449 Parkinson’s Disease and Other Movement Disorders
PARKINSON’S DISEASE AND RELATED DISORDERS
Parkinson’s disease (PD) is the second commonest neurodegenerative disease, exceeded only by Alzheimer’s disease (AD). Its cardinal clinical features were first described by the English physician James Parkinson in 1817. It is noteworthy that James Parkinson was a general physician who captured the essence of this condition based on a visual inspection of a mere handful of patients. It is estimated that approximately 1 million persons in the United States, 1 million in Western Europe, and 5 million worldwide suffer from this disorder. PD affects men and women of all races, all occupations, and all countries. The mean age of onset is about 60 years. The frequency of PD increases with aging, but cases can be seen in patients in their 20s and even younger. Based on the aging of the population and projected demographics, it is estimated that the prevalence of the disease will dramatically increase in the next several decades.
Clinically, PD is characterized by rest tremor, rigidity, bradykinesia (slowing), and gait impairment, known as the “cardinal features” of the disease. Additional features can include freezing of gait, postural instability, speech difficulty, autonomic disturbances, sensory alterations, mood disorders, sleep dysfunction, cognitive impairment, and dementia (Table 449-1).
CLINICAL FEATURES OF PARKINSON’S DISEASE |
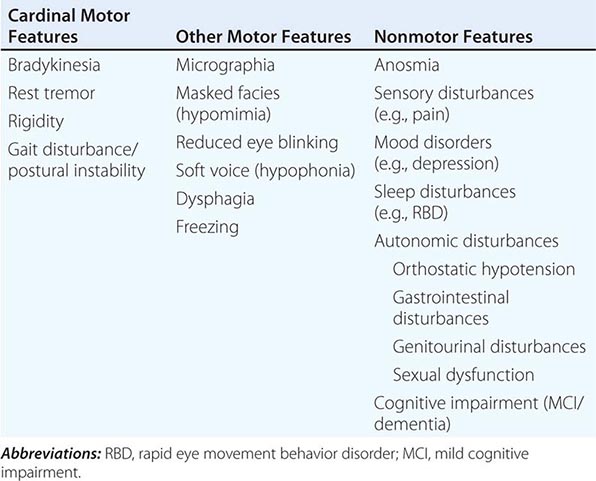
Pathologically, the hallmark features of PD are degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc), reduced striatal dopamine, and intracytoplasmic proteinaceous inclusions known as Lewy bodies that primarily contain the protein alpha synuclein (Fig. 449-1). While interest has primarily focused on the dopamine system, neuronal degeneration with inclusion body formation can also affect cholinergic neurons of the nucleus basalis of Meynert (NBM), norepinephrine neurons of the locus coeruleus (LC), serotonin neurons in the raphe nuclei of the brainstem, and neurons of the olfactory system, cerebral hemispheres, spinal cord, and peripheral autonomic nervous system. This “nondopaminergic” pathology is likely responsible for the development of nondopaminergic clinical features listed in Table 449-1 characterized by their lack of satisfactory response to dopaminergic replacement therapy. There is evidence that Lewy body pathology first begins in the peripheral autonomic nervous system, olfactory system, and dorsal motor nucleus of the vagus nerve in the lower brainstem, and then spreads in a predictable and sequential manner to affect the upper brainstem and cerebral hemispheres (Braak staging). These studies suggest that degeneration of dopamine neurons develops in a mid-stage of the disease. Indeed, epidemiologic studies suggest that clinical symptoms reflecting this nondopaminergic degeneration, such as constipation, anosmia, rapid eye movement (REM) behavior sleep disorder, and cardiac denervation, can precede the onset of the classic motor features of PD.
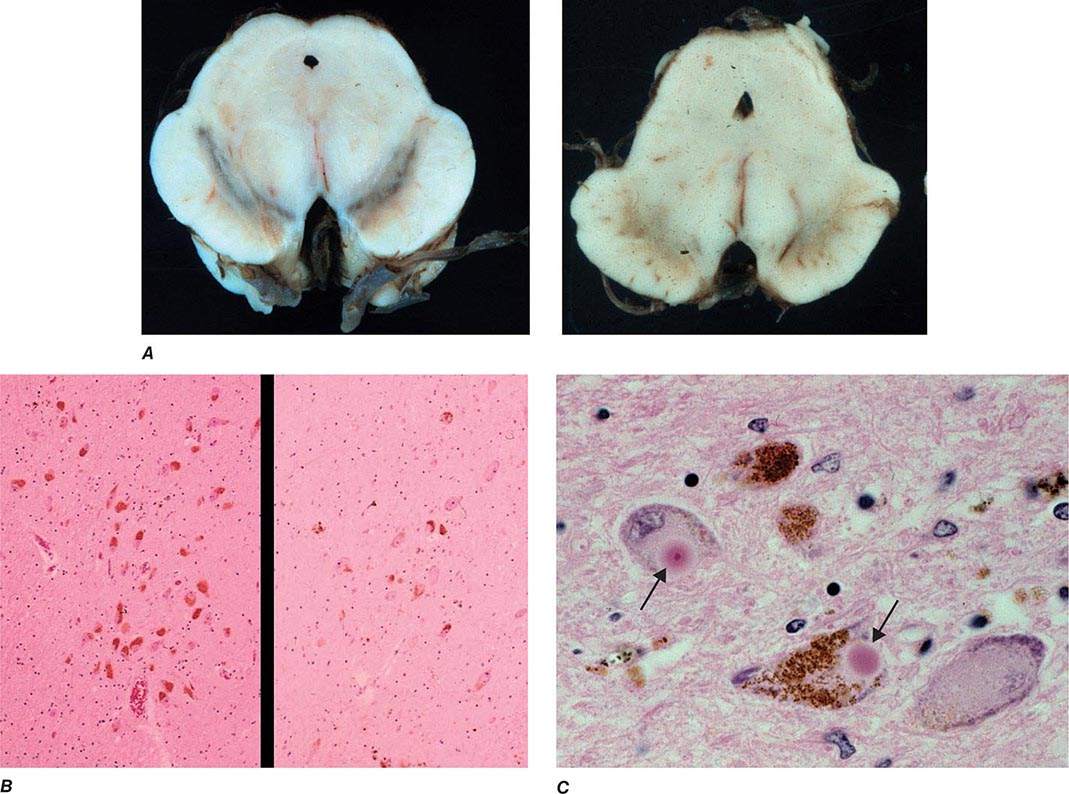
FIGURE 449-1 Pathologic specimens from a patient with Parkinson’s disease (PD) compared to a normal control demonstrating (A) reduction of pigment in SNc in PD (right) versus control (left), (B) reduced numbers of cells in SNc in PD (right) compared to control (left), and (C) Lewy bodies (arrows) within melanized dopamine neurons in PD. SNc, substantia nigra pars compacta.
DIFFERENTIAL DIAGNOSIS
Parkinsonism is a generic term that is used to define a syndrome manifest as bradykinesia with rigidity and/or tremor. It has a differential diagnosis (Table 449-2) that reflects damage to different components of the basal ganglia. The basal ganglia are comprised of a group of subcortical nuclei that include the striatum (putamen and caudate nucleus), subthalamic nucleus (STN), globus pallidus pars externa (GPe), globus pallidus pars interna (GPi), and the SNc (Fig. 449-2). Among the different forms of parkinsonism, PD is the most common (approximately 75% of cases). Historically, PD was diagnosed based on the presence of two of three parkinsonian features (tremor, rigidity, bradykinesia). However, postmortem studies found a 24% error rate when diagnosis was based on these criteria. Clinicopathologic correlation studies subsequently determined that parkinsonism associated with rest tremor, asymmetry, and a good response to levodopa was more likely to predict the correct pathologic diagnosis. With these revised criteria (known as the U.K. Brain Bank Criteria), a clinical diagnosis of PD is confirmed pathologically in as many as 99% of cases. A more complete definition of PD is now needed to incorporate the fact that there is widespread pathology beyond the dopaminergic system, nondopamine and nonmotor clinical features, and a premotor stage of the disease.
