Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, Metabolism, and Elimination
The absorption, distribution, metabolism (biotransformation), and elimination of drugs (ADME) are the processes of pharmacokinetics (Figure 2–1). Understanding and employing pharmacokinetic principles can increase the probability of therapeutic success and reduce the occurrence of adverse drug effects in the body.
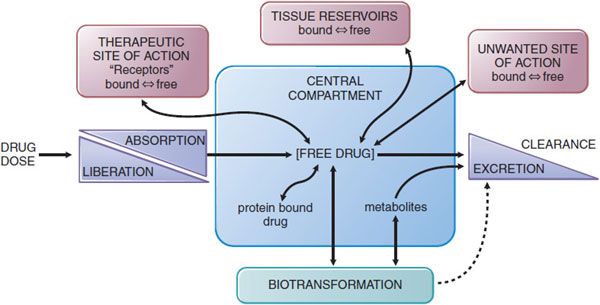
Figure 2-1 The interrelationship of the absorption, distribution, binding, metabolism, and excretion of a drug and its concentration at its sites of action. Possible distribution and binding of metabolites in relation to their potential actions at receptors are not depicted.
PHYSICOCHEMICAL FACTORS IN TRANSFER OF DRUGS ACROSS MEMBRANES
The absorption, distribution, metabolism, excretion, and action of a drug involve its passage across cell membranes. Mechanisms by which drugs cross membranes and the physicochemical properties of molecules and membranes that influence this transfer are critical to understanding the disposition of drugs in the human body. The characteristics of a drug that predict its movement and availability at sites of action are its molecular size and structural features, degree of ionization, relative lipid solubility of its ionized and nonionized forms, and its binding to serum and tissue proteins. Although barriers to drug movement may be a single layer of cells (e.g., intestinal epithelium) or several layers of cells and associated extracellular protein (e.g., skin), the plasma membrane represents the common barrier to drug distribution.
The plasma membrane consists of a bilayer of amphipathic lipids with their hydrocarbon chains oriented inward to the center of the bilayer to form a continuous hydrophobic phase, with their hydrophilic heads oriented outward. Individual lipid molecules in the bilayer vary according to the particular membrane and can move laterally and organize themselves with cholesterol (e.g., sphingolipids), endowing the membrane with fluidity, flexibility, organization, high electrical resistance, and relative impermeability to highly polar molecules. Membrane proteins embedded in the bilayer serve as structural anchors, receptors, ion channels, or transporters to transduce electrical or chemical signaling pathways and provide selective targets for drug actions. Membranes are highly ordered and compartmented. Membrane proteins may be associated with caveolin and sequestered within caveolae, excluded from caveolae, or be organized in signaling domains rich in cholesterol and sphingolipid not containing caveolin or other scaffolding proteins (i.e., lipid rafts). Cell membranes are relatively permeable to water either by diffusion or by flow resulting from hydrostatic or osmotic differences across the membrane, and bulk flow of water can carry with it small drug molecules (<200 Da). Paracellular passage through intercellular gaps is sufficiently large that transfer across capillary endothelium is generally limited by blood flow (Figure 2–2). Capillaries of the central nervous system (CNS) and a variety of epithelial tissues have tight junctions. Bulk-flow transfer is limited when the molecular mass of the solute exceeds 100-200 Da. Accordingly, most large lipophilic drugs must pass through the cell membrane itself (see Figure 2–2) by passive and active processes.
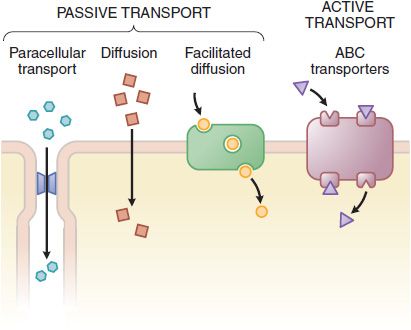
Figure 2-2 The variety of ways drugs move across cellular barriers in their passage throughout the body. See details in Figure 5-4.
