Chapter 20 Antihypertensive Drugs
| Abbreviations | |
|---|---|
| ACE | Angiotensin-converting enzyme |
| ARB | Angiotensin receptor blocker |
| AV | Atrioventricular |
| CNS | Central nervous system |
| CO | Cardiac output |
| Epi | Epinephrine |
| NE | Norepinephrine |
| TPR | Total peripheral resistance |
Therapeutic Overview
upper end of the normal distribution of blood pressure values for their population group. This most common form of hypertension, with no readily identifiable cause, is called essential hypertension. It is usually first diagnosed in middle-aged people but can also be found in children and young adults. Because of its prevalence, it is the disease most often treated with antihypertensive drugs.
Adoption of healthy lifestyles may lower blood pressure as much as some drugs. It may also prevent the onset or progression of hypertension. Patients differ in their sensitivity to these techniques. For example, maintenance of normal body weight and increased physical activity lowers blood pressure in most sedentary and overweight hypertensive individuals, whereas Na+ restriction lowers
| Therapeutic Overview |
|---|
| Hypertension is defined as: |
| Systolic pressure >140 mm Hg and/or diastolic pressure >90 mm Hg |
| Hypertension is a major risk factor for: |
| Atherosclerosis |
| Coronary artery disease |
| Congestive heart failure |
| Diabetes |
| Insulin resistance |
| Stroke |
| Renal disease |
| Retinal disease |
| Hypertension therapy |
| Nonpharmacological |
| Weight reduction, dietary (reduce salt and saturated fat, increase fruits and vegetables, use low-fat dairy products), exercise, smoking cessation, decrease excessive (>30 mL/day) alcohol intake |
| Pharmacological |
| Diuretics, renin-angiotensin inhibitors, sympatholytics, Ca++ channel blockers, direct vasodilators |
blood pressure mainly in hypertensive people categorized as “salt-sensitive.” The major advantage of nonpharmacological therapies is relative safety, as compared with drug therapy. Their principal limitation is the lack of compliance by most people. For most hypertensive patients control of hypertension requires drug treatment to achieve an adequate, sustained blood pressure reduction. Nevertheless, lifestyle modification plays a valuable and important role in management.
Mechanisms of Action
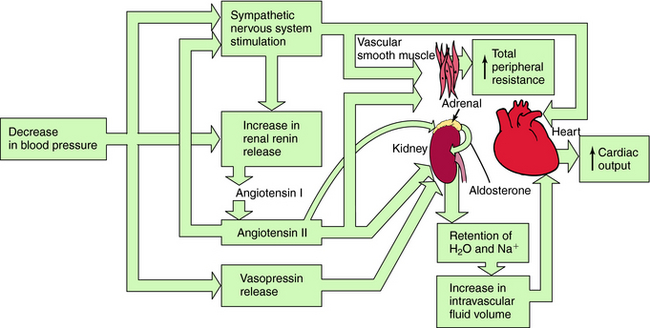
Systemic blood pressure is regulated redundantly by several physiological control systems to ensure optimal tissue perfusion throughout the body. When blood pressure decreases by any means, including antihypertensive drug therapy, one or more of these regulatory mechanisms are activated to compensate for decreases in arterial blood pressure (Fig. 20-1).
The Sympathetic Nervous System
A decrease in blood pressure activates the baroreceptor reflex (Chapter 19), producing increased sympathetic activity, leading to:
Renin-Angiotensin-Aldosterone System
A decrease in arterial pressure produces a decrease in renal perfusion pressure and baroreflex-mediated sympathetic activation of renal β1 adrenergic receptors, inducing the release of renin from the juxtaglomerular cells of the kidney into the blood. Renin cleaves the decapeptide angiotensin I from a circulating glycoprotein, angiotensinogen, which is synthesized mainly in liver. Angiotensin I is converted to the octapeptide angiotensin II by angiotensin–converting enzyme (ACE) present in endothelial cell membranes, especially in the lung. Angiotensin II constricts blood vessels, enhances sympathetic nervous system activity, and causes renal Na+ and H2O retention by direct intrarenal actions and by stimulating the adrenal cortex to release aldosterone (see Fig. 20-1).
