Chapter 2 Pharmacokinetics: Absorption, Distribution, Metabolism, and Elimination
| Abbreviations | |
|---|---|
| CLh | Hepatic clearance |
| CLr | Renal clearance |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| GI | Gastrointestinal |
| Km | Michaelis constant |
| NAD(P) | Nicotinamide adenine dinucleotide (phosphate) |
| pH | Logarithm of the reciprocal of the hydrogen ion concentration |
| pKa | Logarithm of the reciprocal of the dissociation constant |
| UDP | Uridine diphosphate |
| Vmax | Maximum rate of reaction |
WHAT HAPPENS TO DRUGS IN THE BODY?
In nearly all cases drugs must traverse membranes to reach their site of action. The ease by which a compound crosses membranes is key to assessing the rates and extent of absorption and distribution throughout multiple compartments of the body. This chapter considers factors for assessing how specific drugs cross membranes and what variables are most important.
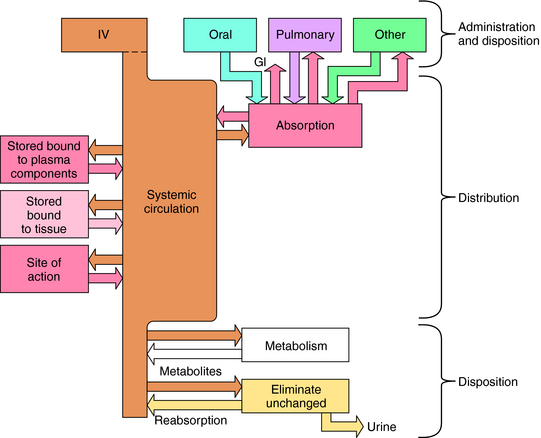
Numerous factors influence the rate of delivery, distribution, and disappearance of drug to and from its site of action. All these processes and variables, depicted in Figure 2-1, are described in this chapter.
Transport of Drugs Across Membranes
some affinity for H2O (i.e., hydrophilicity), or they cannot dissolve and be transported to their sites of action by the blood and other body fluids.
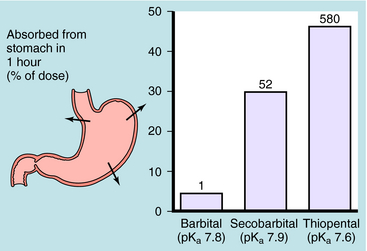
Generally, drugs that have high lipid solubility cross membranes better than those with low lipid solubility. This is exemplified in Figure 2-2 for three different barbiturates. The oil/water equilibrium partition coefficient is a measure of lipid solubility, or hydrophobicity. Drug is added to a mixture of equal volumes of oil and H2O, and the mixture is agitated to promote solubilization of the compound in each phase. When equilibrium is attained, the phases are separated and undergo assay for drug. The ratio of the concentration in the two phases is the partition coefficient. Therefore the larger the partition coefficient is, the greater the lipid solubility is. Figure 2-2 shows that absorption across the stomach wall is greater for the barbiturate with the largest lipid solubility.
Influence of pH on Drug Absorption and Distribution
Passive diffusion of a drug that is a weak electrolyte is generally a function of the pKa of the drug and the pH of the two compartments, because only the uncharged form of the drug can diffuse across membranes. The pH values of the major body fluids, which range from 1 to 8, are shown in Table 2-1. To predict how a drug will be distributed between gastric juice (pH 1.0) on one side of the membrane and blood (pH 7.4) on the other side, the degree of dissociation of the drug at each pH value is determined.
TABLE 2–1 pH of Selected Body Fluids
| Fluids | pH |
|---|---|
| Gastric juice | 1.0-3.0 |
| Small intestine: duodenum | 5.0-6.0 |
| Small intestine: ileum | 8 |
| Large intestine | 8 |
| Plasma | 7.4 |
| Cerebrospinal fluid | 7.3 |
| Urine | 4.0-8.0 |
An acid is defined as a compound that can dissociate and release a hydrogen ion, whereas a base can take up a hydrogen ion. By this definition, RCOOH and RNH3+ are acids and RCOO–and RNH2 are bases. The equilibrium dissociation expression and the equilibrium dissociation constant (Ka) can be described for an acid HA or BH+ and a base A– or B, as shown below. The convention for Ka requires that the acid appear on the left and the base appear on the right of the dissociation equation as:
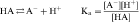
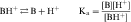
Taking the negative log of both sides yields:
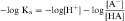
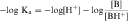
By definition the negative log of [H+] is pH and the negative log of Ka is pKa. Therefore, equations 2-3 and 2-4 can be simplified and rearranged to give:
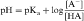
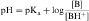
Equations 2-5 and 2-6 are the acid and base forms, respectively, of the Henderson-Hasselbach equation, and they can be used to calculate the pH of the solution when the pKa and the ratios of [A–]/[HA] or [B]/[BH+] are known. In pharmacology it is often of interest to calculate the ratios of [A–]/[HA] or [B]/[BH+] when the pH and the pKa are known. For this calculation, equations 2-5 and 2-6 are rearranged to equations 2-7 and 2-8 as follows:
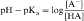
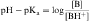
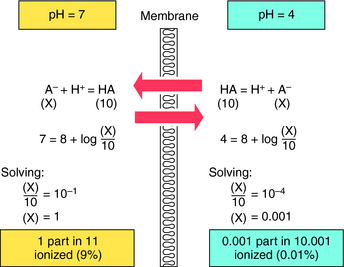
The results are plotted in Figure 2-3 to show the fraction of the nonionized (HA or B) forms. The pKa is the pH when the drug is 50% dissociated. Applying equations 2-7 and 2-8 to an acidic drug with a pKa of 6.0 enables one to calculate the degree of ionization for this drug in the stomach or blood (assuming the blood pH is 7.0 for ease of calculation), as follows:
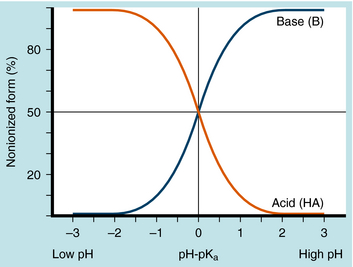
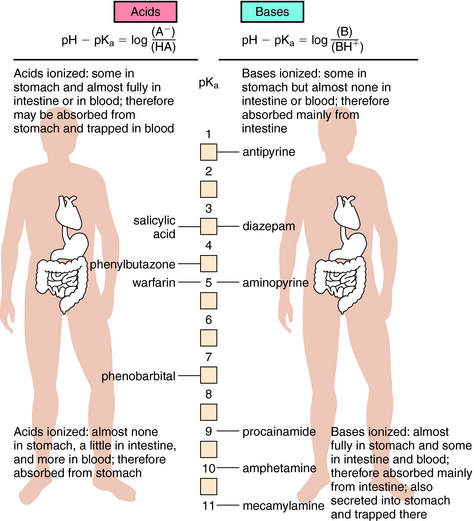
Another example is shown in Figure 2-4 for a basic drug. This approach is particularly useful for predicting whether drugs can be absorbed in the stomach, the upper intestine, or not at all. Figure 2-5 provides a summary of the effects of pH on drug absorption in the GI tract for several acidic and basic drugs. It also assists in predicting which drugs will undergo tubular reabsorption, which is discussed later.
Most drugs are transported across membranes by simple passive diffusion. The concentration gradient across the membrane is the driving force that establishes the rate of diffusion from high to low concentrations. Other mechanisms, including active transport, facilitated diffusion, or pinocytosis, also exist. Active transport involves specific carrier molecules in the membrane that bind to and carry the drug across the lipid bilayer. Because there are a finite number of carrier molecules, they exhibit classical saturation kinetics. Drugs may also compete with a specific carrier molecule for transport, which can lead to drug-drug interactions that modify the time and intensity of action of a given drug. An active transport system may concentrate a drug on one side of a membrane, because cellular energy is used to drive transport, with no dependence on a concentration gradient. The primary active drug transport systems are present in renal tubule cells, biliary tract, blood-brain barrier, and the GI tract.
Distribution to Special Organs and Tissues
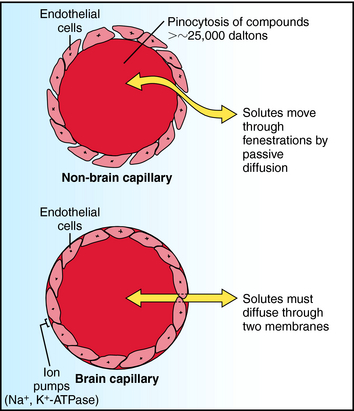
Two compartments of special importance are the brain and the fetus. Many drugs do not readily enter brain. Capillaries in brain differ structurally from those in other tissues, with the result that a barrier exists between blood within brain capillaries and the extracellular fluid in brain tissue. This blood-brain barrier hinders transport of drugs and other materials from blood into brain tissue. The blood-brain barrier is found throughout brain and spinal cord at all regions central to the arachnoid membrane, except for the floor of the hypothalamus and the area postrema. Structural differences between brain and non-brain capillaries, and how these differences influence blood-brain transport of solutes, are shown schematically in Figure 2-6. Non-brain capillaries have fenestrations (openings) between the endothelial cells through which solutes move readily by passive diffusion, with compounds having molecular weights greater than approximately 25,000 daltons (Da) undergoing transport by pinocytosis. In brain capillaries, tight junctions are present because there are no fenestrations, and pinocytosis is greatly reduced. Special transport systems are available at brain capillaries for glucose, amino acids, amines, purines, nucleosides, and organic acids; all other materials must cross two endothelial membranes plus the endothelial cytoplasm to move from capillary blood to tissue extracellular fluid. Thus the main route of drug entry into central nervous system (CNS) tissue is by passive diffusion across membranes, restricting the available compounds used to treat brain disorders. At the same time, the potential deleterious effects of many compounds on the CNS are not realized, because the blood-brain barrier acts as a safety buffer. Generally, only highly lipid-soluble drugs cross the blood-brain barrier, and thus for these drugs no blood-brain barrier exists. In infants and the elderly, the blood-brain barrier may be compromised, and drugs may diffuse into brain.