DIFFERENTIAL DIAGNOSIS OF PARKINSONISM |

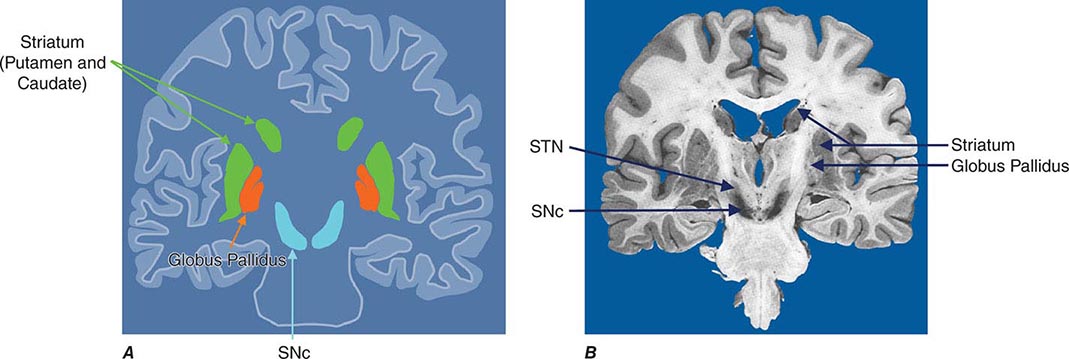
FIGURE 449-2 Basal ganglia nuclei. Schematic (A) and postmortem (B) coronal sections illustrating the various components of the basal ganglia. SNc, substantia nigra pars compacta; STN, subthalamic nucleus.
Imaging of the brain dopamine system in PD with positron emission tomography (PET) or single-photon emission computed tomography (SPECT) shows reduced uptake of striatal dopaminergic markers, particularly in the posterior putamen with relative sparing of the caudate nucleus (Fig. 449-3), reflecting the degeneration of nigrostriatal dopamine neurons. Imaging can be useful in patients where there is diagnostic uncertainty (e.g., dystonic tremor, essential tremor) or in research studies, but is rarely necessary in routine practice because the diagnosis can usually be established on clinical criteria alone. This may change in the future when there is a disease-modifying therapy and it is important to make the diagnosis as early as possible. Genetic testing is not routinely used at present, but can be helpful for identifying at-risk individuals in a research setting. Mutations of the LRRK2 gene (see below) have attracted particular interest because they are the commonest cause of familial PD and are responsible for approximately 1% of typical sporadic cases of the disease. Mutations in LRRK2 are a particularly common cause of PD in Ashkenazi Jews and North African Berber Arabs. The penetrance of the most common LRRK2 mutation ranges from 28 to 74% and is strongly correlated to the age of the carrier, with 50% affected by age 60 years. Mutations in the parkin gene should be considered in patients with onset prior to 40 years of age.
FIGURE 449-3 [11C]Dihydrotetrabenazine positron emission tomography (a marker of VMAT2) in healthy control (A) and Parkinson’s disease (B) patient. Note the reduced striatal uptake of tracer, which is most pronounced in the posterior putamen and tends to be asymmetric. (Courtesy of Dr. Jon Stoessl.)
Atypical and Secondary Parkinsonism Atypical parkinsonism refers to a group of neurodegenerative conditions that usually are associated with more widespread neurodegeneration than is found in PD (often involvement of striatum and/or globus pallidus as well as the SNc). As a group, they present with parkinsonism (rigidity and bradykinesia) but have a slightly different clinical picture than PD, reflecting differences in the underlying pathology. In these conditions, parkinsonism is typically characterized by early speech and gait impairment, absence of rest tremor, no motor asymmetry, poor or no response to levodopa, and an aggressive clinical course. In the early stages, they may show some modest benefit from levodopa and be difficult to distinguish from PD. Pathologically, neurodegeneration typically involves degeneration of the SNc but occurs without Lewy bodies (see below for individual conditions). Neuroimaging of the dopamine system is usually not helpful, because dopamine depletion can be seen in both PD and atypical parkinsonism. By contrast, metabolic imaging of the basal ganglia/thalamus network (using 2-F-deoxiglucose PET) may be helpful, showing a pattern of decreased activity in the GPi with increased activity in the thalamus, the reverse of what is seen in PD.
Multiple-system atrophy (MSA) manifests as a combination of parkinsonian, cerebellar, and autonomic features and can be divided into a predominant parkinsonian (MSA-p) or cerebellar (MSA-c) form. Clinically, MSA is suspected when a patient presents with atypical parkinsonism in conjunction with cerebellar signs and/or early and prominent autonomic dysfunction, usually orthostatic hypotension (Chap. 454). Pathologically, MSA is characterized by degeneration of the SNc, striatum, cerebellum, and inferior olivary nuclei coupled with characteristic glial cytoplasmic inclusions (GCIs) that stain for α-synuclein. Magnetic resonance imaging (MRI) can show pathologic iron accumulation in the striatum on T2-weighted scans, high signal change in the region of the external surface of the putamen (putaminal rim) in MSA-p, or cerebellar and brainstem atrophy (the pontine “hot cross buns” sign [Fig. 454-2]) in MSA-c. Mutations in the CoQ2 gene encoding parahydroxybenzoate-polyprenyl transferase, an enzyme involved in the biosynthesis of coenzyme Q10 (CoQ10), a cofactor of the mitochondrial respiratory chain, have been identified in familial and sporadic forms of MSA.
Progressive supranuclear palsy (PSP) is a form of atypical parkinsonism that is characterized by slow ocular saccades, eyelid apraxia, and restricted eye movements with particular impairment of downward gaze. Patients frequently experience hyperextension of the neck with early gait disturbance and falls. In later stages, speech and swallowing difficulty and cognitive impairment become evident. MRI may reveal a characteristic atrophy of the midbrain with relative preservation of the pons, the “hummingbird sign” on midsagittal images. Pathologically, PSP is characterized by degeneration of the SNc, striatum, subthalamic nucleus, midline thalamic nuclei, and pallidum along with neurofibrillary tangles and inclusions that stain for the tau protein.
Corticobasal ganglionic degeneration is less common and is usually manifest by asymmetric dystonic contractions and clumsiness of one hand coupled with cortical sensory disturbances manifest as apraxia, agnosia, focal limb myoclonus, or alien limb phenomenon (where the limb assumes a position in space without the patient being aware of it). Dementia may occur at any stage of the disease. Both cortical and basal ganglia features are required to make this diagnosis. MRI frequently shows asymmetric cortical atrophy. Pathologic findings include achromatic neuronal degeneration with tau deposits. Because other disorders such as PSP can present with a similar clinical picture, the term corticobasal ganglia syndrome should be used until a precise diagnosis can be confirmed pathologically.
Secondary parkinsonism can occur as a result of drugs, stroke, tumor, infection, or exposure to toxins such as carbon monoxide or manganese. Dopamine-blocking agents such as the neuroleptics are the commonest cause of secondary parkinsonism. These drugs are most widely used in psychiatry, but physicians should be aware that drugs such as metoclopramide and chlorpromazine, which are primarily used to treat gastrointestinal problems, are also neuroleptic agents and common causes of secondary parkinsonism (as well as acute and tardive dyskinesias; see below). Other drugs that can cause secondary parkinsonism include tetrabenazine, calcium channel blockers (flunarizine, cinnarizine), amiodarone, and lithium.
Finally, parkinsonism can be seen as a feature of other degenerative disorders such as Wilson’s disease, Huntington’s disease (especially the juvenile form known as the Westphal variant), dopa-responsive dystonia, and neurodegenerative disorders with brain iron accumulation such as pantothenate kinase (PANK)–associated neurodegeneration (formerly known as Hallervorden-Spatz disease).
Some features that suggest parkinsonism might be due to a condition other than PD are shown in Table 449-3.
