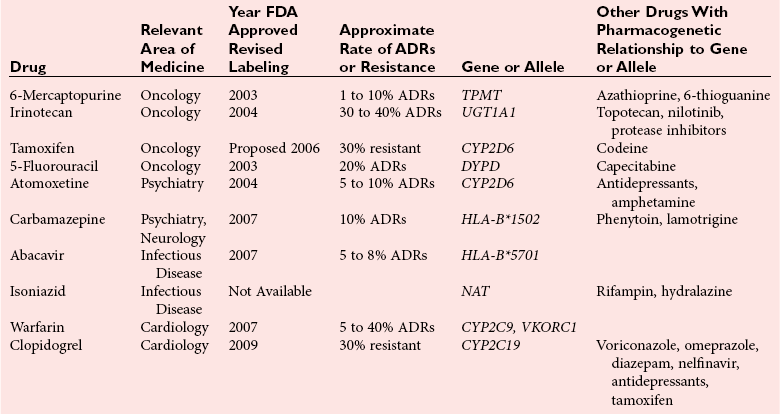
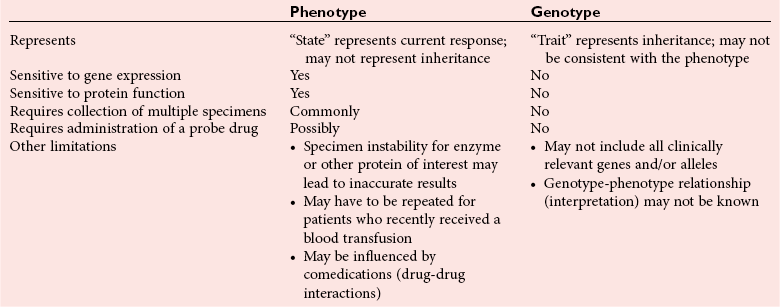
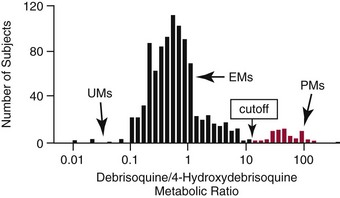
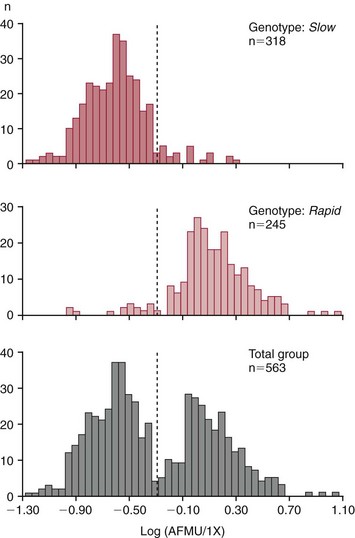
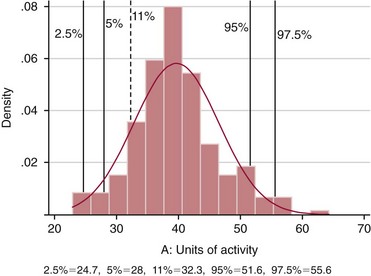
Chapter 43 The term pharmacogenetics comes from merging the names of pharmacology (the study of drugs, particularly drug handling and drug response) and genetics (the study of how traits are inherited). Pharmacogenetic testing is intended to predict or explain how people will respond to medicines and other pharmacologically or toxicologically active compounds, based on discrete genetic characteristics. Pharmacogenetics is a rapidly evolving and expanding field and a critical component of personalized medicine. Related work associating drug handling and response with the entire genome is known as pharmacogenomics, although some use the terms interchangeably. Pharmacogenetics and pharmacogenomics could include human germline mutations, somatic mutations such as those observed in a malignant tumor, gene copy number, or nonhuman (e.g., viral genome) mutations. Pharmacogenetic or pharmacogenomic profiles hold the potential to improve prescribing practices for most if not all drugs.64,213 The goal of pharmacogenetics today is to relate a discrete aspect of a patient’s genetic inheritance (genotype) to phenotype, specifically, one or more aspects of pharmacokinetics (drug handling) and/or pharmacodynamics (drug action). Pretreatment pharmacogenetic testing has the potential to predict a phenotype before a drug is ever administered. Predicted phenotype information can be used to select a specific drug(s) that a patient is likely to respond to or select the optimal dosing interval for that patient, while at the same time avoiding or minimizing the incidence of adverse drug reactions (ADRs), including drug hypersensitivity. ADRs are a leading cause of morbidity and mortality in the United States, and the likelihood of response for many drug classes is less than 50%.26,76,97,165,170 Approximately 10% of all drug labels now include references to pharmacogenetic findings, and several pharmacogenetic tests are available clinically that are designed to identify patients that may be genetically predisposed to ADRs or therapeutic failure.39 However, clinical use of pharmacogenetics in prescribing most drugs requires more specific dosing guidelines and algorithms based on pharmacogenetic information. Such tools are being considered, developed, and/or evaluated to assist with the use of pharmacogenetic data to inform drug and dose selection, and are likely to improve uptake and utility of pharmacogenetic testing in the coming years. Pharmacogenetic testing provides information about the predicted pharmacokinetics and pharmacodynamics of an individual patient that otherwise are not readily measured. Testing can be used to predict unique characteristics of drug handling and drug response that could minimize the traditional trial and error style of dose optimization, particularly when used in combination with biochemical and clinical (measurable) factors. See Chapter 34 of this book for further discussion of pharmacokinetic and pharmacodynamic processes. The first recognized pharmacogenetic targets largely described variation in drug metabolism. The early focus on drug metabolism reflects the fact that measurements of drug and drug metabolite concentrations in biological fluids predate detailed understanding of genetics and development of most biomarkers that describe pharmacodynamics. Distinct metabolic phenotypes were characterized and were subsequently found to cluster within families. For example, the metabolic phenotype for N-acetyltransferase (NAT), attributed to an enzyme originally called isoniazid transacetylase, was recognized in the 1950s with isoniazid (L-isonicotinyl hydrazide), a drug used to treat tuberculosis.11,199 Population studies revealed a bimodal distribution in plasma and urine concentrations of the N-acetylated isoniazid metabolite. The concentration of the parent drug was highly correlated with the prevalence of toxic symptoms, including hepatotoxicity and a painful and progressive peripheral neuropathy that affected up to one third of Caucasian and African American patients. From this initial work, patients were phenotypically described as “fast acetylators” or “slow acetylators.” Slow acetylators excrete large amounts of the parent drug relative to the acetylated metabolite when compared with fast acetylators. Family studies identified a strong genetic linkage of this phenotype and suggested that the phenotype was inherited as an autosomal recessive trait. However, details of the genetics, which are described later in this chapter, were not elucidated until several years later. Clinical use of pharmacogenetic testing requires an understanding of how genetic variation of a metabolic enzyme affects drug response or drug dosing. Drug metabolism can effectively inactivate a substrate or increase water solubility of the substrate to promote its excretion. Conversely, metabolism can activate a substrate or prodrug (e.g., codeine, clopidogrel, tamoxifen). Less commonly, metabolism serves to extend the elimination half-life of a pharmacologically active or potentially toxic metabolite.131 Reactions catalyzed by metabolic enzymes are often categorized as Phase I and Phase II. Phase I reactions convert the parent compound into a more polar metabolite by introducing or removing a single functional group. Examples include (1) oxidation, (2) reduction, and (3) hydrolysis. The metabolites may or may not be pharmacologically or toxicologically active. Most Phase I reactions are oxidative and are mediated by cytochrome P450 isoenzymes (CYPs). Genetic variants of CYPs have been associated with changes in enzyme activity, stability, and/or substrate affinity that can lead to clinically significant phenotypes. In addition to genetic variability, many CYP isoenzymes are susceptible to dramatic differences in expression (>1000-fold) and drug-drug interactions that can change the phenotype from that which might be predicted based on genetics alone. This concept is known as phenocopying. A drug that can inhibit its own metabolism is known to exhibit autophenocopying.134,144 A relatively common fate of Phase I metabolic products is additional modification, such as through Phase II reactions. Phase II reactions may also occur independent of, or before, Phase I reactions. Phase II enzymes are generally transferases that conjugate drugs with acetyl, glucuronyl, amino acyl, or sulfate groups. Many Phase II enzymes exhibit genetic variation that has been associated with clinically significant phenotypical differences caused by changes in enzyme activity, stability, and/or substrate affinity.187 For example, thiopurine S-methyl transferase (TPMT), N-acetyltransferase (NAT1, NAT2), and uridine diphosphate glucuronosyl transferase (UGT1A1) exhibit genetic variation and are discussed later in this chapter primarily relative to metabolism of drugs used in oncology and infectious disease. 