PASSIVE MEMBRANE TRANSPORT. In passive transport, the drug molecule usually penetrates by diffusion along a concentration gradient by virtue of its solubility in the lipid bilayer. Such transfer is directly proportional to the magnitude of the concentration gradient across the membrane, to the lipid-water partition coefficient of the drug, and to the membrane surface area exposed to the drug. At steady state, the concentration of the unbound drug is the same on both sides of the membrane if the drug is a nonelectrolyte. For ionic compounds, the steady-state concentrations depend on the electrochemical gradient for the ion and on differences in pH across the membrane, which will influence the state of ionization of the molecule disparately on either side of the membrane and can effectively trap drug on one side of the membrane.
WEAK ELECTROLYTES AND THE INFLUENCE OF pH. Many drugs are weak acids or bases that are present in solution as both the lipid-soluble and diffusible nonionized form, and the relatively lipid-insoluble nondiffusible ionized species. The transmembrane distribution of a weak electrolyte is influenced by its pKa and the pH gradient across the membrane. The pKa is the pH at which half the drug (weak acid or base electrolyte) is in its ionized form. The ratio of nonionized to ionized drug at any pH may be calculated from the Henderson-Hasselbalch equation:
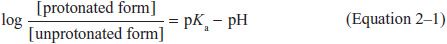
Equation 2–1 relates the pH of the medium around the drug and the drug’s acid dissociation constant (pKa) to the ratio of the protonated (HA or BH+) and unprotonated (A– or B) forms, where HA ↔ A– + H+ (Ka = [A–][H+]/[HA]) describes the dissociation of an acid, and BH+ ↔ B + H+ (Ka = [B][H+]/[BH+]) describes the dissociation of the protonated form of a base. At steady state, an acidic drug will accumulate on the more basic side of the membrane and a basic drug on the more acidic side. This phenomenon, known as ion trapping, is an important process in drug distribution (Figure 2–3).
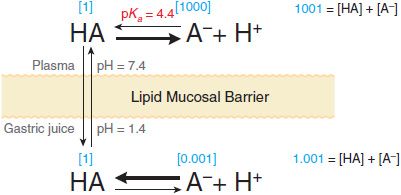
Figure 2-3 Influence of pH on the distribution of a weak acid (pKa = 4.4) between plasma and gastric juice separated by a lipid barrier. Dissociation of the weak acid in plasma (pH 7.4) and gastric acid (pH 1.4). The uncharged form, HA, equilibrates across the membrane. Blue numbers in brackets show relative concentrations of HA and A–, as calculated from Equation 2-1.
CARRIER-MEDIATED MEMBRANE TRANSPORT. Active transport and facilitated diffusion are carrier-mediated processes. Pharmacologically important transporters may mediate either drug uptake or efflux and often facilitate vectorial transport across polarized cells. An important efflux transporter is the P-glycoprotein encoded by the multidrug resistance-1 (MDR1) gene (see Table 5–4). P-glycoprotein localized in the enterocyte limits the absorption of some orally administered drugs because it exports compounds into the lumen of the GI tract subsequent to their absorption. The P-glycoprotein also can confer resistance to some cancer chemotherapeutic agents (see Chapters 60–63). Transporters and their roles in drug action are presented in detail in Chapter 5.
DRUG ABSORPTION, BIOAVAILABILITY, AND ROUTES OF ADMINISTRATION
Absorption is the movement of a drug from its site of administration into the central compartment (see Figure 2–1). For solid dosage forms, absorption first requires dissolution of the tablet or capsule, thus liberating the drug. The clinician is concerned primarily with bioavailability rather than absorption. Bioavailability describes the fractional extent to which a dose of drug reaches its site of action or a biological fluid from which the drug has access to its site of action.
For example, a drug given orally must be absorbed first from the GI tract, but net absorption may be limited by the characteristics of the dosage form, the drug’s physicochemical properties, by intestinal metabolism, and by export into the intestinal lumen. The absorbed drug then passes through the liver, where metabolism and biliary excretion may occur before the drug enters the systemic circulation. Accordingly, a fraction of the administered and absorbed dose of drug will be inactivated or diverted in the intestine and liver before it can reach the general circulation and be distributed to its sites of action. If the metabolic or excretory capacity of the liver and the intestine for the drug is large, bioavailability will be reduced substantially (first-pass effect). This decrease in availability is a function of the anatomical site from which absorption takes place; other anatomical, physiological, and pathological factors can influence bioavailability (described later), and the choice of the route of drug administration must be based on an understanding of these conditions.