FEATURES SUGGESTING AN ATYPICAL OR SECONDARY CAUSE OF PARKINSONISM |
ETIOLOGY AND PATHOGENESIS
Most PD cases occur sporadically (~85–90%) and are of unknown cause. Twin studies suggest that environmental factors likely play an important role in patients older than 50 years, with genetic factors being more important in younger patients. Epidemiologic studies also suggest increased risk with exposure to pesticides, rural living, and drinking well water and reduced risk with cigarette smoking and caffeine. However, no environmental factor has yet been proven to cause typical PD. The environmental hypothesis received support with the demonstration in the 1980s that MPTP (1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine), a byproduct of the illicit manufacture of a heroin-like drug, caused a PD-like syndrome in addicts in northern California. MPTP is transported to the central nervous system, where it is oxidized to form MPP+, a mitochondrial toxin that is selectively taken up by, and damages, dopamine neurons. However, MPTP or MPTP-like compounds have not been linked to sporadic PD.
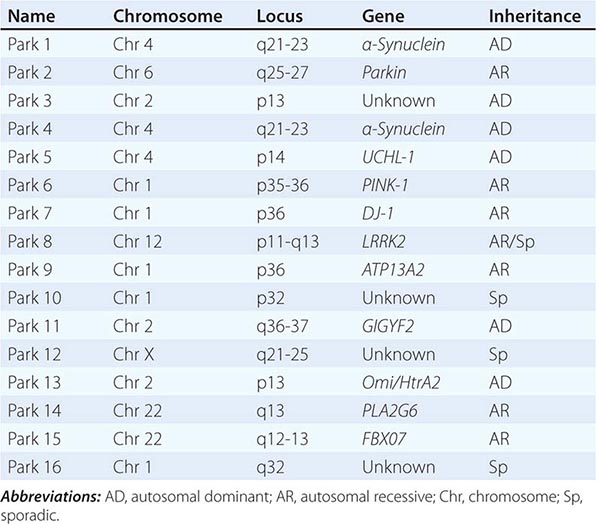
About 10–15% of cases are familial in origin, and multiple specific mutations and gene associations have been identified (Table 449-4). Genetic factors have also been linked to sporadic cases, with several typical PD cases found to carry the LRRK2 mutation, and genome-wide association studies (GWAS) implicating alpha synuclein, tau, and HLA as risk factors. It has been proposed that most cases of PD may be due to a “double hit” involving an interaction between a gene mutation that induces susceptibility coupled with exposure to a toxic environmental factor that may induce epigenetic or somatic DNA alterations. In this scenario, both factors are required for PD to ensue, while the presence of either one alone is not sufficient to cause the disease.
GENETIC CAUSES OF PARKINSON’S DISEASE |
Several factors have been implicated in the pathogenesis of cell death in PD, including oxidative stress, inflammation, mitochondrial dysfunction, and proteolytic stress. Recent studies have demonstrated that with aging dopamine neurons switch from sodium to calcium pacing through calcium channels, potentially making these high-energy neurons vulnerable to calcium-mediated neurotoxicity. Whatever the pathogenic mechanism, cell death appears to occur, at least in part, by way of a signal-mediated apoptotic or “suicidal” process. Each of these mechanisms offers a potential target for neuroprotective drugs. However, it is not clear which of these factors is primary, if the mechanism is the same in each individual case, if they act by way of a network such that a cocktail of agents might be required to provide neuroprotection, or if the findings to date merely represent epiphenomena unrelated to the true cause of cell death that remains undiscovered (Fig. 449-4).
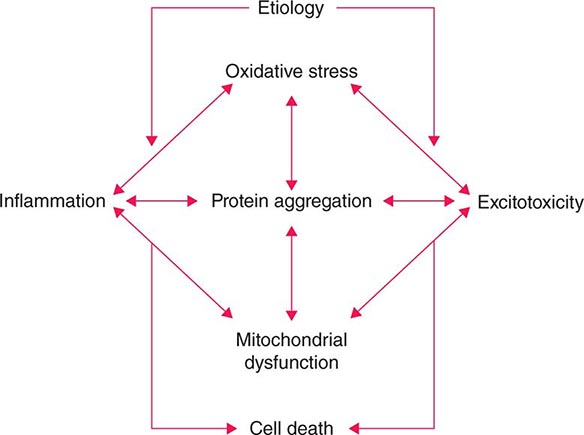
FIGURE 449-4 Schematic representation of how pathogenetic factors implicated in Parkinson’s disease interact in a network manner, ultimately leading to cell death. This figure illustrates how interference with any one of these factors may not necessarily stop the cell death cascade. (Adapted from CW Olanow: Movement Disorders 22:S-335, 2007.)
Gene mutations may not cause all cases of PD, but may be helpful in pointing to specific pathogenic pathways and mechanisms that are central to a neurodegenerative process that might be relevant to all forms of the disease. To date, most interest has focused on pathways implicated by mutations in α-synuclein, LRRK2, and PINK1/Parkin.
Most interest has focused on α-synuclein. Mutations in α-synuclein cause rare familial forms of PD, and α-synuclein constitutes the major component of Lewy bodies in patients with sporadic PD (Fig. 449-1). Furthermore, duplication or triplication of the wild-type α-synuclein can also cause a form of PD, indicating that increased production of the normal protein alone can cause the disease. More recently, Lewy pathology was discovered to have developed in healthy embryonic dopamine neurons that had been implanted into the striatum of PD patients, suggesting that the abnormal protein had transferred from affected cells to healthy unaffected dopamine neurons. Based on these findings, it has been proposed that α-synuclein is a prion and PD is a prion disorder. Here it is proposed that, like the prion protein PrPC, α-synuclein can misfold to form β-rich sheets, generate toxic oligomers and aggregates, polymerize to form amyloid plaques (i.e., Lewy bodies), cause neurodegeneration, and spread to involve unaffected neurons. Indeed, injection of α-synuclein fibrils into the striatum promotes the development of Lewy pathology in host neurons, neurodegeneration, behavioral abnormalities, and the spread of α-synuclein pathology to anatomically connected sites. Further support for this hypothesis comes from the demonstration that inoculation of α-synuclein derived from human Lewy bodies induces widespread Lewy pathology in mice and primates. Collectively, this evidence supports the possibility that neuroprotective therapies for PD might be developed based on inhibiting accumulation or accelerating removal of α-synuclein aggregates.
Mutations in the glucocerebrosidase (GBA) gene associated with Gaucher’s disease numerically represent the most important risk factor for the development of PD. While the responsible mechanism is not precisely known, it is noteworthy that GBA mutations are associated with altered autophagy and lysosomal function and could impair the clearance of α-synuclein.
Six different LRRK2 mutations have been linked to PD, with Gly2019Ser being the most common. The mechanism responsible for cell death with this mutation is not known but is thought to involve changes in kinase activity with altered phosphorylation of target proteins (including autophosphorylation) and possibly lysosomal dysfunction. Kinase inhibitors can block toxicity associated with LRRK2 mutations in laboratory models, and there has been much interest in developing drugs directed at this target. However, kinase inhibitors are likely to be toxic, the physiologic role of LRRK2 is not known, and the large majority of PD patients do not carry a LRRK2 mutation.
Mutations in PINK1 and parkin have implicated mitochondrial dysfunction as a possible cause of PD. Recent studies suggest a role for parkin and PINK1 proteins in the turnover and clearance of damaged mitochondria (mitophagy), and mutations in parkin and PINK1 cause mitochondrial dysfunction in transgenic animals that can be corrected with overexpression of parkin. This is a particularly attractive target because postmortem studies in PD patients show a defect in complex I of the respiratory chain in SNc neurons.
Thus, evidence is accumulating that genetics plays an important role in both familial and “sporadic” forms of PD. It is anticipated that better understanding of the pathways responsible for cell death caused by these mutations will permit the development of more relevant animal models of PD and targets for the development of neuroprotective drugs.
PATHOPHYSIOLOGY OF PD
The classic model of the organization of the basal ganglia in the normal and PD states is provided in Fig. 449-5. With respect to motor function, a series of neuronal circuits or loops link the basal ganglia nuclei with corresponding cortical motor regions in a somatotopic manner. The striatum is the major input region of the basal ganglia, while the GPi and SNr are the major output regions. The input and output regions are connected via direct and indirect pathways that have reciprocal effects on the activity of the output pathway. The output of the basal ganglia provides inhibitory (GABAergic) tone to thalamic and brainstem neurons that in turn connect to motor systems in the cerebral cortex and spinal cord that control motor function. Physiologically, decreased neuronal activity in the GPi/SNr is associated with movement facilitation and vice versa. Dopaminergic projections from SNc neurons serve to modulate neuronal firing and to stabilize the basal ganglia network. The basal ganglia and similar cortical loops are now thought to also play an important role in regulating normal behavioral, emotional, and cognitive functions.
