Grade I
Grade II
Grade III
Grade IV
Angiocentric glioma
Diffuse astrocytoma
Anaplastic astrocytoma
Glioblastoma
Pilocytic astrocytoma
Oligodendroglioma
Anaplastic oligodendroglioma
Giant cell glioblastoma
Subependymal giant cell astrocytoma (SEGA)
Oligoastrocytoma
Anaplastic oligoastrocytoma
Gliosarcoma
Pilomyxoid astrocytoma
Small cell glioblastoma
Pleomorphic xanthoastrocytoma
Glioblastoma with oligodendroglioma features
Specifically, ODG tend to be less diffuse than the other gliomas. Although there are certainly widely infiltrative ODG, these tumors are often confined to the superficial rather than deep portions of the cerebral hemispheres and in some cases, are relatively well demarcated, allowing for more radical surgical debulking compared to other infiltrative gliomas [4, 5] (Fig. 8.1). Another highly favorable property is the chemosensitivity of ODG, particularly those with 1p19q co-deletion. This was first noted more than 20 years ago when AO were shown to be responsive to cytotoxic chemotherapy [6]. These initial observations have matured into multiple randomized controlled clinical trials for AO that have had encouraging results relative to other diffuse or high-grade glioma subtypes [7, 8]. Unfortunately, despite these favorable properties, both LGO and AO remain almost always incurable and inevitably progress to an increasingly aggressive and treatment-resistant disease. This sobering reality is the major driver of the ongoing research into the unique features of ODG that is hoped to result in improved therapies and outcomes for patients with ODG.
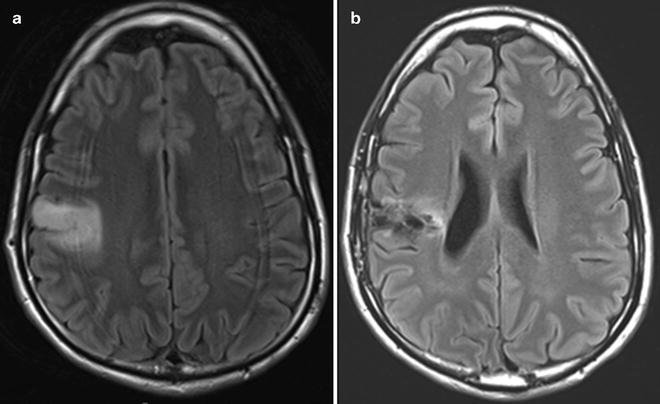
Fig. 8.1
Low-grade oligodendroglioma of the right frontal lobe as depicted in FLAIR brain MRI sequences before (a) and after (b) surgical resection. This is a classic presentation with the predominant involvement being in the cortical rather than deep regions.
Histopathology
ODG is characterized by tumor cells that morphologically and phenotypically resemble oligodendrocytes, which produce myelin that serves to optimize nerve transmission via the insulation of axons. This helps to facilitate saltatory transmission of action potential that greatly expedites nerve impulse speed. They are located within the white matter of the brain, an area that is rich in myelin due to the concentration of axons in this region. ODG display classical histological appearance marked by “back-to-back” tumor cells with regular, round hyperchromatic nuclei in association with clearing of the cytoplasm and close proximity to fine branching vasculature [1]. The former is whimsically referred to as “fried-egg appearance” and the latter as “chicken-wire vasculature.” Nonetheless, these terms accurately describe the classical histological appearance of ODG (Fig. 8.2a). Note that perinuclear clearing is secondary to the “washing out” of cytoplasmic content by organic solvent during the process of formalin-fixed paraffin-embedded (FFPE) specimen preparation and is absent in frozen sections. ODG can also present with subpopulations of gliofibrillary oligodendrocytes and minigemistocytes. Other associated histological features include the frequent presence of microscopic calcospherites which often coalesce into grossly observable calcific deposits that lend themselves to stereotypical radio-dense appearance in computer tomography imaging (Fig. 8.2b). Other histological features include foci of microcystic change and nuclear palisading, which historically was referred to as spongioblastic pattern usually associated with anaplastic ODG (WHO III) [9] (Fig. 8.2c). A feature shared by most ODG is the rather benign nuclear morphology which is frequently lost during malignant transformation to higher grade glioma. However, a focus with low-grade histological feature is sometimes present within glioblastoma suggestive of transformation. ODG tumor cells exhibit a strong physical affinity for non-neoplastic elements of the adjacent neuropil, a common characteristic shared with other infiltrating gliomas. These histological entities, including perineuronal and perivascular satellitoses, as well as subpial and intrafascicular spread, are collectively known as secondary structures of Scherer [10] (Fig. 8.2d).