1. Define a major pathway for metabolism of, or response to, a specific drug, thereby providing clear guidance for drug selection or avoidance. 2. Are associated with a clinically significant and actionable effect on the relationship between dose and plasma drug concentration or a measurable biomarker. 3. Predict dosing for a drug that has a narrow therapeutic index or limited temporal opportunity to exert efficacy, or for a drug that requires a long period of time to establish efficacy. The utility of pharmacogenetic testing may be compromised when (1) no alternative drugs are available for an indication, (2) incorporation of pharmacogenetic testing does not clearly improve patient care, (3) less sophisticated or less expensive tools or tests are sufficient for making prescribing decisions, or (4) accommodations to specific dose or dosing strategies are not available. Some specific gene-drug relationships for which the labeling has been revised to include pharmacogenetic information are shown in Table 43-1. Although these gene-drug examples apply primarily to oncology, psychiatry, neurology, infectious disease, and cardiology, other areas of medicine are likely to benefit from pharmacogenetic testing as well. The examples are intended to provide a foundation of concepts that the reader can translate to future applications as they emerge and develop. Pharmacogenetic testing can predict that a patient is likely to (1) fail therapy, (2) suffer an ADR, or (3) exhibit altered pharmacokinetics. This knowledge then assists in selection of drug, drug dosage, and dosing frequency, thus promoting successful therapy and avoiding undesirable outcomes such as an ADR. Pharmacogenetic testing is designed to detect a phenotype (phenotyping) or a genetic sequence that has been associated with a phenotype (genotyping). Examples of both approaches (phenotyping and genotyping) will be provided; however, more emphasis is given to the genetic approaches, which are currently more common. Neither phenotyping nor genotyping is intended to replace the need for clinical assessments of drug response, or to replace the need for therapeutic drug monitoring. Several considerations of these two testing approaches are summarized in Table 43-2 and are described in greater detail later. A common approach to phenotyping is to administer a probe drug, that is, a drug that is known to be metabolized by the same enzyme or pathway as the intended therapeutic drug. Biological fluid, typically urine and/or blood, is collected at specific times after administration of the probe drug. The concentrations of parent drug and one or more drug metabolites are then determined. The ratios of parent to metabolite or of two metabolite concentrations (e.g., metabolite A/metabolite B) are compared, and a metabolic ratio is calculated. The phenotype associated with CYP2D6 has historically been determined through the use of probe drugs. For this particular enzyme, a continuum of metabolic phenotypes is described, varying from poor (little to no activity) to intermediate, to extensive (normal), to ultra-rapid. Urine collected at a specified time after administration of the probe drug (e.g., 8 hours) is analyzed for the parent drug and a metabolite that is generated primarily via CYP2D6. Example parent-metabolite pairs used to calculate a metabolic ratio for phenotyping of CYP2D6 include dextromethorphan/dextrorphan, debrisoquine/4-hydroxydebrisoquine, sparteine/dehydrosparteine, and nortriptyline/10-hydroxynortriptyline.38 An example of phenotype results generated with the probe drug debrisoquine is shown in Figure 43-1. A “cutoff” metabolic ratio is used to distinguish a normal, extensive metabolizer (EM) from a poor metabolizer (PM). Although the metabolic ratio is theoretically a good indicator of enzyme expression and function at the time of the test, marked variation in metabolic ratio has been demonstrated among the phenotypes.20 Similar variation in metabolic ratio is commonly observed with other probe drugs as well. Another example of a probe drug is caffeine, which is used to determine acetylator phenotype mediated by NAT.21,72,121 To determine the NAT phenotype with this probe drug, the patient is given 200 mg caffeine after an overnight fast. Urine is collected 4 and 5 hours later. The 4-hour specimen is discarded, and the 5-hour specimen is analyzed for caffeine metabolites 5-acetylamino-6-formylamino-3-methyluracil (AFMU) and 1-methylxanthine (1X). See Figure 43-2 for sample data. Cutoffs for acetylator phenotypes are based on the ratio of AFMU/1X. Considering a bimodal distribution of acetylators, a ratio of less than 0.66 is used to define slow acetylators. Because a trimodal distribution has now been described and may be of interest, a ratio higher than 3.0 could define “ultrarapid” acetylators; a ratio between 0.66 and 3.0 would define an “intermediate” acetylator phenotype.57 Phenotyping is also performed in vitro by measuring the activity of a particular metabolic enzyme using peripheral blood cells as a surrogate to metabolism by the whole patient. For example, a common method of phenotyping TPMT activity is to isolate red blood cells, lyse the cells, and incubate the lysate with substrate 6-mercaptopurine (6-MP) and a methyl donor such as S-adenosyl-L-methionine for 1.5 hours at 37 °C.74,201 The concentration of methylated product, 6-methyl mercaptopurine (6-MMP), is measured and compared with a reference interval. An example of TPMT phenotype data generated in a healthy adult population is shown in Figure 43-3. Reduced or deficient TPMT activity is detected when low 6-MMP concentrations are measured. The clinical significance of low TPMT activity is high risk