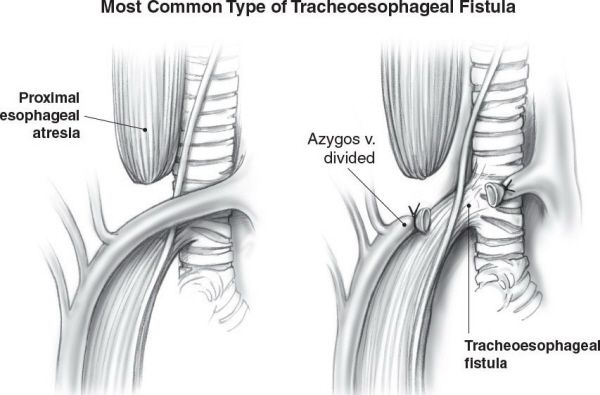
Depicted are the types of congenital esophageal atresia and tracheoesophageal fistula. (A) Proximal esophageal atresia with distal tracheoesophageal fistula (Gross Type C). (B) Isolated esophageal atresia without tracheoesophageal fistula. (C) Tracheoesophageal fistula without esophageal atresia. (D) Esophageal atresia with proximal and distal tracheoesophageal fistula (rare). (E) Proximal tracheoesophageal fistula with distal esophageal atresia (rare). (With permission from Mulholland MW, Lillemoe KD, Doherty GM, et al., eds. Greenfield’s Surgery. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.)
•There are five types of TEF:
•Proximal esophageal atresia with distal TEF (85%)
•Isolated esophageal atresia
•H-type TEF without esophageal atresia
•Proximal TEF with distal esophageal atresia
•Proximal and distal TEF
•Inability to pass a nasogastric tube is suspicious for TEF
•Chest X-ray can help determine the length of the esophageal gap
•Abdominal X-ray with air in the stomach excludes an isolated esophageal atresia
•Treatment
•Right extra-pleural thoracotomy through the fourth intercostal space or thoracoscopic trans-pleural repair
•The proximal esophagus is richly supplied by vessels from the thyrocervical trunk
•The distal esophagus has a more tenuous segmental supply from the intercostal arteries
•The operation includes TEF ligation and transection, and restoration of esophageal continuity via an end-to-end anastomosis
•On postoperative day 5 to 7, an esophagram is obtained and if no leak is identified, then oral feeds are started and the chest drain is removed
•Early complications (5% to 15%)
•Anastomotic disruption (secondary to poor blood supply and tension)
•Recurrent TEF
•Tracheomalacia
•Late complications
•Anastomotic stricture (25%)
•Gastroesophageal reflux (50%)
•Esophageal dysmotility (100%)
Proximal esophageal atresia with distal TEF is the most common type of TEF.
Which immunoglobulin is secreted in breast milk?
IgA is the most common antibody in breast milk, the gut, saliva, and most bodily secretions.
Which immunoglobulin does not cross the blood-placenta barrier?
IgM is large and does not cross the placenta (remember M for Magnum).
An 8-year-old boy presents following a bicycle crash with a ruptured spleen. What is the best indicator of early shock?
Tachycardia in childhood is defined as a heart rate >150 for a neonate, >120 in the first year, and >100 after 1 year. It is the best indicator of shock.
Fluid Resuscitation in Children
•20 cc/kg of crystalloid bolus for trauma
•If shock persists after a second bolus, then administer blood (10 cc/kg)
•An acceptable urine output is 2 to 4 cc/kg/hr in children
•Children have a lower glomerular filtration rate compared to adults
A healthy infant presents with bilious emesis, abdominal distention, and shock. What is the most likely diagnosis?
The presentation with shock suggests malrotation with midgut volvulus—a surgical emergency. In fact, bilious emesis in a newborn is malrotation until proven otherwise.