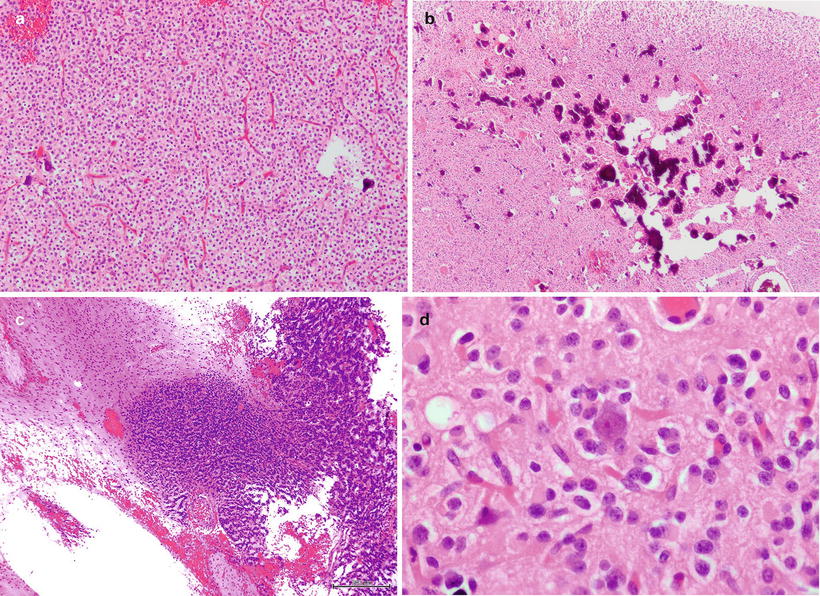
Fig. 8.2
(a) ODG displays classical appearance of perinuclear clearing and fine branching vasculatures, (b) frequent deposits of microcalcifications are common in ODG, (c) nuclear palisading is sometimes seen in ODG and especially associated with WHO III anaplastic ODG, (d) perineuronal satellitoses by ODG tumor cells is a feature shared with other infiltrating gliomas
Immunohistochemically, ODG tumor cells, especially those exhibiting classical morphology, are negative for glial fibrillary acidic protein (GFAP) expression. Morphological variants such as gliofibrillary oligodendrocytes and minigemistocytes display GFAP immunopositive cytoplasm and processes. Nonetheless, there is frequent background of GFAP reactivity due to the infiltrative nature of the tumor. Also, one must be cognizant of the reactive astrocytes admixed with tumor cells (Fig. 8.3a). Ki67 specific antibody highlights the proliferative fraction of the tumor. Immunoexpression of P53 is not very informative given TP53 mutation is a rare event in ODG (Fig. 8.3b) [11]. The clinical introduction of IDH1 R132H mutant specific antibody (clone H-09) has dramatically altered the daily practice of neuropathology (Fig. 8.3c) [12]. Since a majority of WHO II/III infiltrating gliomas exhibit IDH1 mutations, and R132H variant is the dominant form, the antibody is very useful in pinpointing tumor cells, even in small and challenging specimens.