of myelosuppression due to an accumulation of 6-thioguanines (6-TGNs) when patients are treated with 6-MP or azathioprine (AZA), a related prodrug that is metabolized to 6-MP. Patients with impaired TPMT activity require lower doses of these drugs and close clinical monitoring to minimize the risk of toxicity and ensure efficacy of the drug.7,36 This application is discussed in greater detail in the following section on TPMT genetics. Other examples of enzymes that have been successfully evaluated for in vitro phenotypes include (1) pseudocholinesterase, for detecting individuals with susceptibility to prolonged apnea when treated with succinylcholine or other similar medications; (2) glucose-6-phosphate dehydrogenase (G6PD), for detecting susceptibility to hemolysis in response to drugs such as primaquine, sulfonamides, chloramphenicol, and vitamin K; and (3) NAT, for detecting poor metabolism of drugs such as sulfonamides, isoniazid, and clonazepam.33,50 With each, a substrate for the enzyme is incubated with the isolated cells under conditions (temperature, time, cofactors, pH, etc.) optimal for the enzyme of interest. The reaction is stopped and the product is measured with an analytical technique such as spectrophotometry or liquid chromatography with ultraviolet or mass spectrometric detection. The in vitro phenotype approach has several limitations, for example, data are not valid if the patient from whom cells are collected is receiving the same drug used by the laboratory as a substrate in the phenotyping assay for that drug, at the time of specimen collection. Factors such as (1) recent blood transfusions, (2) hormonal fluctuations, (3) handling of the blood before and after isolation of cells, and (4) inadequate environmental control during the assay also introduce variability.81 In addition, many enzymes are expressed in variable quantities with age, leading to the need for age- or population-specific reference intervals to accurately interpret results. For example, a study of 60 full-term newborns demonstrated that TPMT activity was higher in the newborns than in race-matched healthy adults, but displayed the same tri-modal distribution observed in adults. This observation may lead to inappropriate interpretation of newborn phenotype results, if results are compared with an adult reference interval.118 Finally, unrecognized or unrecorded confounding factors, such as diet, comedications, and consumption of alcohol or over-the-counter medications, may compromise interpretation of in vitro phenotype results as well.165 The second major approach to detecting and evaluating pharmacogenetic variability is through genotyping. Genetic testing may be useful for predicting specific pharmacokinetic and/or pharmacodynamic variables that are well characterized. The value of genotyping depends very much on a strong genotype-phenotype correlation. In the simplest scenario, a genotype result will account for 100% of the anticipated phenotype. Completely definitive genotype-phenotype relationships (100% certainty) are rare—a fact that creates controversy surrounding the value of a single-gene association. It is estimated, for example, that genotyping of TPMT can reliably predict the phenotype 80 to 90% of the time.151 However, the complexity of the metabolism surrounding thiopurine drugs (see TPMT discussion later) raises questions regarding other potentially important genes that contribute to the overall pharmacokinetics of these drugs. For example, variants in three other genes have recently been demonstrated to influence metabolism of azathioprine, and a polymorphism in the inosine trisphosphatase gene (ITPA) appears to be responsible for ADRs experienced by some patients treated with AZA.168,193 When a good genotype-phenotype relationship exists, genotyping assays have advantages over phenotyping because they (1) can be less expensive than phenotyping, (2) are more reproducible between laboratories, and (3) are not subject to intraindividual variability caused by a specific set of clinical conditions or expected biological variations that complicate phenotyping assays. Genotyping assays are also less invasive and less time-consuming for the patient. Thus, the single sample required for genotyping may be collected anytime, anywhere. Saliva-based testing, which generates an adequate quantity and quality of DNA for most genetic testing, would make at-home collections particularly convenient.66 Despite advantages of genotyping over phenotyping, genotyping has limitations that must be recognized. For example, some of the assays using target amplification are prone to false negatives because they rely on the presence or absence of a polymerase chain reaction (PCR) product. The specific allelic variants detected by a given analytical method vary, and, short of sequencing the entire gene, currently available genotyping techniques cannot detect all possible variants. Because of the large amount of variation in many genes, suites of variants are screened, potentially missing some important genetic variations because the patient possesses a rare or novel polymorphism that is not detected by the screen. Further, polymorphisms with unknown clinical significance may be identified by some techniques. Extreme care must be taken to ensure that these data are not misinterpreted, and that the laboratory test employed provides sufficient coverage of genetic variants common to the ethnic population of interest. Another disadvantage of genotyping is that it may not accurately predict function of the expressed protein. Using CYP2D6 as an example, it has been shown that patients of a single common genotype have metabolic ratios (with probe drug dextromethorphan or debrisoquine) that span a 1000- to 10,000-fold range. The exact reason for this broad range is unknown, but it is probably related to (1) individual differences in expression of the CYP2D6 gene, (2) inherent alternative metabolic pathways, (3) significant interlaboratory and intraindividual variability in phenotype measurements, and (4) possible combinations of other minor undetected genetic polymorphisms in the CYP2D6 gene. Finally, specific interactions between the target enzyme or protein and other drugs, chemicals, or some foods can change the phenotype, such as by converting a “normal” or extensive metabolizer phenotype to a poor metabolizer