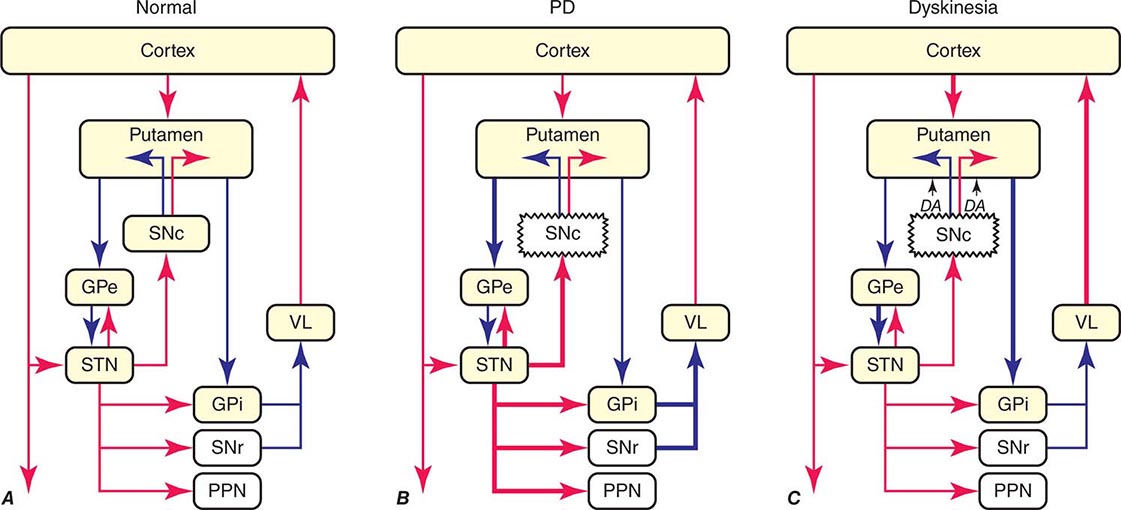
FIGURE 449-5 Basal ganglia organization. Classic model of the organization of the basal ganglia in the normal (A), Parkinson’s disease (PD) (B), and levodopa-induced dyskinesia (C) state. Inhibitory connections are shown as blue arrows and excitatory connections as red arrows. The striatum is the major input region and receives its major input from the cortex. The GPi and SNr are the major output regions, and they project to the thalamocortical and brainstem motor regions. The striatum and GPi/SNr are connected by direct and indirect pathways. This model predicts that parkinsonism results from increased neuronal firing in the STN and GPi and that lesions or DBS of these targets might provide benefit. This concept led to the rationale for surgical therapies for PD. The model also predicts that dyskinesia results from decreased firing of the output regions, resulting in excessive cortical activation by the thalamus. This component of the model is not completely correct because lesions of the GPi ameliorate rather than increase dyskinesia in PD, suggesting that firing frequency is just one of the components that lead to the development of dyskinesia. DBS, deep brain stimulation; GPe, external segment of the globus pallidus; GPi, internal segment of the globus pallidus; PPN, pedunculopontine nucleus; SNc, substantia nigra, pars compacta; SNr, substantia nigra, pars reticulata; STN, subthalamic nucleus; VL, ventrolateral thalamus. (Derived from JA Obeso et al: Trends Neurosci 23:S8, 2000.)
In PD, dopamine denervation with loss of dopaminergic tone leads to increased firing of neurons in the STN and GPi, excessive inhibition of the thalamus, reduced activation of cortical motor systems, and the development of parkinsonian features (Fig. 449-5). The current role of surgery in the treatment of PD is based on this model, which predicted that lesions or high-frequency stimulation of the STN or GPi might reduce this neuronal overactivity and improve PD features.
TREATMENT | PARKINSON’S DISEASE |
LEVODOPA
Since its introduction in the late 1960s, levodopa has been the mainstay of therapy for PD. Experiments in the late 1950s by Carlsson demonstrated that blocking dopamine uptake with reserpine caused rabbits to become parkinsonian; this could be reversed with the dopamine precursor, levodopa. Subsequently, Hornykiewicz demonstrated a dopamine deficiency in the striatum of PD patients and suggested the potential benefit of dopaminergic replacement therapy. Dopamine does not cross the blood-brain barrier (BBB), so clinical trials were initiated with levodopa, a precursor of dopamine. Studies over the course of the next decade confirmed the value of levodopa and revolutionized the treatment of PD.
Levodopa is routinely administered in combination with a peripheral decarboxylase inhibitor to prevent its peripheral metabolism to dopamine and the development of nausea and vomiting due to activation of dopamine receptors in the area postrema that are not protected by the BBB. In the United States, levodopa is combined with the decarboxylase inhibitor carbidopa (Sinemet), whereas in many other countries, it is combined with benserazide (Madopar). Levodopa is also available in controlled-release formulations as well as in combination with a catechol-O-methyltransferase (COMT) inhibitor (see below). Levodopa remains the most effective symptomatic treatment for PD and the gold standard against which new therapies are compared. No current medical or surgical treatment provides antiparkinsonian benefits superior to what can be achieved with levodopa. Levodopa benefits the classic motor features of PD, prolongs independence and employability, improves quality of life, and increases life span. Almost all PD patients experience improvement, and failure to respond to an adequate trial should cause the diagnosis to be questioned.
There are, however, important limitations of levodopa therapy. Acute dopaminergic side effects include nausea, vomiting, and orthostatic hypotension. These are usually transient and can generally be avoided by gradual titration. If they persist, they can be treated with additional doses of a peripheral decarboxylase inhibitor (e.g., carbidopa) or a peripheral dopamine-blocking agent such as domperidone (not available in the United States). More important are motor complications (see below) that develop in the majority of patients treated long-term with levodopa. In addition, the disease continues to progress, and features such as falling, freezing, autonomic dysfunction, sleep disorders, and dementia may emerge with disease progression that are not adequately controlled by levodopa. Indeed, these nondopaminergic features (especially falling and dementia) are the primary source of disability and the main reason for nursing home placement for patients with advanced PD
Levodopa-induced motor complications consist of fluctuations in motor response (“on” episodes when the drug is working and “off” episodes when parkinsonian features return) and involuntary movements known as dyskinesias (Fig. 449-6). When patients initially take levodopa, benefits are long-lasting (many hours) even though the drug has a relatively short half-life (60–90 min). With continued treatment, however, the duration of benefit following an individual dose becomes progressively shorter until it approaches the half-life of the drug. This loss of benefit is known as the wearing-off effect. In more severe cases, patients may experience a delay in turning on (delayed-on) or no response at all to a given dose (no-on). Dyskinesias tend to occur at the time of levodopa peak plasma concentration and maximal clinical benefit (peak-dose dyskinesia). They are usually choreiform in nature but can manifest as dystonic movements, myoclonus, or other movement disorders. They are not troublesome when mild, but can be disabling when severe, and can limit the ability to fully use levodopa to control PD features. In more advanced states, patients may cycle between “on” periods complicated by disabling dyskinesias and “off” periods in which they suffer from severe parkinsonism and painful dystonic postures. Patients may also experience “diphasic dyskinesias,” which occur as the levodopa dose begins to take effect and again as it wears off. These dyskinesias typically consist of transient, stereotypic, rhythmic movements that predominantly involve the lower extremities and are frequently associated with parkinsonism in other body regions. They can be relieved by increasing the dose of levodopa, although higher doses may induce more severe peak-dose dyskinesia.