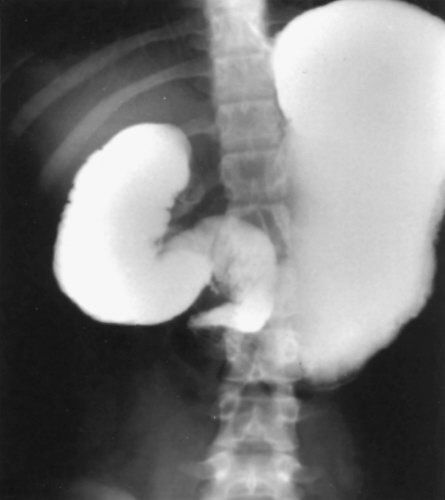
Malrotation. An upper gastrointestinal imaging study is shown that demonstrates contrast in the stomach and proximal duodenum. The distal duodenum does not cross midline and there is an abrupt cut-off consistent with the diagnosis of malrotation with midgut volvulus. (With permission from Mulholland MW, Lillemoe KD, Doherty GM, et al., eds. Greenfield’s Surgery. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.)
Malrotation
•During the 6 to 12th week of gestation, the intestine undergoes evisceration, elongation, and eventual return to the peritoneum in a 270 degree counterclockwise rotation with fixation
•Malrotation is associated with abnormal rotation and fixation
•Ladds bands extend from the colon to the duodenum, causing duodenal obstruction and biliary emesis
•Midgut volvulus refers to the narrow-based mesentery twisting around the superior mesenteric artery (usually clockwise)
•This results in intestinal obstruction and vascular occlusion
•Most develop symptoms in the first month of life
•If the patient is stable and the diagnosis is in question, then perform an UGI (gold standard test), which will demonstrate:
•Bird’s beak appearance of third part of duodenum
•Ligament of Treitz is to the right of midline
•Midgut volvulus is a surgical emergency
•Volume resuscitation is essential
•If the patient is in shock, no diagnostic studies are warranted
•Immediate surgical exploration is required to avoid loss of small intestine and possible death
•Surgical treatment is the Ladd’s procedure
•Division of abnormal peritoneal bands
•Correction of malrotation
•Restoration of a broad-based mesentery
•Appendectomy (because cecum is in the left upper quadrant)
An upper GI contrast series should only be performed if malrotation is suspected in the absence of midgut volvulus or shock—conditions which mandate emergent surgical exploration because of possible vascular compromise.
A full-term neonate with Down syndrome has bilious emesis during the first day of life. The newborn’s abdominal exam is normal. What is the most likely diagnosis?
Duodenal atresia is most likely in this patient.
Duodenal Atresia
•Failure of recanalization during the 8th to 10th week of gestation
•Characterized by bilious emesis
•No abdominal distention
•Usually presents within the first 24 hours of life
•Trisomy 21 is present in approximately 25% of infants with duodenal atresia
•85% of atresias are distal to the ampulla of Vater
•Rule out an anorectal malformation and check for patent anus
•“Double bubble” sign on abdominal X-ray
•Air within the stomach and first and second portions of duodenum
•If there is no distal air, the diagnosis is secure and no further studies are indicated
•If there is distal air, an urgent UGI should be performed to rule out midgut volvulus
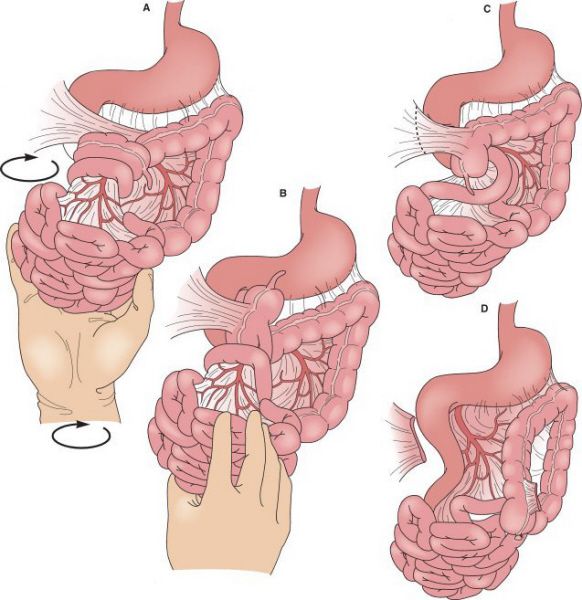
Ladd procedure. (A and B) Detorsion of midgut. (C and D) Division of Ladd’s bands (peritoneal attachments) of cecum to abdominal cavity. (With permission from Mulholland MW, Lillemoe KD, Doherty GM, et al., eds. Greenfield’s Surgery. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.)