phenotype. This type of phenocopying is observed with grapefruit juice, which has been shown to inhibit CYP3A4, and many antidepressant medications (e.g., fluoxetine), which are known to act as both substrates and inhibitors of CYP2D6.63,135,144 Such drug and food interactions can have extremely important consequences for the phenotype and can be difficult to both recognize and monitor. Pharmacogenetic testing is clinically useful only when information is sufficient to allow interpretation of the results. This information must be derived from in vivo human studies. Many examples of this type of information can be found in the peer-reviewed literature. One limitation of existing literature, however, is that most data are based on retrospective studies, and there is no single printed source in which all of this information has been collated. The Pharmacogenetics and Pharmacogenomics Knowledge Base is a publicly available Internet research tool developed by Stanford University with funding from the National Institutes of Health (NIH) and is part of the NIH Pharmacogenetics Research Network (PGRN), a nationwide collaborative research consortium. Its aim is to aid researchers in understanding how genetic variation among individuals contributes to differences in reactions to drugs. This regularly updated database is an excellent source of genetic and clinical information derived from research studies at various medical centers in the PGRN (www.pharmgkb.org, accessed June 28, 2011). Several of the figures illustrating pharmacogenetically important drug metabolism pathways are found in this chapter, with permission from PharmGKB and Stanford University.91 Many ongoing clinical trials designed to study the efficacy and toxicity of new pharmaceutical products or new indications for previously developed pharmaceuticals have employed pharmacogenetic testing, or have incorporated pharmacogenetic testing into the study protocol (for a current listing, see www.clinicaltrials.gov, accessed June 28, 2011). Thus it is anticipated that pharmacogenetic guidelines, and possibly new companion diagnostics that would be simultaneously released to market with the pharmaceutical, will be available in the coming years. In addition, drugs that were previously removed from development because of ADRs may be reconsidered if a genetic test can be demonstrated to identify individuals at high risk for ADRs, that would then avoid use of that drug. A discussion of pharmacogenetic testing of clinical relevance to the areas of oncology, psychiatry and neurology, cardiology, and infectious disease is provided in the next section. For each of the major genes discussed in this chapter, Table 43-1 provides a list of common drugs for which pharmacogenetic associations with these genes have been made and drug labels revised. Because many other drug-gene combinations are recognized that may lead to additional drug label revisions, the discussion provided here is in no way comprehensive. In addition, many of the specific genes discussed later have applications to other areas of medicine, some of which are mentioned briefly. The purpose of the discussion in this chapter is to provide examples against which additional pharmacogenetic tests and applications can be compared, critiqued, and understood, and to guide involvement of the clinical laboratorian in this emerging new field of laboratory medicine. Oncology is an area of medicine in which correct or incorrect choice of therapy can lead to a life or death situation. The temporal opportunity for a therapy to exert effect is narrow, the incidence of ADRs with many available drugs is high, and the overall rates of efficacy are low. It is estimated that as many as 40% of oncology patients receive the wrong drug.27,198 Many of the tests routinely used to characterize tumor tissue are based on detecting overexpression of specific proteins (e.g., HER2/neu) or the presence of somatic gene variants in a tumor (e.g., K-ras). Tumor profiling tests, which evaluate the expression of many genes (e.g., Oncotype DX), can be used to assess the prognosis of certain cancers. Results of these tests certainly guide therapeutic decisions but are not discussed here. The germline pharmacogenetics that is the focus of this chapter is complementary to these tumor-based tests. Some pharmacogenetic tests that predict metabolism are potentially applicable to cancer chemotherapy because metabolic enzymes are required to activate many drugs used in oncology (e.g., tamoxifen, cyclophosphamide). Metabolic enzymes are also required to inactivate some drugs commonly used in oncology (e.g., irinotecan, 6-MP, 5-fluorouracil). Impairment or altered expression of a key metabolic enzyme could make the difference between therapeutic success and ADRs.70 In theory, optimal dose could be determined for these and all other drugs, based on understanding and coordinated application of pretherapeutic pharmacogenetics with post-therapeutic pharmacokinetic and pharmacodynamic monitoring. Table 43-3 lists examples of genes that are potentially important in selecting and dosing cancer therapies. TABLE 43-3 Pharmacogenetics has also been applied to risk stratification; this is particularly relevant for cancers that could be triggered by environmental or dietary exposure to a wide variety of xenobiotics.15,164 Also shown in Table 43-3 are some of the genetic targets and known substrates that have been studied with regard to risk of various cancers. Genes that encode drug-metabolizing enzymes are of greatest interest for this application because it is through these enzymes that xenobiotics become activated or inactivated. For example, several substrates of NAT1 and NAT2 are potentially cancer-causing. In addition to NAT1 and NAT2, the glutathione S-transferases (GSTs) are important contributors in establishing the risk of cancer triggered by xenobiotic exposures. At least 16 GSTs have been identified, and these are divided into eight classes. The key members are listed in Table 43-3: alpha (GSTA1, GSTA2), mu (GSTM1), pi (GSTP1) and theta (GSTT1). Additional classes are called kappa, sigma, zeta, and omega. The GSTM1-deficient trait has, for example, been associated with higher risks of lung and bladder cancer, although the specific trigger or substrate is not known.60 With additional study and validation of these and other genetic markers, testing