinosis when severe), and defective bone mineralization. Preventing calcitriol intoxication in these mice by dietary or genetic manipulations reveals that FGF-23 deletion impairs modulation of renal phosphate clearance in response to high-phosphate and low-phosphate diets. Other genetic manipulations, including disruption of FGF-23 glycosylation or inactivation of FGF-23 receptors, produce a similar effect.
When humans are fed high-phosphate or low-phosphate diets, their renal clearance of phosphate changes as described above. However, their blood levels of FGF-23 change less than expected, or sometimes not at all. Whether this discordance reflects the importance of other phosphatonins, or other variables, is not yet clear. It is also unclear whether serum inorganic phosphate regulates FGF-23 secretion and/or catabolism, because changes in serum inorganic phosphate are not consistently correlated with changes in serum FGF-23 levels. Although 1,25(OH)2D can increase FGF-23 secretion and blood levels, other mechanisms must be more important regulators of serum FGF-23, because serum FGF-23 is poorly correlated with serum 1,25(OH)2D.
Calcitonin, Glucocorticoids, Thyroid Hormone, and Gonadal Steroids
PTH, vitamin D, and FGF-23 are the primary regulators of calcium and phosphate homeostasis, but several other endogenous hormones also have important effects on bone mineral metabolism. These hormones include calcitonin, glucocorticoids, thyroid hormone, estrogens, and androgens.
Calcitonin is important to calcium homeostasis in some animals but less important in humans. This hormone is a 32-amino acid peptide that is synthesized and released by parafollicular C cells of the thyroid gland in response to hypercalcemia. Calcitonin binds directly to receptors on osteoclasts; this binding inhibits the resorptive activity of the osteoclasts and thereby decreases bone resorption and plasma calcium levels. In adult humans, endogenous calcitonin has only weak effects on plasma calcium levels, and the elimination of calcitonin secretion after thyroidectomy generally causes no significant changes in plasma calcium levels. Nevertheless, exogenous calcitonin is useful in the emergency treatment of certain forms of hypercalcemia, as discussed below.
Pharmacologic doses of glucocorticoids promote osteocyte and osteoblast apoptosis and inhibit osteoblast maturation and osteoblast activity, thereby decreasing bone formation and, to a lesser extent, bone resorption. Chronic glucocorticoid use is a common cause of iatrogenic bone loss, osteoporosis, and fractures. When taking the history of a patient such as RS, it is important to determine whether she has ever taken glucocorticoids for months at a time, because this would be a significant risk factor for osteoporosis. Pharmacologic doses of glucocorticoids also decrease intestinal absorption of calcium and (at high doses) renal tubular reabsorption of calcium. The latter effects would tend to lower plasma calcium levels; however, glucocorticoid use is not associated with hypocalcemia or changes in blood PTH, presumably because glucocorticoid-induced bone loss releases compensating amounts of skeletal calcium.
Excess thyroid hormone also increases bone turnover. By stimulating bone resorption more than bone formation, prolonged high levels of thyroid hormone can cause bone loss. In fact, low bone mass is a common manifestation of hyperthyroidism. Therefore, the evaluation of RS’s osteoporosis should include assessment of her thyroid status and measurement of her serum TSH level to rule out hyperthyroidism (see Chapter 28, Pharmacology of the Thyroid Gland).
Estrogens and androgens inhibit osteoclastic activity and thereby slow the rate of bone turnover and bone loss. Among other effects, these gonadal steroids inhibit the production of RANKL by immune cells and the production by osteoblasts of cytokines such as interleukin-6 that recruit and activate osteoclasts. Estrogen also has a pro-apoptotic effect on osteoclasts and an anti-apoptotic effect on osteoblasts and osteocytes. As described in more detail in Chapter 30, Pharmacology of Reproduction, estrogen exerts its actions principally by binding to the estrogen receptor (ER), which is a nuclear transcription factor. Binding of estrogen facilitates dimerization of the ER, allowing the estrogen–ER complex to recruit coactivator or corepressor molecules and bind to promoter regions of target genes. In this way, estrogen regulates the transcription of target genes encoding, for example, the cytokines that are important in bone turnover.

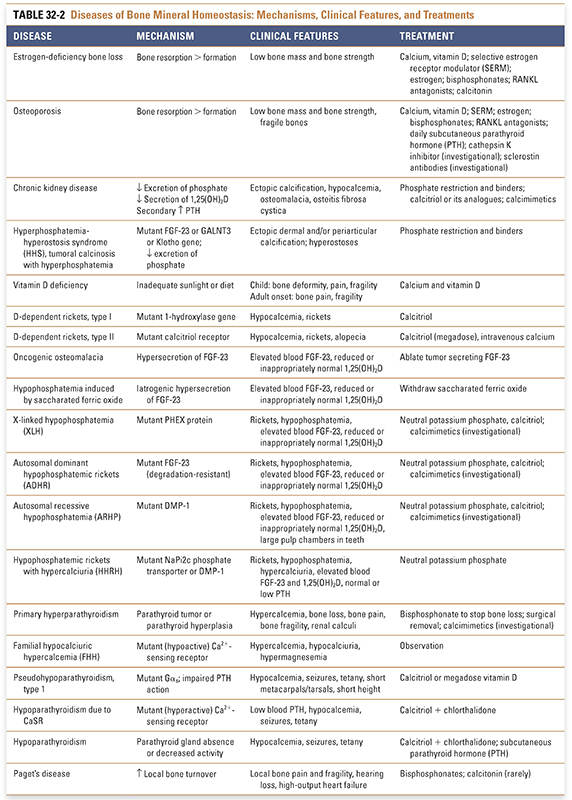
Bone turnover, including repeated cycles of bone resorption and bone formation, is required to maintain the integrity of the skeleton. Osteoporosis and chronic kidney disease are two common disorders of bone mineral homeostasis. In osteoporosis, bone turnover is disrupted such that bone resorption exceeds bone formation. In chronic kidney disease, the pathophysiology involves a complex interplay between decreased mineral absorption and secondary hyperparathyroidism. A summary of these and related diseases of bone mineral homeostasis—including their mechanisms, clinical features, and treatments—is provided in Table 32-2.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