•Surgical treatment is a duodenoduodenostomy. The repair may be performed by upper transverse laparotomy incision or laparoscopic technique.
Duodenal atresia often presents in the first 24 hours of life as bilious emesis without abdominal distention.
A 3-day-old full-term infant has bilious emesis and abdominal distention. What is the differential diagnosis?
The differential diagnosis includes intestinal atresia, malrotation, meconium ileus, Hirschsprung disease, and imperforate anus.
Jejunoileal Atresia
•Caused by an in utero mesenteric vascular accident
•Usually presents with bilious emesis within the first 2 to 3 days of life
•Associated with cystic fibrosis in 10% of cases
•Abdominal distention is usually present with a distal atresia
•Abdominal X-ray demonstrates multiple distended loops of bowel with air–fluid levels
•Contrast enema demonstrates a microcolon and no reflux into dilated intestines
•Contrast enema can also show multiple areas of involvement (10% of cases)
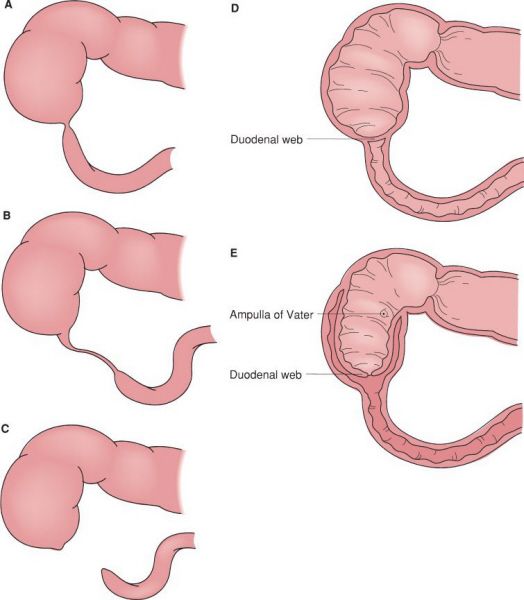
Anatomic forms of duodenal atresia. (A) Short segment atresia with patent lumen. (B) Long segment atresia with patent lumen. (C) Complete atresia with discontinuity. (D) Distal duodenal web. (E) Proximal duodenal web. (With permission from Mulholland MW, Lillemoe KD, Doherty GM, Maier RV, Upchurch GR, eds. Greenfield’s Surgery. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.)