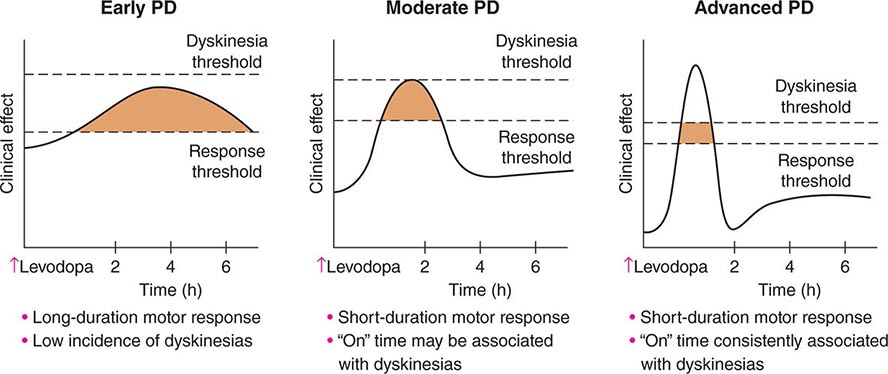
FIGURE 449-6 Changes in motor response associated with chronic levodopa treatment. Levodopa-induced motor complications. Schematic illustration of the gradual shortening of the duration of a beneficial motor response to levodopa (wearing off) and the appearance of dyskinesias complicating “on” time. PD, Parkinson’s disease.
The cause of levodopa-induced motor complications is not precisely known. They are more likely to occur in females, younger individuals with more severe disease, and with the use of higher doses (mg/kg) of levodopa. The classic model of the basal ganglia has been useful for understanding the origin of motor features in PD, but has proved less valuable for understanding levodopa-induced dyskinesias (Fig. 449-5). The model predicts that dopamine replacement might excessively inhibit the pallidal output system, thereby leading to increased thalamocortical activity, enhanced stimulation of cortical motor regions, and the development of dyskinesia. However, lesions of the pallidum that completely destroy its output are associated with amelioration rather than induction of dyskinesia as suggested by the classic model. It is now thought that dyskinesia results from levodopa-induced alterations in the GPi neuronal firing pattern (pauses, bursts, synchrony, etc.) and oscillatory activity, and not simply the firing frequency alone. This in turn leads to the transmission of misinformation from pallidum to thalamus/cortex, resulting in dyskinesia. Surgical lesions or high-frequency stimulation might ameliorate dyskinesia by interfering with (blocking or masking) this abnormal neuronal activity and preventing the transfer of misinformation to motor systems.
Current information suggests that altered neuronal firing patterns and motor complications relate to nonphysiologic levodopa replacement. Striatal dopamine levels are normally maintained at a relatively constant level. In PD, dopamine neurons degenerate and striatal dopamine is dependent on the peripheral availability of levodopa. Intermittent doses of short-acting levodopa result in fluctuating plasma levels because of variability in transit of the drug from the stomach to the duodenum where it is absorbed and the short half-life of the drug. This variability results in exposure of dopamine receptors to pathologically high and low concentrations of dopamine. It has been hypothesized that more continuous delivery of levodopa might prevent the development of motor complications. Indeed, a recent controlled study demonstrated that continuous intraintestinal infusion of levodopa/carbidopa intestinal gel (Duodopa) is associated with significant improvement in “off” time and in “on” time without dyskinesia in advanced PD patients compared with optimized standard oral levodopa.
Behavioral alterations can also be encountered in levodopa-treated patients. A dopamine dysregulation syndrome has been described where patients have a craving for levodopa and take frequent and unnecessary doses of the drug in an addictive manner. PD patients taking high doses of levodopa can develop purposeless, stereotyped behaviors such as the meaningless assembly and disassembly or collection and sorting of objects. This is known as punding, a term taken from the Swedish description of the meaningless behaviors seen in chronic amphetamine users. Hypersexuality and other impulse-control disorders are occasionally encountered with levodopa, although these are more commonly seen with dopamine agonists.
DOPAMINE AGONISTS
Dopamine agonists are a diverse group of drugs that act directly on dopamine receptors. Unlike levodopa, they do not require metabolism to an active product and do not undergo oxidative metabolism. Initial dopamine agonists were ergot derivatives (e.g., bromocriptine, pergolide, cabergoline) and were associated with ergot-related side effects, including cardiac valvular damage. They have largely been replaced by a second generation of nonergot dopamine agonists (e.g., pramipexole, ropinirole, rotigotine). In general, dopamine agonists do not have comparable efficacy to levodopa. They were initially introduced as adjuncts to levodopa to enhance motor function and reduce “off” time in fluctuating patients. Subsequently, it was shown that dopamine agonists, possibly because they are relatively long-acting, are less prone than levodopa to induce dyskinesia. For this reason, many physicians initiate therapy with a dopamine agonist, although supplemental levodopa is eventually required in virtually all patients. Both ropinirole and pramipexole are available as orally administered immediate (tid) and extended-release (qd) formulations. Rotigotine is administered as a once-daily transdermal patch. Apomorphine is a dopamine agonist with efficacy comparable to levodopa, but it must be administered parenterally and has a very short half-life and duration of activity (45 min). It is generally administered by injection as a rescue agent for the treatment of severe “off” episodes. Apomorphine can also be administered by continuous subcutaneous infusion and has been demonstrated to reduce both “off” time and dyskinesia in advanced patients. However, this approach has not been approved in the United States.
Dopamine agonist use is associated with a variety of side effects. Acute side effects are primarily dopaminergic and include nausea, vomiting, and orthostatic hypotension. As with levodopa, these can usually be avoided by slow titration. Side effects associated with chronic use include hallucinations and cognitive impairment. Sedation with sudden unintended episodes of falling asleep while driving a motor vehicle have been reported. Patients should be informed about this potential problem and should not drive when tired. Dopamine agonists can also be associated with impulse-control disorders, including pathologic gambling, hypersexuality, and compulsive eating and shopping. The precise cause of these problems, and why they appear to occur more frequently with dopamine agonists than levodopa, remains to be resolved, but reward systems associated with dopamine and alterations in the ventral striatum and orbitofrontal regions have been implicated. In general, chronic side effects are dose-related and can be avoided or minimized with lower doses. Injections of apomorphine and patch delivery of rotigotine can be complicated by development of skin lesions at sites of administration.
MAO-B INHIBITORS
Inhibitors of monoamine oxidase type B (MAO-B) block central dopamine metabolism and increase synaptic concentrations of the neurotransmitter. Selegiline and rasagiline are relatively selective suicide inhibitors of the MAO-B enzyme. Clinically, MAO-B inhibitors provide antiparkinsonian benefits when used as monotherapy in early disease and reduced “off” time when used as an adjunct to levodopa in patients with motor fluctuations. MAO-B inhibitors are generally safe and well tolerated. They may increase dyskinesia in levodopa-treated patients, but this can usually be controlled by down-titrating the dose of levodopa. Inhibition of the MAO-A isoform prevents metabolism of tyramine in the gut, leading to a potentially fatal hypertensive reaction known as a “cheese effect” because it can be precipitated by foods rich in tyramine such as some cheeses, aged meats, and red wine. Selegiline and rasagiline do not functionally inhibit MAO-A and are not associated with a cheese effect with doses typically used in clinical practice. There are theoretical risks of a serotonin reaction in patients receiving concomitant selective serotonin reuptake inhibitor (SSRI) antidepressants, but these are rarely encountered.
Interest in MAO-B inhibitors has also focused on their potential to have disease-modifying effects. MPTP toxicity can be prevented experimentally by coadministration of an MAO-B inhibitor that blocks its conversion to the toxic pyridinium ion MPP+. MAO-B inhibitors also have the potential to block the oxidative metabolism of dopamine and prevent oxidative stress. In addition, both selegiline and rasagiline incorporate a propargyl ring within their molecular structure that provides antiapoptotic effects in laboratory models. The DATATOP study showed that selegiline significantly delayed the time until the emergence of disability, necessitating the introduction of levodopa, in untreated PD patients. However, it could not be determined whether this was due to a neuroprotective effect that slowed disease progression or a symptomatic effect that merely masked ongoing neurodegeneration. More recently, the ADAGIO study demonstrated that early treatment with rasagiline 1 mg/d, but not 2 mg/d, provided benefits that could not be achieved when treatment with the same drug was initiated at a later time point, consistent with a disease-modifying effect; however, the long-term significance of these findings is uncertain.