ORAL (ENTERAL) VERSUS PARENTERAL ADMINISTRATION. Some characteristics of the major routes employed for systemic drug effect are compared in Table 2–1.
Table 2–1
Some Characteristics of Common Routes of Drug Administrationa
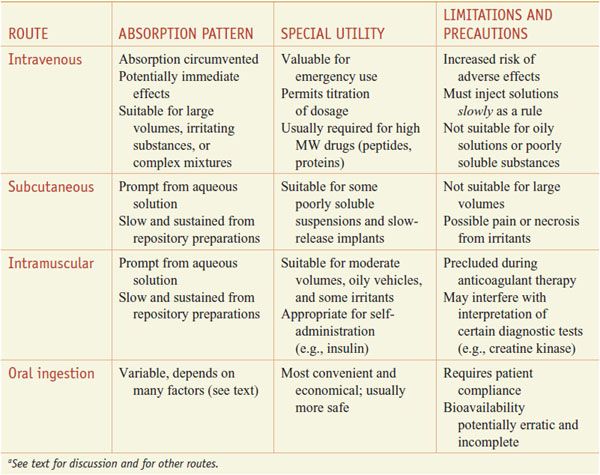
Oral ingestion is the most common method of drug administration. It also is the safest, most convenient, and most economical. Its disadvantages include limited absorption of some drugs because of their physical characteristics (e.g., low water solubility or poor membrane permeability), emesis as a result of irritation to the GI mucosa, destruction of some drugs by digestive enzymes or low gastric pH, irregularities in absorption or propulsion in the presence of food or other drugs, and the need for cooperation on the part of the patient. In addition, drugs in the GI tract may be metabolized by the enzymes of the intestinal flora, mucosa, or liver before they gain access to the general circulation.
Parenteral injection of drugs has distinct advantages over oral administration. In some instances, parenteral administration is essential for the drug to be delivered in its active form, as in the case of monoclonal antibodies. Availability usually is more rapid, extensive, and predictable when a drug is given by injection; the effective dose can be delivered more accurately. In emergency therapy and when a patient is unconscious, uncooperative, or unable to retain anything given by mouth, parenteral therapy may be necessary. Parenteral administration also has its disadvantages: asepsis must be maintained, especially when drugs are given over time (e.g., intravenous or intrathecal administration); pain may accompany the injection; and it is sometimes difficult for patients to perform the injections themselves if self-medication is necessary.
ORAL ADMINISTRATION. Absorption from the GI tract is governed by factors such as surface area for absorption, blood flow to the site of absorption, the physical state of the drug (solution, suspension, or solid dosage form), its water solubility, and the drug’s concentration at the site of absorption. For drugs given in solid form, the rate of dissolution may limit their absorption. Because most drug absorption from the GI tract occurs by passive diffusion, absorption is favored when the drug is in the nonionized, more lipophilic form. Based on the pH-partition concept (Figure 2–3), one would predict that drugs that are weak acids would be better absorbed from the stomach (pH 1-2) than from the upper intestine (pH 3-6), and vice versa for weak bases. However the epithelium of the stomach is lined with a thick mucus layer, and its surface area is small; by contrast, the villi of the upper intestine provide an extremely large surface area (~200 m2). Accordingly, the rate of absorption of a drug from the intestine will be greater than that from the stomach even if the drug is predominantly ionized in the intestine and largely nonionized in the stomach. Thus, any factor that accelerates gastric emptying will generally increase the rate of drug absorption, whereas any factor that delays gastric emptying is expected to have the opposite effect. Gastric emptying rate is influenced by numerous factors, including the caloric content of food; volume, osmolality, temperature, and pH of ingested fluid; diurnal and interindividual variation; metabolic state (rest or exercise); and the ambient temperature. Gastric emptying is influenced in women by the effects of estrogen (i.e., compared to men, it is slower for premenopausal women and those taking estrogen replacement therapy).
Drugs that are destroyed by gastric secretions and low pH or that cause gastric irritation sometimes are administered in dosage forms with an enteric coating that prevents dissolution in the acidic gastric contents. Enteric coatings are useful for drugs such as aspirin, which can cause gastric irritation, and for presenting a drug such as mesalamine to sites of action in the ileum and colon (see Figure 47–4).