•Surgical correction involves an end-to-end anastomosis
•A tapering enteroplasty can be used for a bowel size discrepancy
•Resection of the bulbous proximal end may be necessary
•Preserve length to avoid short gut syndrome
Colonic Atresia
•Caused by an in utero mesenteric vascular accident
•Similar in presentation to jejunoileal atresia
•Abdominal distention is present
•X-ray shows multiple dilated loops of intestine with air–fluid levels
•It can be difficult to distinguish between small and large intestine
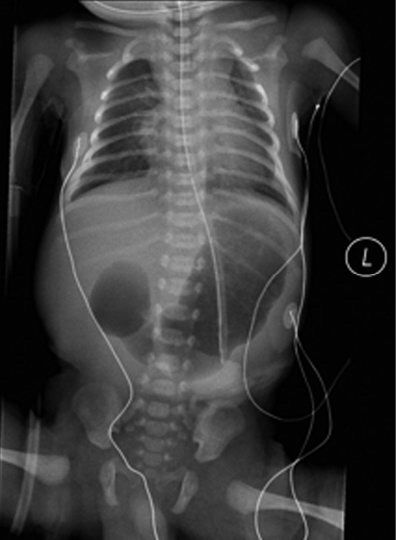
Plain abdominal radiograph of an infant with duodenal atresia. Air is seen in a dilated stomach and proximal duodenum. This finding is commonly referred to as a “double bubble sign”. It is important to note that air is not seen in the distal gastrointestinal tract. If air is seen in the distal gastrointestinal tract then duodenal stenosis or malrotation with midgut volvulus should be considered and a contrast imaging study may be indicated.
•Contrast enema demonstrates microcolon with a cutoff usually in the proximal colon
•Surgical correction involves an end-to-end anastomosis as above
Intestinal atresia can be associated with gastroschisis.
A newborn with cystic fibrosis presents with mild abdominal distention. An X-ray demonstrates a “ground glass” appearing mass on the right side of the abdomen. What is the next step in management?
A gastrografin enema is usually successful in treating simple meconium ileus (MI). Complicated cases may require surgery.
Meconium Ileus
•Obstruction of the terminal ileum by a highly viscid and tenacious meconium
•Approximately 15% of neonates with meconium ileus have cystic fibrosis
•A chloride sweat test should be performed to rule out cystic fibrosis
•Simple meconium ileus can usually be treated with:
•Gastrografin enemas
•N-Acetylcysteine enemas
•Rectal irrigation
•Complex meconium ileus (with atresia, perforation, or volvulus) or patients refractory to conservative therapy require surgical treatment
•Enterotomy or limited intestinal resection with evacuation of meconium
•An enterostomy may also be necessary
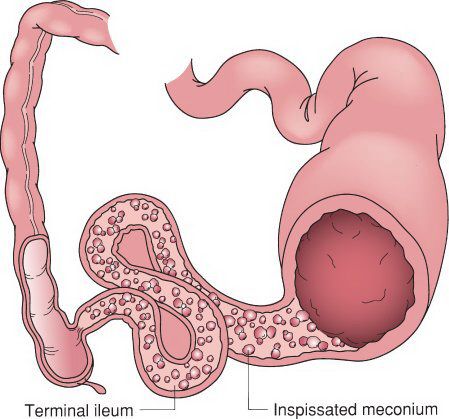
Uncomplicated meconium ileus. The proximal intestinal lumen is obstructed with the abnormal inspissated meconium which obstructs the distal ileum. The colon is small due to the in-utero proximal obstruction. (With permission from Mulholland MW, Lillemoe KD, Doherty GM, Maier RV, Upchurch GR, eds. Greenfield’s Surgery. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.)
N-Acetylcysteine enemas can be used for meconium ileus.
A full-term neonate has bilious emesis during the first and second days of life. The newborn’s abdominal exam reveals marked distention. An abdominal X-ray is significant for dilated loops of small bowel. A contrast enema reveals a narrow rectum, compared to the sigmoid. The baby failed to evacuate the contrast the following day. What is the next step in management?
A bedside suction rectal biopsy at least 2 cm above the dentate line is the gold standard test for infants suspected to have Hirschsprung disease.