COMT INHIBITORS
When levodopa is administered with a decarboxylase inhibitor, it is primarily metabolized in the periphery by COMT. Inhibitors of COMT increase the elimination half-life of levodopa and enhance its brain availability. Combining levodopa with a COMT inhibitor reduces “off” time and prolongs “on” time in fluctuating patients while enhancing motor scores. Two COMT inhibitors have been approved, tolcapone and entacapone. There is also a combination tablet of levodopa, carbidopa, and entacapone (Stalevo).
Side effects of COMT inhibitors are primarily dopaminergic (nausea, vomiting, increased dyskinesia) and can usually be controlled by down-titrating the dose of levodopa by 20–30%. Severe diarrhea has been described with tolcapone, and to a lesser degree with entacapone, and necessitates stopping the medication in 5–10% of individuals. Cases of fatal hepatic toxicity have been reported with tolcapone, and periodic monitoring of liver function is required. This problem has not been encountered with entacapone. Discoloration of urine can be seen with both COMT inhibitors due to accumulation of a metabolite, but it is of no clinical concern.
It has been proposed that initiating levodopa in combination with a COMT inhibitor to enhance its elimination half-life could provide more continuous levodopa delivery if administered at frequent intervals and reduce the risk of motor complications. While this result has been demonstrated in a preclinical MPTP model, and continuous infusion reduces both “off” time and dyskinesia in advanced PD patients, no benefit of initiating levodopa with a COMT inhibitor compared to levodopa alone was detected in early PD patients in the STRIDE-PD study. This may have been because the combination was not administered at frequent enough intervals to provide continuous levodopa availability. For now, the main value of COMT inhibitors continues to be in patients who experience motor fluctuations.
OTHER MEDICAL THERAPIES
Centrally acting anticholinergic drugs such as trihexyphenidyl and benztropine were used historically for the treatment of PD, but they lost favor with the introduction of dopaminergic agents. Their major clinical effect is on tremor, although it is not certain that this benefit is superior to what can be obtained with agents such as levodopa and dopamine agonists. Still, they can be helpful in individual patients with severe tremor. Their use is limited particularly in the elderly, due to their propensity to induce a variety of side effects including urinary dysfunction, glaucoma, and particularly cognitive impairment.
Amantadine also has historical importance. Originally introduced as an antiviral agent, it was appreciated to also have antiparkinsonian effects that are now thought to be due to N-methyl-D-aspartate (NMDA) receptor antagonism. While some physicians use amantadine in patients with early disease for its mild symptomatic effects, it is most widely used as an antidyskinesia agent in patients with advanced PD. Indeed, it is the only oral agent that has been demonstrated in controlled studies to reduce dyskinesia without worsening parkinsonian features, although benefits may be relatively transient. Cognitive impairment is a major concern. Other side effects include livido reticularis and weight gain. Amantadine should always be discontinued gradually because patients can experience withdrawal-like symptoms.
Several new classes of drug are currently being investigated in an attempt to enhance antiparkinsonian effects, reduce off time, and treat or prevent dyskinesia. These include adenosine A2A antagonists, nicotinic agonists, glutamate antagonists, and 5-HT1A agonists.
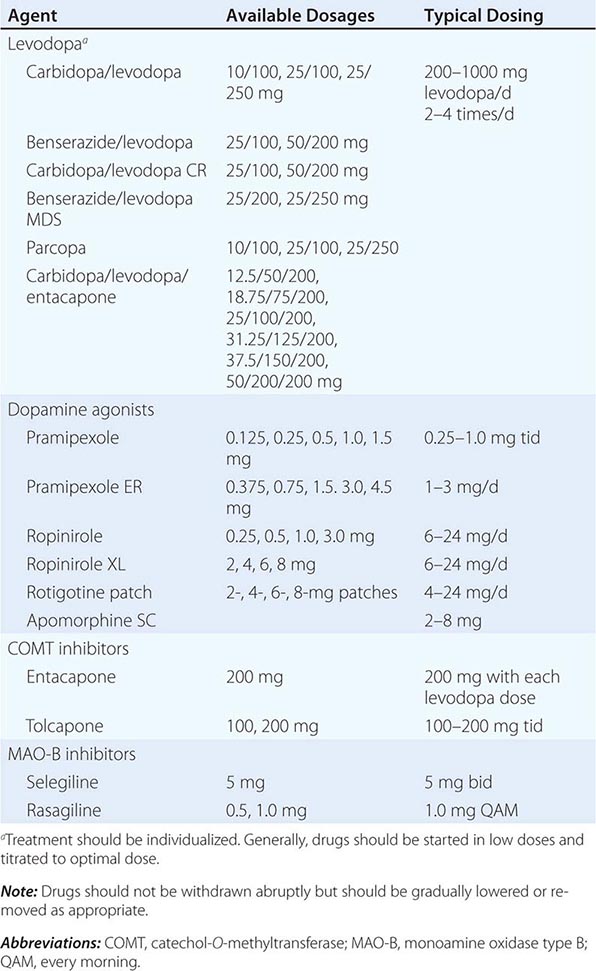
A list of the major drugs and available dosage strengths is provided in Table 449-5.
DRUGS COMMONLY USED FOR TREATMENT OF PARKINSON’S DISEASEa |
NEUROPROTECTION
Despite the many therapeutic agents available for the treatment of PD, patients continue to experience disease progression with intolerable disability. A neuroprotective therapy that slows or stops disease progression remains the major unmet therapeutic need in PD. As noted above, trials of certain drugs (e.g., selegiline and rasagiline) have provided positive results consistent with a disease-modifying effect. However, it is not possible to determine if the positive results were due to neuroprotection with slowing of disease progression or confounding symptomatic effects that mask ongoing progression. CoQ10, a mitochondrial bioenhancer and antioxidant, attracted attention with a positive preliminary trial, but this was not replicated in larger double-blind studies.
SURGICAL TREATMENT
Surgical treatments for PD have been used for more than a century. Lesions placed in the motor cortex improved tremor but were associated with motor deficits, and this approach was abandoned. Subsequently, it was appreciated that lesions placed into the ventral intermediate (VIM) nucleus of the thalamus reduced contralateral tremor without inducing hemiparesis, but these lesions did not meaningfully help other more disabling features of PD. In the 1990s, it was shown that lesions placed in the posteroventral portion of the GPi (motor territory) improved rigidity and bradykinesia as well as tremor. Importantly, pallidotomy was also associated with marked improvement in contralateral dyskinesia. This procedure gained favor with greater understanding of the pathophysiology of PD (see above). However, this procedure is not optimal for patients with bilateral disease, because bilateral lesions are associated with side effects such as dysphagia, dysarthria, and impaired cognition, and has largely been replaced by deep brain stimulation (DBS). Unilateral lesions of the STN are associated with a larger antiparkinsonian benefit and reduced levodopa requirement, but there is a concern about the risk of hemiballismus, and this procedure is not commonly performed.
Most surgical procedures for PD performed today use DBS. Here, an electrode is placed into the target area and connected to a stimulator inserted SC over the chest wall. DBS simulates the effects of a lesion without necessitating making a brain lesion. The precise mechanism whereby DBS works is not fully resolved but may act by disrupting the abnormal signal associated with PD and motor complications. The stimulation variables can be adjusted with respect to electrode configuration, voltage, frequency, and pulse duration in order to maximize benefit and minimize adverse side effects. In cases with intolerable side effects, stimulation can be stopped and the system removed. The procedure does not require making a lesion in the brain and is thus suitable for performing bilateral procedures with relative safety.