to identify individuals with high susceptibility to chemicals may become important, particularly in high-risk occupational settings such as agriculture and manufacturing. Pharmacogenetics testing has shown promise in selecting or optimizing drug therapy for depression, schizophrenia, pain, and addictive behaviors. Many drugs used in psychiatry and neurology have low (<50%) efficacy rates, high interindividual variation in pharmacokinetics, and life-threatening ADRs. In addition, the response to drugs used for depression and schizophrenia cannot be adequately assessed for weeks to months after initiation. Standard-of-care practices today are based on the traditional trial-and-error approach. Pharmacogenetic tests could contribute to drug and dose selection by evaluating patient candidacy for specific drugs, and by allowing pretherapeutic identification of individual differences in pharmacokinetics or pharmacodynamics, particularly for drugs with a narrow therapeutic index.28,112 It is no surprise that initial applications of pharmacogenetics in these clinical areas were related to thoroughly-characterized metabolic phenotypes. Of particular interest have been drug substrates of CYP2D6, CYP2C19, and CYP2C9. Dosing guidelines for many antidepressant and antipsychotic medications based on predicted metabolic phenotypes have been published.88,89 A more conservative recommended strategy is to avoid drugs that are associated with extreme phenotypes (e.g., UM, PM) or high risk of ADRs, unless alternative therapies are not available.29 Risk of ADRs may be associated with inappropriate drug concentrations, as a result of impaired metabolism, or may be unrelated to concentration and dose. For example, severe cutaneous reaction syndromes (SCARS), such as Stevens-Johnson syndrome and toxic epidermal necrolysis, lead to life-threatening and extremely painful rashes. Approximately one third of all drug-induced SCARS are associated with the antiepileptic drug carbamazepine. The occurrence of SCARS in patients treated with carbamazepine has been associated with a specific allele of human leukocyte antigen (HLA), HLA-B*1502.207 This allele occurs most commonly in patients of Asian ancestry. In the Han Chinese population, the odds ratio for carbamazepine-induced SCARS is 2504 when the HLA-B*1502 allele is present. Associations of this allele with SCARS induced by phenytoin and lamotrigine have also been reported.111 Detection of HLA alleles associated with drug-induced SCARS before therapy is initiated could allow drug avoidance and substantially reduced risk of the associated ADR. Resistance to drugs used in psychiatry and neurology is common and is not necessarily related to drug concentration or dose of drug. Genes related to pharmacodynamics of these drugs primarily code for various components within the neurotransmitter systems on which the drugs act. Genes associated with the serotoninergic (e.g., SLC6A4), noradrenergic (SLC6A2), and dopaminergic (e.g., DRD2) neurotransmitter systems are best characterized. Serotonin-receptor variants have been correlated with response to antidepressants such as escitalopram, fluoxetine, and paroxetine. In addition, the S allele of the serotonin transporter gene (SLC6A4), also known as the short form of the long/short polymorphism, predicts poor response and increased risk of ADRs with fluvoxamine, mirtazapine, and paroxetine. Variants in the glucocorticoid receptor gene (NR3C1) have also been proposed to predict response to nortriptyline and escitalopram. Time to response to the antipsychotics risperidone and olanzapine has been associated with variants in the promoter region of the dopamine D2 receptor gene (DRD2). Genes associated with the biosynthesis and disposition of neurotransmitters (MAOA, TPH) may be important as well.100,129,159,189 Optimal dosing of the anticonvulsant drug phenytoin can be predicted by testing for variation in CYP2C9, CYP2C19, and MDR1; the latter encodes the p-glycoprotein. CYP2C9 is estimated to contribute to 90% of phenytoin metabolism, and CYP2C19 is thought to contribute to 10% of phenytoin metabolism, in persons with EM phenotypes for both enzymes. In patients who are CYP2C9 PM (poor metabolizers), CYP2C19 is the primary route of phenytoin elimination, and hence the importance of testing for CYP2C19 increases.46,84,110 However, pharmacogenetics has not been helpful in elucidating resistance mechanisms in epilepsy.82,166 Procainamide is a classic antiarrhythmic, the metabolism of which is primarily mediated by N-acetylation. The rate of acetylation is genetically determined, based on NAT, and shows a bimodal (possibly trimodal) distribution of slow and fast acetylators that correlates to genetic variants in NAT1 or NAT2. The N-acetylated metabolite, known commonly as NAPA, is routinely monitored with parent drug to optimize dose. Many CYP substrates have cardiovascular applications as well. For example, clopidogrel is a commonly prescribed CYP2C19 substrate. Clopidogrel is a prodrug and, much like tamoxifen and codeine, will exert little to no benefit unless metabolically activated. Indeed, approximately one third of patients are classified as resistant to clopidogrel, in part because of this pharmacokinetic variation. Pharmacodynamic variability can also explain drug resistance and toxicity. For example, drug transporters that facilitate uptake and elimination of statins may have pharmacogenetic implications. Variants of the OATP1B1, encoded by SLCO1B1, and the ABCB1, encoded by the ABCB1, are associated with pharmacokinetic variability and statin-induced myopathy.103,138 In the PROSPER study, Iakoubova and colleagues73 have reported that elderly carriers of the KIF6 719Arg variant with prior vascular disease receive significant benefit from pravastatin therapy; however, no benefit was observed in noncarriers with prior disease or in those without prior disease (carriers or noncarriers). Pharmacogenomics has been applied to infectious disease, primarily to understand organism susceptibility, to study comparative genomics, and to target drug development efforts. This growing area of research will be complementary to host pharmacogenetics, the focus of this chapter. In patients treated with interferon, overlap in the relationship between organism and host has been exemplified by the association of clearance of hepatitis C virus and variants