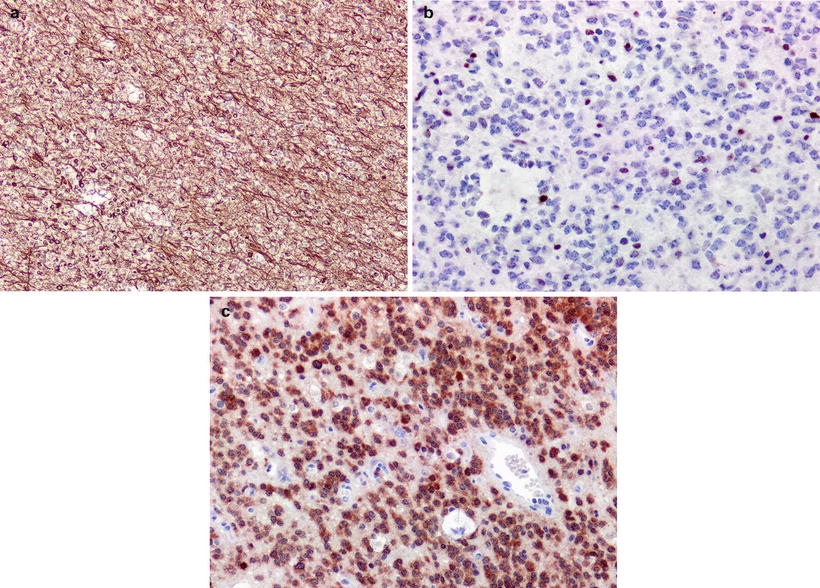
Fig. 8.3
(a) ODG is generally immunonegative for GFAP except for tumor cells with gliofibrillary oligodendrocytic and minigemistocytic morphology. There is strong background immunopositivity consistent with infiltrative nature of tumor cells, (b) ODG is typically negative for mutations in TP53 and is reflected in “non-neoplastic” pattern of TP53 reactivity, (c) ODG tumor cells display strong cytoplasmic reactivity for IDH1 R132H mutant protein
As mentioned previously, the WHO grading scheme divides ODG with predominantly malignant oligodendroglial morphology into grades II and III, with the latter known as anaplastic oligodendroglioma (AO). The principal features of WHO III AO include predominance of hypercellular foci, worsening cytological pleomorphism, tumor necrosis, conspicuous microvascular proliferation, and significantly elevated mitotic activity. The latter two are particularly associated with aggressive behavior [13]. Presence of necrosis, even of the pseudopalisading variant, might not portend a worse prognosis as long as there is a predominance of tumor cells with classic ODG morphology [14]. Conversely, necrosis in tumors with mixed oligoastrocytic populations is associated with poor outcome.
Given the considerable subjectivity in the histological differentiation of WHO grade II and III oligodendroglial neoplasms, there is an even greater challenge in differentiating between pure ODG and mixed oligoastrocytomas (WHO grade II and III). Recently, this conundrum has extended further to clinical diagnosis of glioblastoma with oligodendroglioma component, or GBMO, vs. glioblastoma, or GBM—both of which are WHO grade IV malignant gliomas with poor prognosis. GBMO, when using strict diagnostic criteria, certainly presents distinctly from GBM under the microscope. Recent studies have also highlighted the uniqueness of the molecular underpinnings in GBMO [15]. It remains contentious whether GBMO is associated with a more favorable outcome compared to GBM, and this is exacerbated by the relatively loose definition of this entity along with the absence of agreed upon molecular biomarkers [16].
Inclusion of subjective histomorphological criteria presents significant problems in ensuring diagnostic uniformity among neuropathologists. This is especially problematic in accounting for the malignant oligodendroglial component in an infiltrating glioma and whether to diagnose a tumor as pure ODG or a mixed oligoastrocytoma. However, the most important issue arises from the vagaries of glioma cell morphology, which can lead to over-diagnosis of oligodendroglial neoplasms, pure or mixed. Strong correlation between 1p19q co-deletions and the classical ODG morphology described above highlights association between underlying glioma genetics and histopathology [17]. This is an especially powerful finding given the survival benefits observed in 1p19q co-deleted WHO grade II and III oligodendroglial neoplasms [18, 19].
Clinical Behavior of Oligodendroglial Tumors
The clinical behavior of ODG is characterized by slow progression. LGO have a peak incidence commiserate with all low-grade gliomas, in the third decade of life. There is no clear gender or race distribution. The most common clinical presentation is seizures. Seizures, rather than focal neurologic deficits such as hemiplegia, are thought to be the presenting symptom due to the slow growth rate and infiltrative pattern of LGO, leading to brain irritation rather than to mass effect (source?). However, in some cases of AO where the tumor may grow more rapidly, patients may present with focal neurologic symptoms such as weakness, sensory alteration, vision change or personality change depending on the specific location of the tumor. Alternatively, patients with rapidly growing AO may present with more general symptoms of elevated intracranial pressure such as new and persistent headache or confusion. However, as above, new onset seizure is the most common presentation for ODG (50–90 % of patients) [2, 20].