Controlled-Release Preparations. The rate of absorption of a drug administered as a tablet or other solid oral dosage form is partly dependent on its rate of dissolution in GI fluids. This is the basis for controlled-release, extended-release, sustained-release, and prolonged-action pharmaceutical preparations that are designed to produce slow, uniform absorption of the drug for 8 h or longer. Potential advantages of such preparations are reduction in the frequency of administration compared with conventional dosage forms (often with improved compliance by the patient), maintenance of a therapeutic effect overnight, and decreased incidence and/or intensity of undesired effects (by dampening of the peaks in drug concentration) and nontherapeutic blood levels of the drug (by elimination of troughs in concentration) that often occur after administration of immediate-release dosage forms. Controlled-release dosage forms are most appropriate for drugs with short half-lives (t1/2 <4 h) or in selected patient groups such as those receiving antiepileptics.
Sublingual Administration. Venous drainage from the mouth is to the superior vena cava, bypassing the portal circulation and thereby protecting the drug from rapid intestinal and hepatic first-pass metabolism. For example, nitroglycerin (see Chapter 27) is effective when retained sublingually because it is nonionic and has very high lipid solubility.
TRANSDERMAL ABSORPTION. Absorption of drugs able to penetrate the intact skin is dependent on the surface area over which they are applied and their lipid solubility (see Chapter 65). Systemic absorption of drugs occurs much more readily through abraded, burned, or denuded skin. Toxic effects result from absorption through the skin of highly lipid-soluble substances (e.g., a lipid-soluble insecticide in an organic solvent). Absorption through the skin can be enhanced by suspending the drug in an oily vehicle and rubbing the resulting preparation into the skin. Hydration of the skin with an occlusive dressing may be used to facilitate absorption. Controlled-release topical patches have become increasingly available, including nicotine for tobacco-smoking withdrawal, scopolamine for motion sickness, nitroglycerin for angina pectoris, testosterone and estrogen for replacement therapy, various estrogens and progestins for birth control, and fentanyl for pain relief.
RECTAL ADMINISTRATION. Approximately 50% of the drug that is absorbed from the rectum will bypass the liver; thus reducing hepatic first-pass metabolism. However, rectal absorption can be irregular and incomplete, and certain drugs can cause irritation of the rectal mucosa.
PARENTERAL INJECTION. The major routes of parenteral administration are intravenous, subcutaneous, and intramuscular. Absorption from subcutaneous and intramuscular sites occurs by simple diffusion along the gradient from drug depot to plasma. The rate is limited by the area of the absorbing capillary membranes and by the solubility of the substance in the interstitial fluid. Relatively large aqueous channels in the endothelial membrane account for the indiscriminate diffusion of molecules regardless of their lipid solubility. Larger molecules, such as proteins, slowly gain access to the circulation by way of lymphatic channels. Drugs administered into the systemic circulation by any route, excluding the intraarterial route, are subject to possible first-pass elimination in the lung prior to distribution to the rest of the body. The lungs also serve as a filter for particulate matter that may be given intravenously and provide a route of elimination for volatile substances.
Intravenous. Factors limiting absorption are circumvented by intravenous injection of drugs in aqueous solution because bioavailability is complete and rapid. Also, drug delivery is controlled and achieved with an accuracy and immediacy not possible by any other procedure. Certain irritating solutions can be given only in this manner because the drug, when injected slowly, is greatly diluted by the blood.
There are advantages and disadvantages to intravenous administration. Unfavorable reactions can occur because high concentrations of drug may be attained rapidly in plasma and tissues. There are therapeutic circumstances where it is advisable to administer a drug by bolus injection (e.g., tissue plasminogen activator) and other circumstances where slower administration of drug is advisable (e.g., antibiotics). Intravenous administration of drugs warrants close monitoring of the patient’s response; once the drug is injected, there is often no retreat. Repeated intravenous injections depend on the ability to maintain a patent vein. Drugs in an oily vehicle, those that precipitate blood constituents or hemolyze erythrocytes, and drug combinations that cause precipitates to form must not be given by this route.