DBS for PD primarily targets the STN or the GPi. It provides dramatic results, particularly with respect to reducing “off” time and dyskinesias, but does not improve or prevent the development of features that fail to respond to levodopa such as freezing, falling, and dementia. The procedure is thus primarily indicated for patients who suffer disability resulting from severe tremor, or levodopa-induced motor complications that cannot be satisfactorily controlled with drug manipulation. In such patients, DBS has been shown to improve quality of life in comparison to best medical therapy. Side effects can be seen with respect to the surgical procedure (hemorrhage, infarction, infection), the DBS system (infection, lead break, lead displacement, skin ulceration), or the stimulation itself (ocular and speech abnormalities, muscle twitches, paresthesias, depression, and rarely suicide). Recent studies indicate that benefits following DBS of the STN and GPi are comparable, but that GPi stimulation may be associated with a reduced frequency of depression. Although not all PD patients are candidates, the procedure is profoundly beneficial for many. Studies of DBS in early PD patients show benefits in comparison to medical therapy, but this must be weighed against the cost of the procedure and the risk of side effects. Long-term studies demonstrate continued benefits with respect to the classical motor features of PD, but DBS does not prevent the development of nondopaminergic features, which continue to be a source of disability. Studies continue to evaluate the optimal way to use DBS (low- vs high-frequency stimulation, closed systems, etc.). Comparison of DBS to other therapies aimed at improving motor function without causing dyskinesia, such as Duodopa and apomorphine infusions, remains to be performed. Studies are examining additional DBS targets that might benefit gait dysfunction, depression, and cognitive impairment in PD patients.
EXPERIMENTAL THERAPIES FOR PD
There has been considerable scientific and public interest in a number of novel interventions that are being investigated as possible treatments for PD. These include cell-based therapies (such as transplantation of fetal nigral dopamine cells or dopamine neurons derived from stem cells), gene therapies, and trophic factors. Transplant strategies are based on the concept of implanting dopaminergic cells into the striatum to replace degenerating SNc dopamine neurons. Fetal nigral mesencephalic cells have been demonstrated to survive implantation, re-innervate the striatum in an organotypic manner, and restore motor function in PD models. However, two double-blind studies failed to show significant benefit of fetal nigral transplantation in comparison to a sham operation with respect to their primary endpoints. Additionally, grafting of fetal nigral cells is associated with a previously unrecognized form of dyskinesia that persists after lowering or even stopping levodopa. This has been postulated to be related to unregulated release of dopamine from serotonin neurons. In addition, there is evidence that after many years, transplanted healthy embryonic dopamine neurons from unrelated donors develop PD pathology and become dysfunctional, suggesting transfer of α-synuclein from affected to unaffected neurons in a prion-like manner (see discussion above). Perhaps most importantly, it is not clear how replacing dopamine cells alone will improve nondopaminergic features such as falling and dementia, which are the major sources of disability for patients with advanced disease. These same concerns apply to dopamine neurons derived from stem cells, which have not yet been properly tested in PD patients and bear the additional concern of tumors and unanticipated side effects. The short-term future for this technology as a treatment for PD, at least in its current state, is therefore not promising, and there is no scientific basis to warrant routine treatment with stem cells as is being marketed in some countries.
Trophic factors are a series of proteins that enhance neuronal growth and restore function to damaged neurons. There are several different trophic factors that have been demonstrated to have beneficial effects on dopamine neurons in laboratory studies. Glial-derived neurotrophic factor (GDNF) and neurturin have attracted particular attention as possible therapies for PD. However, double-blind trials of intraventricular and intraputaminal infusions of GDNF failed to show benefits compared to placebo in PD patients, possibly because of inadequate delivery of the trophic molecule throughout the target region.
Gene delivery offers the potential of providing widespread delivery throughout a target region and long-term expression of a therapeutic protein with a single procedure. Gene therapy involves placing the DNA of a therapeutic protein into a viral vector that can then be delivered to specific target regions. The DNA of the therapeutic protein is then incorporated into the genome of the host cells and released on a continual basis. The AAV2 virus has been most often used as the viral vector because it does not promote an inflammatory response, is not incorporated into the host genome, and is associated with long-lasting transgene expression. Clinical trials of AAV2 delivery of the trophic factor neurturin showed promising results in open label trials but failed in double-blind trials, possibly because axonal damage in PD prevented retrograde transport of the protein to dopamine neurons in the SNc where it is required to induce upregulation of repair genes required for the trophic response. However, a subsequent double-blind trial of AAV2-neurturin delivered into both the putamen and SNc also failed.
Gene delivery is also being explored as a means of delivering aromatic amino acid decarboxylase with or without tyrosine hydroxylase to the striatum to facilitate dopamine production and glutamic acid decarboxylase to the STN to inhibit overactive neuronal firing. None of these procedures has been established to be effective in PD patients. Furthermore, although gene delivery technology has great potential, this approach also carries the risk of unanticipated side effects, and current approaches directed at the nigrostriatal system do not address the nondopaminergic features of the illness.
MANAGEMENT OF THE NONMOTOR AND NONDOPAMINERGIC FEATURES OF PD
Although PD management has primarily focused on the dopaminergic features of the disease, management of the nondopaminergic features should not be ignored. Some nonmotor features, although not thought to reflect dopaminergic pathology, nonetheless benefit from dopaminergic drugs. For example, problems such as anxiety, panic attacks, depression, sweating, sensory problems, freezing, and constipation all tend to be worse during “off” periods and may improve with better dopaminergic control. Approximately 50% of PD patients suffer depression during the course of the disease, and depression is frequently underdiagnosed and undertreated. Antidepressants should not be withheld, particularly for patients with major depression. Serotonin syndromes have been a theoretical concern with the combined use of SSRIs and MAO-B inhibitors but are rarely encountered. Anxiety can be treated with short-acting benzodiazepines.
Psychosis can be a problem for some PD patients. In contrast to AD, hallucinations are typically visual, formed, and nonthreatening. Importantly, they can limit the use of dopaminergic agents to obtain satisfactory motor control. Psychosis in PD often responds to low doses of atypical neuroleptics and permits higher doses of levodopa to be tolerated. Clozapine is the most effective drug, but it can be associated with agranulocytosis, and regular monitoring is required. For this reason, many physicians start with quetiapine even though it has not been established to be effective in placebo-controlled trials. Hallucinations in PD patients are often a harbinger of a developing dementia.
Dementia in PD (PDD) is common, ultimately affecting as many as 80% of patients. Its frequency increases with aging and, in contrast to AD, primarily affects executive functions and attention, with relative sparing of language, memory, and calculations. When dementia precedes, or develops within 1 year after, the onset of motor dysfunction, it is by convention referred to as dementia with Lewy bodies (DLB; Chap. 448). These patients are particularly prone to have hallucinations and diurnal fluctuations. Pathologically, DLB is characterized by Lewy bodies distributed throughout the cerebral cortex (especially the hippocampus and amygdala) and is often also associated with AD pathology. It is likely that DLB and PDD represent a PD spectrum rather than separate disease entities. Mild cognitive impairment (MCI) frequently precedes the onset of dementia and is a more reliable index of impending dementia in PD than in the general population. Dopaminergic drugs can worsen cognitive function in demented patients and should be stopped or reduced to try and provide a compromise between antiparkinsonian benefit and preserved cognitive function. Drugs are usually discontinued in the following sequence: anticholinergics, amantadine, dopamine agonists, COMT inhibitors, and MAO-B inhibitors. Eventually, patients with cognitive impairment should be managed with the lowest dose of standard levodopa that provides meaningful antiparkinsonian effects and does not worsen mental function. Anticholinesterase agents such as rivastigmine and donepezil reduce the rate of deterioration of measures of cognitive function and can improve attention, but do not typically improve cognitive function in any meaningful way.
Autonomic disturbances are common and frequently require attention. Orthostatic hypotension can be problematic and contribute to falling. Initial treatment should include adding salt to the diet and elevating the head of the bed to prevent overnight sodium natriuresis. Low doses of fludrocortisone (Florinef) or midodrine provide control for most cases. Vasopressin, erythropoietin, and the norepinephrine precursor 3-0-methylDOPS can be used in more severe or refractory cases. If orthostatic hypotension is prominent in early disease, MSA should be considered. Sexual dysfunction can be helped with sildenafil or tadalafil. Urinary problems, especially in males, should be treated in consultation with a urologist to exclude prostate problems. Anticholinergic agents, such as oxybutynin (Ditropan), may be helpful. Constipation can be a very important problem for PD patients. Mild laxatives or enemas can be useful, but physicians should first ensure that patients are drinking adequate amounts of fluid and consuming a diet rich in bulk with green leafy vegetables and bran. Agents that promote gastrointestinal (GI) motility can also be helpful.