The event of a first time seizure in an adult often leads to urgent medical evaluation including a computed tomography (CT) of the head or a magnetic resonance imaging (MRI) of the brain. Head CT will commonly show an area of hypodensity and in the case of more chronic tumors, may show punctate hyperdensities consistent with calcification or microhemorrhages (Fig. 8.4a). The typical MRI features for a LGO include diffuse hyperintensity on T2-weighted or fluid attenuated inverse recovery (FLAIR) imaging sequences, a lack of gadolinium contrast enhancement, and normal diffusion weighted imaging (Fig. 8.4b). In comparison, the characteristic MRI features for AO include a mixed-intensity central core with diffuse surrounding signal hyperintensity on T2-weighted or FLAIR sequences and hypointensity on T1-weighted sequences, with some scattered hyperintensity due to calcification or microhemorrhage, as well as heterogeneous enhancement after administration of gadolinium [21–23] (Fig. 8.4c). Importantly, the presence of contrast enhancement does not uniformly predict a higher histological grade. In fact, in a recent study of MRI features of ODG, 28 % of AO did not have gadolinium (Gd) enhancement whereas 20 % of LGO did demonstrate Gd enhancement [24].
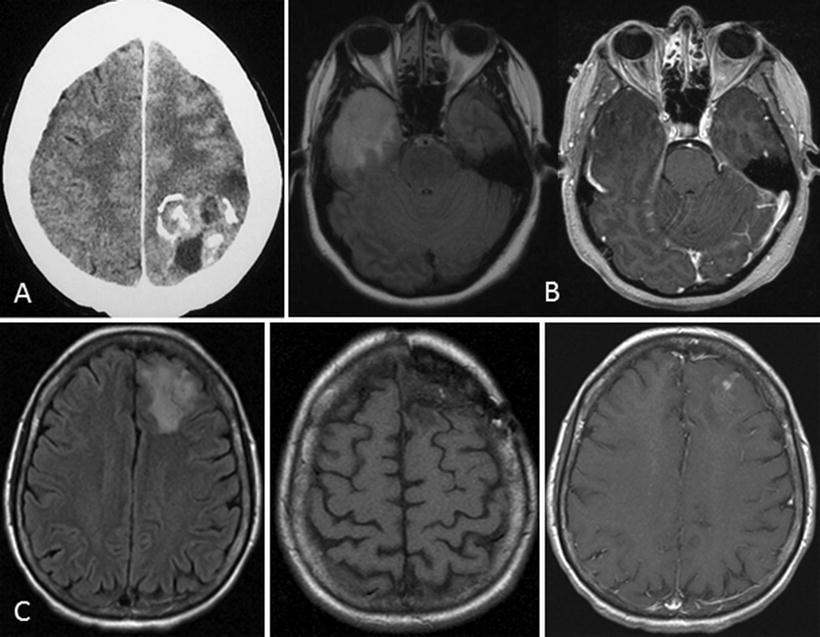
Fig. 8.4
Classic imaging features of ODG. (a) computed tomography scan showing hyperdensities consistent with calcification and possibly, microhemorrhages; magnetic resonance imaging (MRI) of AO with (b) FLAIR, (c) T1w, and (d) T1wGd sequences
Functional MRI has recently been applied to ODG for the purpose of predicting grade as well as allele status (1p19q co-deletion vs. intact), since 1p19q status has been confirmed as both a prognostic and predictive marker [25, 26]. For example, perfusion-weighted imaging and proton MR spectroscopy added to standard anatomical imaging result in 72 % accuracy (83 % sensitivity and 65 % specificity) for distinguishing tumors with intact vs. co-deleted 1p/19q [25]. Although applying functional MRI and metabolic imaging remains experimental, interesting observations have been made that may offer insights into the molecular behavior of ODG subtypes in vivo. Specifically, when the metabolic radiolabelled tracers 201Thallium (201Tl) and 18Ffluorodeoxyglucose (18FF-FDG) were assessed in patients with LGO, AO, and mixed oligoastrocytoma (with and without 1p19q co-deletion), 80 % of high-grade tumor had markers of elevated metabolism as might be expected [27]. Interestingly, LGO with 1p19q co-deletion also had an elevated metabolism profile compared to tumors without this molecular marker. In summary, the unique features of ODG tumors are increasingly being explored with specialized imaging techniques in the context of molecular discoveries, with the goal to better understand disease biology of these tumors and aid in the clinical management.