of the gene for interleukin 28B (IL28B).44 Regarding more traditional host pharmacogenetics of infectious disease, several targets related to pharmacokinetics are substrates of NAT: isoniazid, dapsone, and the sulfonamides. Although pharmacogenetic testing for NAT1 and NAT2 is not routine, variation in these genes has the potential to influence metabolism and steady-state concentrations of these drugs. Concentrations of proguanil are affected by variant CYP2C19, and CYP2C8 is associated with risk of toxicity from amodiaquine. Other pharmacogenetic associations include (1) CYP2B6 alleles and risk of central nervous system ADRs from efavirenz,19 (2) UGT1A gene variants and severe hyperbilirubinemia associated with protease inhibitors,95 and (3) variants of the CYP3A family and rates of clarithromycin clearance.140 Pharmacodynamic targets that are unrelated to the desirable response of the drug represent a unique opportunity for pharmacogenetic testing. An example of a very successful pharmacogenetic test for predicting risk of ADRs is detection of HLA-B*5701, which predicts abacavir-induced hypersensitivity reaction. Genotyping for HLA-B*5701 before an abacavir-containing regimen is prescribed was recommended in the 2008 guidelines on the use of antiretroviral agents in HIV-1–infected adults and adolescents, which were developed through the Office of AIDS Research Advisory Council and have now become routine clinical practice.109,147,188 As stated earlier, most Phase I reactions are oxidative and are mediated by cytochrome P450 enzymes (CYPs). CYPs are heme-containing enzymes that are synthesized from a superfamily of CYP genes and are classified into families, and further into subfamilies, based on amino acid homology. The major CYPs that are believed to contribute to drug metabolism are CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and the CYP3A family. Examples of drug substrates, inhibitors, and inducers are shown in Table 43-4. Although the pharmacogenetics of only CYP2C9, CYP2C19, and CYP2D6 are specifically discussed here, all the CYP families exhibit genetic variation that could influence optimal prescribing practices for drug substrates.187 Alleles for all CYP genes are described according to international consensus.77 In brief, the *1 (“star 1”) allele is the normal (EM) allele that codes for a protein predicted to function with full enzymatic activity and to be expressed in typical quantities. Often *1 describes a scenario wherein no variant alleles are detected. Therefore, the true accuracy of a *1 allele designation is dependent on whether the assay used to make that call is comprehensive. All alleles are described with a star followed by a number, and possibly a letter to describe an allele subtype. Subtypes have not been shown to influence the phenotype, and identification of them probably is not important clinically but could be important for haplotype assignment or to support research studies. Numerical assignments of other alleles are based largely on chronology of identification, wherein alleles identified earliest in time have the lowest numbers. Some alleles are associated with normal protein function and expression, others with impaired function, altered expression, or no predicted enzyme activity at all (e.g., truncated protein, complete gene deletion). Common alleles for CYP2C9, CYP2C19, and CYP2D6 are shown in Table 43-5. A common single-nucleotide polymorphism (SNP) used to detect each allele is bolded, but in many cases detection of other SNPs is required to accurately classify a genotype. The assignment of phenotype predictions from genotype is described in detail for CYP2D6 (Figure 43-4) but applies generally to other CYPs as well. TABLE 43-4 Examples of Drug Substrates, Inhibitors, and Inducers of Cytochrome P450 Subfamilies Modified from http://medicine.iupui.edu/flockhart/table.htm (accessed on November 28, 2009), with the permission of David A. Flockhart, MD, PhD. TABLE 43-5 Common CYP2D6, CYP2C9, and CYP2C19 Alleles* *Commonly used analytical target for genotyping is bolded. Modified from the Human Cytochrome P450 (CYP) Allele Nomenclature Committee. Available at: http://www.imm.ki.se/CYPalleles (accessed on June 28, 2011). Reproduced with permission of Sarah C. Sim, Webmaster. No information regarding enzyme activity is listed in the above table when none was present at this reference. Cytochrome P450 2D6 (CYP2D6), originally named debrisoquine hydroxylase, is a Phase I enzyme known to metabolize more than 100 drugs and environmental toxins.8,51,144 As shown in Table 43-4, CYP2D6 is inhibited by several compounds, some of which are substrates. When a CYP2D6 inhibitor is administered along with a CYP2D6 substrate, the phenotype for the patient may appear impaired. This concept, known as phenocopying, is of significant concern with CYP2D6. Also, CYP2D6 is frequently implicated in ADRs and has been the subject of many U.S. Food and Drug Administration (FDA)-issued public health advisories. Metabolic phenotypes of CYP2D6 are best described as a continuum that varies from no activity to very high activity. For ease of characterization, a tetra-modal distribution of CYP2D6 activity is described containing the four groups of people called ultra-rapid metabolizers (UMs), extensive (normal) metabolizers (EMs), intermediate metabolizers (IMs), and poor metabolizers (PMs). Translation of a phenotype to a clinical application can be somewhat difficult, as the actual phenotype is dependent not only on the genetic makeup of CYP2D6, but also on (1) expression of CYP2D6, (2) whether drug-drug interactions affect the phenotype, and (3) whether CYP2D6 activates or inactivates the drug of interest.34 The pharmacogenetics of CYP2D6 and/or phenotype testing can potentially be applied to improve prescribing of any medications known to be transformed by this enzyme. Two clinical applications for CYP2D6 pharmacogenetics include tamoxifen (described later), neuroleptics (some described in the psychiatry section of this chapter), and codeine. The relationship between the CYP2D6 enzyme metabolic rate (phenotype) and the CYP2D6 genotype has been extensively characterized.3,98,127,132,149 The CYP2D6 gene contains 4408 bases and is located on chromosome 22q13.2, near two pseudogenes with greater than 90% homology. More than 90 allelic variants have been described in the CYP2D6 