Sleep disturbances are common in PD patients, with many experiencing fragmented sleep with excess daytime sleepiness. Restless leg syndrome, sleep apnea, and other sleep disorders should be treated as appropriate. REM behavior disorder (RBD) is a syndrome comprised of violent movements and vocalizations during REM sleep, possibly representing acting out of dreams due to a failure of the normal inhibition of motor movements that typically accompanies REM sleep. Many PD patients have a history of antecedent RBD preceding the onset of the classic motor features of PD, and most cases of RBD go on to develop an α-synucleinopathy (PD or MSA). Low doses of clonazepam (0.5–1 mg at bedtime) are usually effective in controlling this problem. Consultation with a sleep specialist and polysomnography may be necessary to identify and optimally treat sleep problems.
NONPHARMACOLOGIC THERAPY
Gait dysfunction with falling is an important cause of disability in PD. Dopaminergic therapies can help patients whose gait is worse in “off” time, but there are currently no specific therapies available. Canes and walkers may become necessary to increase stability and reduce the risk of falling.
Freezing, where patients suddenly become stuck in place for seconds to minutes as if their feet were glued to the ground, is a major cause of falling. Freezing may occur during “on” or “off” periods. Freezing during “off” periods may respond to dopaminergic therapies, but there are no specific treatments for “on” period freezing. Some patients will respond to sensory cues such as marching in place, singing a song, or stepping over an imaginary line.
Exercise, with a full range of active and passive movements, has been shown to maintain and even improve function for PD patients, and active and passive exercises with full range of motion reduce the risk of arthritis and frozen joints. Some laboratory studies suggest the possibility that exercise might also have neuroprotective effects, but this has not been confirmed in PD. Exercise is generally recommended for all PD patients. It is less clear that physical therapy or specific exercises such as tai chi are required. It is important for patients to maintain social and intellectual activities to the extent possible. Education, assistance with financial planning, social services, and attention to home safety are important elements of the overall care plan. Information is available through numerous PD foundations and on the web, but should be reviewed with physicians to ensure accuracy. The needs of the caregiver should not be neglected. Caring for a person with PD involves a substantial work effort and there is an increased incidence of depression among caregivers. Support groups for patients and caregivers may be useful.
CURRENT MANAGEMENT OF PD
The management of PD should be tailored to the needs of the individual patient, and there is no single treatment approach that is universally accepted and applicable to all individuals. Clearly, if an agent could be demonstrated to have disease-modifying effects, it should be initiated at the time of diagnosis. Indeed, constipation, RBD, and anosmia may represent premotor features of PD and could permit the initiation of a disease-modifying therapy prior to the onset of the classical motor features of the disease. However, no therapy has yet been proved to be disease modifying. For now, physicians must use their judgment in deciding whether or not to introduce rasagiline (see above) or other drugs for their possible disease-modifying effects.
The next important issue to address is when to initiate symptomatic therapy. Several studies now suggest that it may be best to start therapy at the time of diagnosis (or soon after) in order to preserve beneficial compensatory mechanisms and possibly provide functional benefits even in the early stage of the disease. Levodopa remains the most effective symptomatic therapy for PD, and some recommend starting it immediately using low doses (≤400 mg/d), but others prefer to delay levodopa treatment, particularly in younger patients, in order to reduce the risk of inducing motor complications. An alternate approach is to begin with an MAO-B inhibitor and/or a dopamine agonist, and reserve levodopa for later stages when these drugs can no longer provide satisfactory control. In making this decision, the age, degree of disability, and side effect profile of the drug must all be considered. In patients with more severe disability, the elderly, those with cognitive impairment, or those in whom the diagnosis is uncertain, most physicians would initiate therapy with levodopa. Regardless of initial choice, it is important not to deny patients levodopa when they cannot be adequately controlled with alternative medications.
If motor complications develop, patients can initially be treated by manipulating the frequency and dose of levodopa or by combining lower doses of levodopa with a dopamine agonist, a COMT inhibitor, or an MAO-B inhibitor. Amantadine is the only drug that has been demonstrated to treat dyskinesia without worsening parkinsonism, but benefits may be short-lasting, and there are important side effects related to cognitive function. In advanced cases, it may be necessary to consider a surgical therapy such as DBS if the patient is a suitable candidate, but as described above, these procedures have their own set of complications. Continuous intraintestinal infusion of levodopa/carbidopa intestinal gel (Duodopa) appears to offer similar benefits to DBS, but also requires a surgical intervention with potentially serious complications. Continuous infusion of apomorphine is another treatment option and does not require surgery but is associated with potentially troublesome skin nodules. Comparative studies of these approaches in more advanced patients are awaited. There are ongoing efforts aimed at developing a long-acting oral or transdermal formulation of levodopa that mirrors the pharmacokinetic properties of a levodopa infusion. Such a formulation might provide all of the benefits of levodopa without motor complications and avoid the need for polypharmacy and surgical intervention.
A decision tree that considers the various treatment options and decision points for the management of PD is provided in Fig. 449-7.
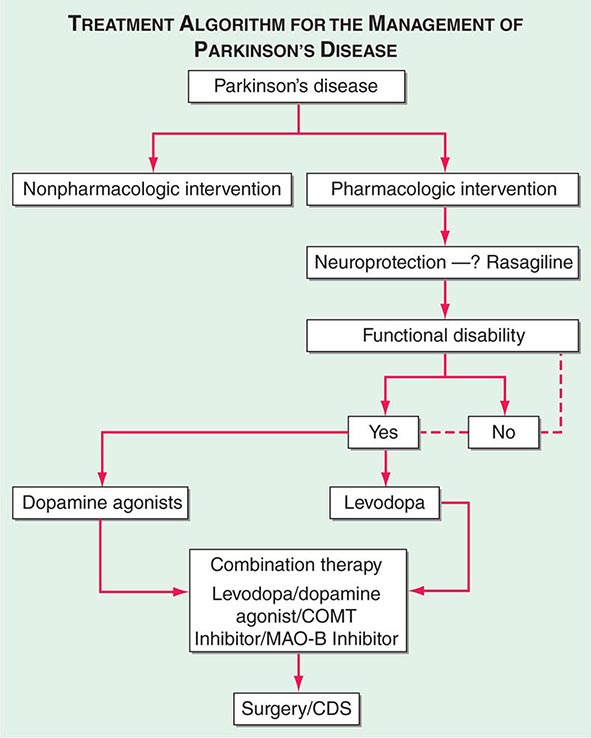
FIGURE 449-7 Treatment options for the management of Parkinson’s disease (PD). Decision points include: (1) Introduction of a neuroprotective therapy: No drug has been established to have or is currently approved for neuroprotection or disease modification, but there are several agents that have this potential based on laboratory and preliminary clinical studies (e.g., rasagiline 1 mg/d, coenzyme Q10 1200 mg/d, the dopamine agonists ropinirole, and pramipexole). (2) When to initiate symptomatic therapy: There is a trend toward initiating therapy at the time of diagnosis or early in the course of the disease because patients may have some disability even at an early stage, and there is the possibility that early treatment may preserve beneficial compensatory mechanisms; however, some experts recommend waiting until there is functional disability before initiating therapy. (3) What therapy to initiate: Many experts favor starting with a monoamine oxidase type B (MAO-B) inhibitor in mildly affected patients because of the good safety profile of the drug and the potential for a disease-modifying effect; dopamine agonists for younger patients with functionally significant disability to reduce the risk of motor complications; and levodopa for patients with more advanced disease, the elderly, or those with cognitive impairment. Recent studies suggest the early employment of polypharmacy using low doses of multiple drugs to avoid side effects associated with high doses of any one agent. (4) Management of motor complications: Motor complications are typically approached with combination therapy to try and reduce dyskinesia and enhance the “on” time. When medical therapies cannot provide satisfactory control, surgical therapies such as DBS or continuous infusion of levodopa/carbidopa intestinal gel can be considered. (5) Nonpharmacologic approaches: Interventions such as exercise, education, and support should be considered throughout the course of the disease. CDS, continuous dopaminergic stimulation; COMT, catechol-O-methyltransferase. (Adapted from CW Olanow et al: Neurology 72:S1, 2009.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