The time of disease progression for both LGO and AO is highly variable. Traditionally, factors such as age at diagnosis, degree of surgical resection, and tumor grade were considered the major predictors. Recently, based on data from the European Organization for Research and Treatment of Cancer (EORTC) study 26951 for AO [28], nine variables including adjuvant treatment received, age, tumor location (frontal or other), extent of resection, WHO performance status, the presence of endothelial abnormalities or necrosis, and finally, 1p/19q co-deletion and IDH1 mutation status, were shown to be important prognostic factors. Based on these factors, they divided patients into low, intermediate, and high risk groups, with the best prognosis being associated with patients with age of diagnosis <40 years, a good performance status, tumor location in the frontal lobe, confirmed extensive resection without residual tumor on imaging, absence of endothelial abnormalities or necrosis, and 1p/19q co-deletion with IDH1 mutation. For patients with AO who met all of these criteria, the median overall survival (OS) was 127 months (95 % CI: 95 months—not reached).
Treatment of Oligodendroglial Tumors
The first step in establishing a treatment plan for any patient with a suspected diagnosis of glioma is surgery. Surgery serves the role of both obtaining tissue to confirm the histology and molecular subtype and offering an opportunity for cytoreduction. Although the advanced imaging techniques discussed above may help in distinguishing tumor subtypes with increasing confidence, tissue is required for diagnostic confirmation. As discussed below, it is now widely accepted that initial molecular studies in any tumor suspected of being ODG should include analysis of 1p19q status. Studies such as IDH1/2 status and MGMT promoter methylation are also commonly obtained at the time of initial diagnosis, as these allow for molecular subclassification and have impact on both overall prognosis and treatment decisions.
The first therapeutic intervention for all ODG is maximum safe surgical resection. There are a series of single center studies that all support the conclusion that gross total resection of the entire area of tumor involvement based on abnormal MRI signal is optimal as long as this can be accomplished safely without causing significant postoperative neurologic deficits [5, 29–31]. If significant tumor debulking is not feasible due to tumor location and risk for neurologic injury, the options are biopsy to confirm diagnosis, or if LGO is suspected, it may be reasonable to proceed with cautious surveillance in select patients until clinical or radiologic evidence of tumor progression. In widely infiltrative tumors where there is no clearly optimal location for diagnostic sampling, recent studies suggest that functional imaging techniques (diffusion tensor imaging, MR spectroscopy, perfusion imaging) may help determine the optimal location of tissue sampling [32, 33].
When the diagnosis of an AO is confirmed histologically, the next question to address is 1p19q status. For patients with 1p19q co-deleted AO, there is level one evidence in support of the use of procarbazine, carmustine (CCNU), vincristine (PCV) either before or after radiation therapy [7, 8]. This is based on two independent studies, EORTC 26951 and RTOG 9402, that had very similar conclusions despite slightly different designs. In EORTC 26951, patients with newly diagnosed AO were treated with RT alone vs. RT followed by up to six cycles of PCV [8]. At median 5-year follow-up, there was a significant difference in progression free survival (PFS), but not OS between the RT + PVC group and the RT alone group regardless of 1p19q status. However, at median 140 months follow-up, in patients with 1p19q co-deleted AO treated with RT + PCV median OS had not been reached vs. median OS of 9 years in patients treated with RT alone. In the RTOG 9402 trial, patients with AO were randomized to either four cycles of PCV followed by RT or RT alone. Similar to the EORTC study, there was no statistically significant difference in median OS at 5 years or at 12 years when all AO patients are analyzed in aggregate. However, at 12 year follow-up, the subpopulation of patients with 1p19q co-deleted AO treated with PCV + RT had a median OS of 14.7 vs. 7.3 months for those treated with RT alone [7] In contrast, there was no statistically significant difference in median OS for patients without 1p19q co-deletion treated with RT alone vs. RT + PCV. In summary, for patients with 1p19q co-deleted AO, there is strong evidence in support of a treatment paradigm that incorporates PCV and RT, however, there is no clear data to support a specific sequence (Table 8.2).
Table 8.2
Evidence for PCV chemotherapy for 1p19q co-deleted anaplastic oligodendrogliomas.