gene. Alleles, listed with associated nucleotide changes and known effects on enzyme activity for CYP2D6, are shown in Table 43-5. Effects on enzyme activity are used to group the alleles according to the predicted phenotype, assuming that two copies of that allele were inherited: PM, IM, EM, and UM. However, the actual phenotype will reflect a sum of the alleles. A summary of CYP2D6 alleles and assignment of phenotypes are shown in Figure 43-4. Among Caucasians, approximately 1 to 10% are genetically UM, and 5 to 10% are PM. Only 1 to 3% of African Americans and Asians are PM, but many are IM.75 The CYP2D6 phenotype that is most simple to characterize is the PM phenotype, for which essentially no CYP2D6 activity is anticipated. The PM phenotype is expected when two or more copies of PM alleles are inherited. The sensitivity and specificity of the genotype to predict the PM phenotype are estimated to be 100%, but are far less for the other phenotypes. Other phenotype assignments are based largely on the highest functioning phenotype prediction. For example, at least one functional EM allele is thought to generate a phenotype that falls within the limits of normal. The UM phenotype is assigned when more than two copies of functional EM alleles are identified; this is often referred to as a gene duplication. Duplication of PM alleles has no effect on the phenotype, and duplication of IM alleles may or may not affect the phenotype.210 An activity score has been proposed to help translate genotype to phenotype.41 Based on the AmpliChip CYP2D6 test (Roche Molecular Diagnostics, Pleasanton, California), which is an FDA-cleared in vitro diagnostic test that detects 33 allelic variants, the EM genotype predicts an EM phenotype with 95% sensitivity and 47% specificity. For the IM phenotype, the sensitivity of genotyping is estimated at 42% and specificity at 97%. For the UM phenotype, the sensitivity of genotyping is estimated at 6% and specificity at 99%.142 These results illustrate that difficulties exist in detection and in incorporation of nongenetic factors when CYP2D6 phenotypes are assigned for all but the PM, based on genotype data alone. To separate CYP2D6 from structurally similar pseudogenes, CYP2D6 genotyping protocols often employ long PCR or nested PCR strategies with a first PCR step designed to amplify a large CYP2D6-specific region.61 Small nucleotide changes within this region, including SNPs and smaller insertions/deletions of one or a few bases, are then detected in a second amplification step designed as a PCR restriction fragment length polymorphism (RFLP) assay or as an allele-specific PCR without subsequent digestion. By evaluating the entire gene, such as through use of the single-strand conformation polymorphism technique described by Broly and colleagues, both known and unknown variants in the CYP2D6 gene can be detected.16 Other technologies used for genotyping CYP2D6 include real-time PCR methods and microarrays.22,62,130,142 It is important that an assay identify a sufficient number of variants to accurately classify an allele. For example, both the CYP2D6*10 and CYP2D6*4 alleles contain the 100C→T variant. Because the phenotype predictions differ for the two, it is clinically relevant to determine which allele is present. The pattern of additional SNPs unique to each allele helps distinguish the two and suggests the likelihood that both alleles could be present. Because complete gene deletion (CYP2D6*5) and duplication events (CYP2D6*1XN) occur relatively commonly and have significant effect on the phenotype, analytical techniques that can detect and quantify these events are preferred. Successful published methods have utilized RFLP, long-product PCR assays, microarrays, or real-time quantitative PCR of CYP2D6, sometimes in relation to an internal reference gene.69,104,152 Tamoxifen is an antiestrogenic prodrug widely used to treat and prevent breast cancer; it is a good example of a drug for which pharmacogenetic testing of CYP2D6 is applied clinically.67 Tamoxifen mediates its therapeutic effects through modulation of estrogen receptors (ERs), leading to suppression of estrogen-mediated cell proliferation. Therefore, hormone-sensitive breast tumors (ER-positive) are most likely to respond to tamoxifen. A meta-analysis of the Early Breast Cancer Trialist Collaborative Group (EBCTCG) study at the 15-year follow-up period for ER-positive breast cancer showed that 5 years of tamoxifen reduced the recurrence rate by 50% and the mortality rate by a third.1 However, the success of tamoxifen therapy is variable, and 30 to 45% of patients on tamoxifen relapse or die from recurrent cancer.133 Tamoxifen is extensively metabolized by many Phase I and Phase II enzymes (Figure 43-5). The lack of efficacy with tamoxifen can be explained, in part, by interindividual differences in the metabolic activation of tamoxifen. Tamoxifen is a prodrug and must be metabolized to the active principles to elicit the desired therapeutic effect. The most potent antiestrogenic metabolites are 4-hydroxy tamoxifen and endoxifen, each shown to exhibit approximately 100-fold greater affinity for the ER than the parent drug tamoxifen. The concentrations of these metabolites compared with the concentrations of N-desmethyl tamoxifen, an inactive metabolite, and tamoxifen itself, are shown in Table 43-6. Because the amounts of endoxifen are greater than the amounts of 4-hydroxy tamoxifen, endoxifen is largely credited with producing antiestrogenic effects of tamoxifen.14 Considerable evidence suggests that CYP2D6 is a major route for production of endoxifen. Thus, patients with impaired CYP2D6 have been shown to produce less endoxifen than patients with a CYP2D6 EM phenotype. In addition, a PM phenotype and lower endoxifen concentrations are observed in patients with EM genotype, who are coprescribed known strong inhibitors of CYP2D6, such as fluoxetine or paroxetine. It is interesting to note that patients treated with CYP2D6 strong inhibitors and patients who are known CYP2D6 PMs have been shown to exhibit fewer of the common antiestrogenic ADRs such as hot flashes.14,49,128 TABLE 43-6 Involvement of CYP2D6 in Steady-State Concentrations of Tamoxifen and Major Tamoxifen Metabolites Modified from Brauch H, Mürdter TE, Eichelbaum M, Schwab M. Pharmacogenomics of tamoxifen therapy. Clin Chem 2009;55:1770-82.