Subcutaneous. Injection into a subcutaneous site can be done only with drugs that are not irritating to tissue; otherwise, severe pain, necrosis, and tissue sloughing may occur. The rate of absorption following subcutaneous injection of a drug often is sufficiently constant and slow to provide a sustained effect. Moreover, altering the period over which a drug is absorbed may be varied intentionally, as is accomplished with insulin for injection using particle size, protein complexation, and pH. The incorporation of a vasoconstrictor agent in a solution of a drug to be injected subcutaneously also retards absorption. Absorption of drugs implanted under the skin in a solid pellet form occurs slowly over a period of weeks or months; some hormones (e.g., contraceptives) are administered effectively in this manner.
Intramuscular. Drugs in aqueous solution are absorbed rapidly after intramuscular injection depending on the rate of blood flow to the injection site. This may be modulated to some extent by local heating, massage, or exercise. Generally, the rate of absorption following injection of an aqueous preparation into the deltoid or vastus lateralis is faster than when the injection is made into the gluteus maximus. The rate is particularly slower for females after injection into the gluteus maximus. This has been attributed to the different distribution of subcutaneous fat in males and females and because fat is relatively poorly perfused. Slow, constant absorption from the intramuscular site results if the drug is injected in solution in oil or suspended in various other repository (depot) vehicles.
Intraarterial. Occasionally, a drug is injected directly into an artery to localize its effect in a particular tissue or organ, such as in the treatment of liver tumors and head and neck cancers. Diagnostic agents sometimes are administered by this route (e.g., technetium-labeled human serum albumin).
Intrathecal. The blood-brain barrier and the blood-cerebrospinal fluid (CSF) barrier often preclude or slow the entrance of drugs into the CNS. Therefore, when local and rapid effects of drugs on the meninges or cerebrospinal axis are desired, drugs sometimes are injected directly into the spinal subarachnoid space. Brain tumors also may be treated by direct intraventricular drug administration.
PULMONARY ABSORPTION. Gaseous and volatile drugs may be inhaled and absorbed through the pulmonary epithelium and mucous membranes of the respiratory tract. Access to the circulation is rapid by this route because the lung’s surface area is large. In addition, solutions of drugs can be atomized and the fine droplets in air (aerosol) inhaled. Advantages are the almost instantaneous absorption of a drug into the blood, avoidance of hepatic first-pass loss, and in the case of pulmonary disease, local application of the drug at the desired site of action (see Chapters 19 and 36).
TOPICAL APPLICATION
Mucous Membranes. Drugs are applied to the mucous membranes of the conjunctiva, nasopharynx, oropharynx, vagina, colon, urethra, and urinary bladder primarily for their local effects.
Eye. Topically applied ophthalmic drugs are used primarily for their local effects (see Chapter 64).
BIOEQUIVALENCE
Drug products are considered to be pharmaceutical equivalents if they contain the same active ingredients and are identical in strength or concentration, dosage form, and route of administration. Two pharmaceutically equivalent drug products are considered to be bioequivalent when the rates and extents of bioavailability of the active ingredient in the 2 products are not significantly different under suitable test conditions. However, brand name and generic forms of the same drug are not always legally equivalent; law suits that have succeeded against the makers of brand name drugs have failed against the producers of the equivalent generic forms (see recent cases involving Phenergan and generic promethazine). Generic versus brand name prescribing is further discussed in connection with drug nomenclature and the choice of drug name in writing prescription orders (see Appendix I).
DISTRIBUTION OF DRUGS
Following absorption or systemic administration into the bloodstream, a drug distributes into interstitial and intracellular fluids depending on the particular physicochemical properties of the individual drug. Cardiac output, regional blood flow, capillary permeability, and tissue volume determine the rate of delivery and potential amount of drug distributed into tissues. Initially, liver, kidney, brain, and other well-perfused organs receive most of the drug; delivery to muscle, most viscera, skin, and fat is slower. This second distribution phase may require minutes to several hours before the concentration of drug in tissue is in equilibrium with that in blood. The second phase also involves a far larger fraction of body mass (e.g., muscle) than does the initial phase and generally accounts for most of the extravascularly distributed drug. With exceptions such as the brain, diffusion of drug into the interstitial fluid occurs rapidly because of the highly permeable nature of the capillary endothelial membrane. Thus, tissue distribution is determined by the partitioning of drug between blood and the particular tissue.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