Study | Tumor grade | Treatment | Molecular subtype | mPFS (years) | mOS (years) |
|---|---|---|---|---|---|
RTOG 9402 [7] | AO | PCV + RT | 1p/19q co-deleted (n = 59) | 8.4 | 14.7 |
All others (n = 89) | 1.2 | 2.6 | |||
RT | 1p/19q co-deleted (n = 67) | 2.9 | 7.3 | ||
All others (n = 76) | 1.0 | 2.7 | |||
EORTC 26951 [8] | AO | PCV + RT | 1p/19q co-deleted (n = 43) | 13.1 | Not reached |
All others (n = 114) | 1.2 | 2.1 | |||
RT | 1p/19q co-deleted (n = 37) | 4.1 | 9.3 | ||
All others (n = 122) | 0.73 | 1.8 |
Despite the compelling data using PCV, many neuro-oncologists advocate for consideration of RT and temozolomide (TMZ) as first line therapy for patients with newly diagnosed AO. This practice is based on the fact that as alkylator-based chemotherapies, PCV and TMZ have similar mechanisms of action and that PCV is much more toxic, resulting in only 42 % of the patients in RTOG 9402 and 30 % of the patients in EORTC 26951 receiving all prescribed chemotherapy [7, 8]. When the initial reports of these studies were published in 2006, the high rates of toxicity and lack of OS benefit led many neuro-oncologists to apply the regimen of RT + TMZ followed by six cycles of TMZ to AO based on the proven benefit of this regimen in glioblastoma [34–36]. However, despite evidence that TMZ does have activity in AO (newly diagnosed and recurrent), it remains to be proven that TMZ plus RT has similar efficacy to PCV plus RT in patients with newly diagnosed AO [37–41]. Ongoing studies that assess the benefit of RT + TMZ in patients with anaplastic gliomas segregated based on 1p19q status will help further clarify this issue. Specifically, the Chemoradiation and Adjuvant Temozolomide in Non-deleted anaplastic glial tumors (CATNON, NCT00626990) trial is addressing the question of whether adding TMZ to RT improves OS for non-deleted anaplastic gliomas. A concurrent study, a randomized trial of Chemoradiation vs. Radiation vs. Temozolomide in 1p/19q Co-deleted anaplastic gliomas (CODEL, NCT 00887146) was initially designed to assess the impact of RT alone, vs. RT + TMZ, vs. TMZ alone on OS in the setting of 1p19q co-deleted anaplastic gliomas. However, in the wake of the long-term results of the RTOG 9402 and EORTC 26951 studies, it was amended to include three comparator arms: RT + PCV, RT + TMZ and adjuvant TMZ, and TMZ alone. In summary, to date the only level 1 evidence to guide treatment recommendations is for the use of RT + PCV for patients with newly diagnosed 1p19q co-deleted AO, while there remains no agreed upon optimal therapy for non-deleted AO.
There is also no agreed upon optimal therapy for LGO. Even more so than for high-grade tumors, the extent of resection has been associated with better PFS and OS [42]. After maximal safe surgical resection, the treatment options include surveillance, RT, or for patients with 1p19q co-deletion, possibly chemotherapy. RT has been the traditional first line therapy for all low-grade gliomas, including LGO. However, data shows that there is no difference in OS between patients who received RT at the time of initial diagnosis or at the time of progression [43]. To help clarify which patients may benefit from early RT treatment, the definition of high and low risk groups for low-grade glioma has been proposed [44]. The high risk group is defined as having tumor dimension ≥6 cm and astrocytoma histology. Low risk gliomas were defined as tumor <6 cm with oligodendroglioma histology. Additional prognostic factors include 1p19q status, Mini Mental Status Examination score and extent of resection. Another unanswered question is whether there is benefit to adding chemotherapy to RT for LGO. RTOG 98-02 assessed the effect of RT + PCV vs. RT alone on OS. There was no statistically significant difference between the RT + PCV and RT groups, although there was an improvement for the combined therapy group in PFS (5-year PFS 63 % RT + PCV vs. 46 % RT) alone (HR, 0.6; 95 % CI, 0.41–0.86; p = 0.06; log-rank p = 0.005) [45]. The ongoing Eastern Oncology Group E3F05 study: Radiation Therapy with or without Temozolomide in Treating Patients With Low-Grade Glioma (NCT 00978458) is assessing the benefit of RT alone vs. RT + TMZ as first line therapy in patients with symptomatic or progressive low-grade gliomas. Finally, there remains the question about whether RT should be deferred for LGO with chemotherapy being offered as first line therapy. A series of studies have suggested that TMZ shows significant activity in patients with LGO, with the most robust effects seen in patients with 1p19q co-deletion, IDH1 mutation status, and MGMT methylation [46–49].