Hirschsprung Disease
•Failure of the normal migration of neural crest cells
•Absent ganglia in the myenteric and submucosal plexus
•The absence of ganglia always begins in the distal rectum and extends proximally
•80% to 85% of cases are limited to the rectosigmoid
•Presents as bilious emesis, feeding intolerance, and abdominal distention
•Associated with failure to pass meconium within the first 48 hours of life
•A contrast enema demonstrates a contracted rectum with proximally dilated large bowel
•Failure to evacuate rectal contrast at 24 hours can be diagnostic
•Suction rectal biopsy is required to confirm absence of ganglion cells and nerve hypertrophy
•Surgical treatment
•Soave endorectal pull through with removal of the diseased aganglionic distal bowel and a coloanal anastomosis
•Children who present acutely ill may need a staged procedure with a diverting colostomy
•Remember to do frozen intraoperative biopsies to help determine the anatomic location of the transition zone
•Even though the diseased bowel has been removed, enterocolitis may still occur, requiring rectal irrigations and broad spectrum antibiotics
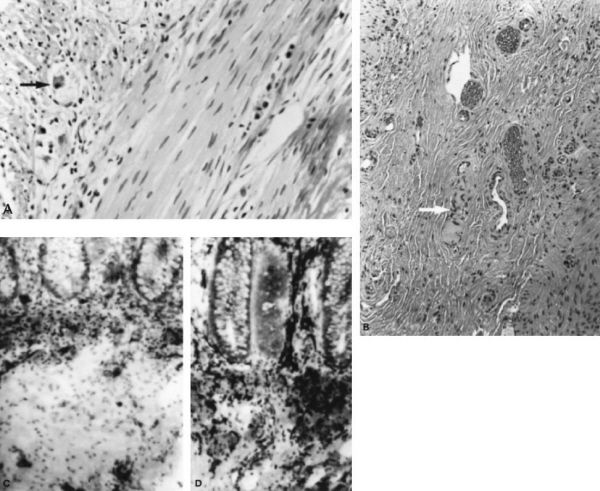
Rectal biopsy in patient with Hirschsprung disease. (A) Normal rectal biopsy with ganglion cells indicated by arrow (hematoxylin-eosin). (B) Rectal biopsy specimen with aganglionosis (hematoxylin-eosin). Note the characteristic thickened nerve fiber (arrow). (C) Normal rectal biopsy using acetylcholinesterase histochemical staining. (D) Similarly stained specimen from a patient with Hirschsprung disease. Many thickened submucosal nerve fibers stain densely black. (With permission from Mulholland MW, Lillemoe KD, Doherty GM, Maier RV, Upchurch GR, eds. Greenfield’s Surgery. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.)
Hirschsprung disease often presents in the first 24 hours of life as bilious emesis with abdominal distention.
Imperforate Anus
•Congenital defect with the anus absent or misplaced
•Usually forms an anterior fistulous tract
•May be associated with a cloacal deformity
•Divided into “high” and “low” malformations (relative to levator muscle)
•High/cloacal anomalies (fistula to bladder, vagina, or prostatic urethra) are initially treated with an end sigmoid colostomy, followed by a posterior sagittal anorectoplasty (PSARP) and genitourinary reconstruction if a cloacal anomaly is present
•Low anomalies are treated with a PSARP
•Preoperative anal dilatation may be required to avoid stricture
A colostomy is not generally needed to treat a low (below levator muscle) imperforate anus.
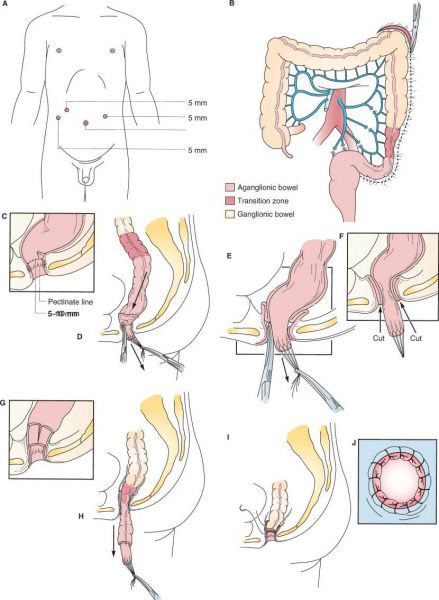
Laparoscopic-assisted pull-through for Hirschsprung
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