Diuretics cause Na+ excretion and reduce fluid volume by inhibiting electrolyte transport in the renal tubules. The diuretics can be classified into three broad categories related to their sites and mechanisms of action (see Chapter 21). Thiazide diuretics inhibit the Na+/Cl− cotransporter principally in the distal convoluted tubules and produce a relatively sustained diuresis, natriuresis, and kaliuresis. These diuretics are most effective in patients with adequate renal function.
Drugs Affecting the Sympathetic Nervous System
Adrenergic Receptor Antagonists
There is wide diversity in the pharmacological profile of adrenergic β receptor antagonists (see Chapter 11). Some of these compounds, such as propranolol and pindolol, are nonselective and antagonize both β1 and β2 receptors, whereas others, like atenolol, are selective for the β1 receptor subtype. In addition, some β receptor blockers such as pindolol have modest intrinsic sympathomimetic activity, while others including labetalol and carvedilol are competitive antagonists at α1, β1, and β2 adrenergic receptors. Despite these differences, all β receptor blockers used for the treatment of hypertension share the common characteristic of competitively antagonizing the effects of norepinephrine (NE) and epinephrine (Epi) on β1 adrenergic receptors in the heart and renin-secreting cells of the kidney. Furthermore, clinically useful α1 adrenergic receptor antagonists lower blood pressure by blocking α1 receptors on vascular smooth muscle.
Centrally Acting Sympatholytics
The antihypertensive effects of the prodrug α-methyldopa are attributed to its conversion in the brain by l-aromatic amino acid decarboxylase and dopamine-β-hydroxylase to α-methyl-NE, which is a preferential agonist at α2 adrenergic receptors (see Chapter 11, Fig. 11-12). Clonidine, guanfacine, and guanabenz, which readily enter the brain after systemic administration, are selective agonists at central α2 receptors (see Chapter 11). In addition, because these drugs are taken orally for the treatment of hypertension, activation of presynaptic α2 adrenergic receptors on peripheral sympathetic nerve terminals may inhibit the release of NE and potentially contribute to their antihypertensive action.
The central site(s) where α2 receptor agonists act to lower blood pressure have not been completely identified and characterized but may include the nucleus of the solitary tract and the C1 neurons of the rostral ventrolateral medulla (see Chapter 19).
Voltage-dependent Ca++ channels play important roles in the excitation-contraction-relaxation cycle (see Chapter 22 and Chapter 24). Under resting conditions, when intracellular Ca++ is low, regulatory proteins prevent actin and myosin filaments from interacting with each other, and muscle is relaxed. When intracellular Ca++ concentrations increase by influx or release from internal stores, Ca++ occupies binding sites on Ca++-binding regulatory proteins, such as troponin C (in cardiac and skeletal muscle) and calmodulin (in vascular smooth muscle). These proteins then interact with other proteins and enzymes (e.g., troponin I in cardiac and skeletal muscle and myosin light-chain kinase in smooth muscle), facilitating cross-bridge formation between actin and myosin, which underlies contraction. When Ca++ channels inactivate, Ca++ is pumped out of the cell, activation of contractile proteins is reversed, actin dissociates from myosin, and the muscle relaxes.
The direct-acting vasodilators are among the most powerful drugs used to lower blood pressure and include hydralazine, minoxidil, diazoxide, nitroprusside, and fenoldopam. As indicated in Chapter 24, several purported mechanisms have been proposed to mediate the ability of hydralazine to dilate arterioles, whereas minoxidil and pinacidil bind to ATP-sensitive K+ channels, causing them to open. This allows K+ to partially equilibrate along its concentration gradient, which shifts the membrane potential toward the K+
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