Pharmacogenetics
Defining Pharmacogenetic Targets
Approaches to Pharmacogenetic Testing
Clinical Application of Pharmacogenetic Testing
Pharmacogenetics in Oncology
Gene or Protein Target
Chemotherapeutic Drug
Carcinogen Substrates
CD117 (C-KIT)
Imatinib
CYP2B6
Cyclophosphamide, ifosfamide
CYP2C9
Cyclophosphamide, ifosfamide
CYP2C19
Tamoxifen
CYP2D6
Tamoxifen
CYP3A4
Cyclophosphamide, etoposide
Aflatoxin B1
Ifosfamide, teniposide, vindesine, vinblastine, vincristine
6-Aminochrysene
DPYD
5-Fluorouracil, capecitabine
EGFR
Erlotinib, cetuximab, gefitinib
GSTA1, GSTA2
Cyclophosphamide
Cumene hydroperoxides
GSTM1
Aflatoxin B1 epoxides
Benzo(a)pyrene 4,5 oxide
Trans-stilbene oxide
GSTP1
Thiotepa, cyclophosphamide, ethacrynic acid
4-Nitrochinolone 1-oxide
GSTT1
Ethylene oxide, methyl chloride, dichloromethane
Her2/neu
Trastuzumab, lapatinib
KRAS
Cetuximab, panitumumab
MTHFR
Methotrexate, 5-fluorouracil
NAT2
Amonafide
p-Aminobenzoic acid, 4-aminobiphenyl, 2-aminofluorene, p-aminosalicylic acid, benzidine, toluidine
Philadelphia chromosome
Busulfan, dasatinib, nilotinib
PML/RAR (alpha) fusion
Tretinoin, arsenic oxide
TPMT
6-Mercaptopurine, azathioprine, 6-thioguanine
TYMS
5-Fluorouracil, capecitabine
UGT1A1
Irinotecan, nilotinib
UGT2B15
Tamoxifen
Pharmacogenetics in Psychiatry and Neurology
Pharmacogenetics in Cardiology
Pharmacogenetics in Infectious Disease
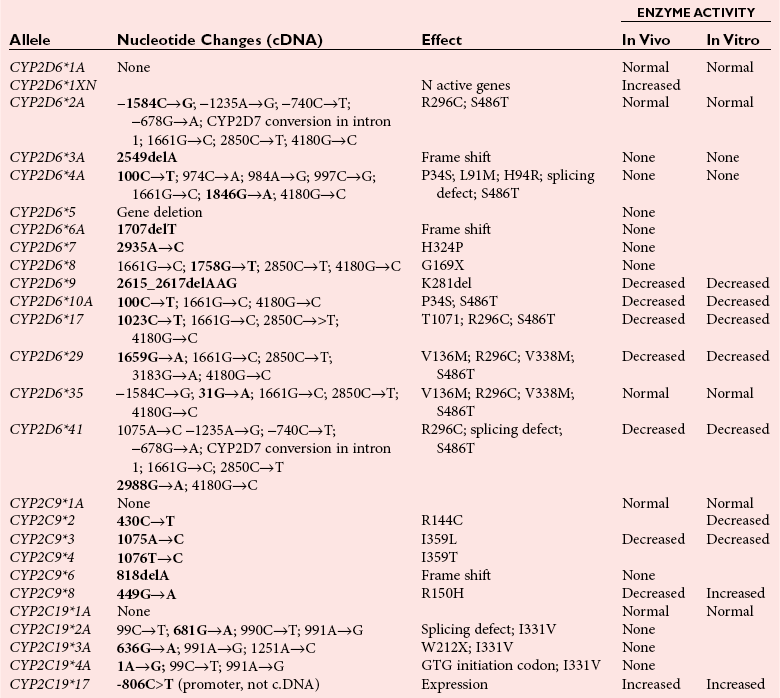
Phase I Metabolic Enzymes: Cytochrome P450 Isozymes
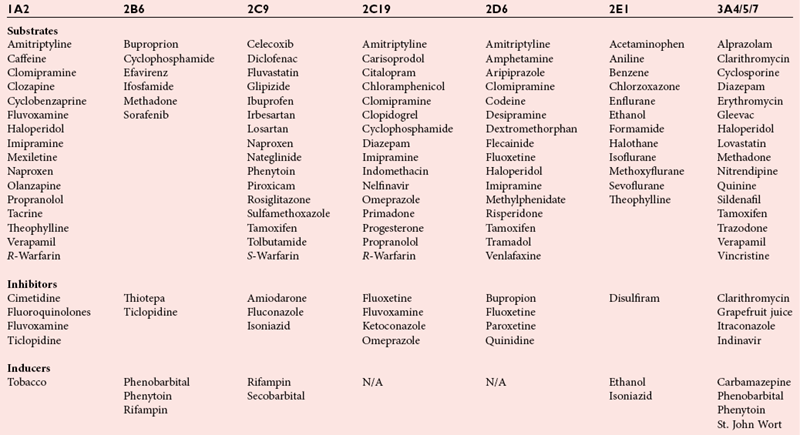
Cytochrome P450 2D6 (CYP2D6)
Genotype to Phenotype
Testing
Tamoxifen Application

![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pharmacogenetics