Molecular Genetics of ODG: Predictive, Prognostic, and the Impact on Treatment Decisions
The discovery of associations between ODG histomorphology, 1p19q co-deletion, and clinical outcome resulting from slow and predictable natural history and chemosensitivity remains a landmark in modern neuro-oncology and pathology [50]. Initial observations of a favorable response to the PCV regimen have been extended to TMZ [48, 51]. Due to its strong association with chemoresponsiveness, molecular testing for 1p19q co-deletion, either via fluorescence in situ hybridization (FISH) or loss of heterozygosity (LOH) assays, in gliomas with oligodendroglial features on histology has become a standard diagnostic adjunct in modern neuro-oncology (Fig. 8.5) [52, 53]. The unique yet simple histology of ODG also belies other distinctive molecular features that span the genomic, transcriptomic, and epigenomic realms, which will be explored in detail below [54–58]. A recent report highlighted how a genetic variant rs55705857 in 8q24.21 is associated with ODG development in a background of mutant IDH1/2 [59]. This study suggests that germline variants may be important factors in acquiring OGD, and perhaps other gliomas as well.
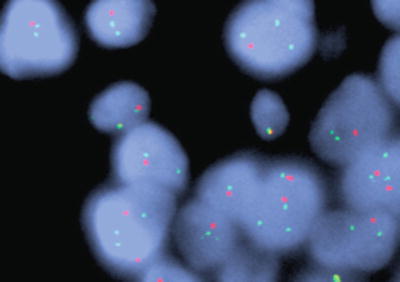
Fig. 8.5
1p FISH shows individual ODG tumor cells contain relatively more 1q chromosomes (green probe) compared to 1p chromosomes (red probe) consistent with 1p deletion.
Fundamentally, ODG is defined by 1p19q co-deletion, or more specifically, unbalanced translocations of chromosomes 1p and 19q [60, 61]. This essentially leads to loss of one copy each of 1p and 19q, resulting in loss of heterozygosity (LOH) affecting the regions of numerical loss. Moreover, 1p19q co-deleted anaplastic ODG that exhibits polysomy for chromosomes 1p and 19q has intermediate survival between 1p19q retained tumors and 1p19q co-deleted ODGs in a euploidy background [62, 63]. This suggests that accumulation of additional genetic aberrations, such as those regulating cell cycling and homologous recombination that contribute to the polysomy state, might contribute to earlier recurrence. An issue raised by these data is the importance of the specific assay used for determination of 1p19q status, since only FISH (and not LOH PCR) permits the enumeration of absolute numbers of chromosomes and determination of ploidy status.
In addition to 1p19q co-deletion as a unique biomarker of ODG chemoresponsiveness, early studies have identified somatic mutations in TP53 as virtually restricted to astrocytomas and not occurring in ODG [11, 64]. Whole exome sequencing has confirmed this early finding in untreated ODG [65]. Another significant molecular feature of OGD is the strong association with mutations of IDH1/2 [66]. In the same study, 99 of 107 WHO II/III pure ODG tumors showed 1p19q co-deletion or loss of heterozygosity of which 90 contain IDH1 and three contain IDH2 mutations [67]. In a review of published studies by Kloosterhof and colleagues, they found 76 % and 67 % IDH1 and 4 % and 5 % IDH2 mutations in LGO (WHO II) and AO (WHO III), respectively [68]. These percentages are similar to WHO II/III infiltrating astrocytomas and mixed oligoastrocytomas. Heterozygous neomorphic mutations in IDH genes result in the generation of the onco-metabolite 2-hydroxyglutarate or 2HG [69, 70]. 2HG has significant pleiotropic effects on the tumor epigenome, leading to aberrant histone regulation and development of the glioma hypermethylator phenotype [71–73]. This important discovery has generated leads for practical applications in improved diagnosis and treatment of IDH mutated gliomas [74–76]. Prior to the discovery of recurrent mutations in the IDH genes, the sole focus was on uncovering genetic candidates within the 1p and 19q LOH regions and particular effort on the former given its stronger association with the ODG phenotype and superior clinical performance [77]. Obvious candidates such as NOTCH2, located at 1p11 have been extensively investigated [78, 79]. Identification of recurrent somatic mutations in CIC, a gene that resides in chromosome 19q, is 69 % of ODG with 1p19q co-deletions and IDH gene mutation using next generation sequencing (NGS) [66, 80]. NGS excels at the unprecedented depth and breadth of profiling of the transcriptome, exome, and genome of cancer. NGS has refined many long-standing molecular findings of ODG at basepair resolution (Fig. 8.6). Data from subsequent studies have added to these initial findings [81, 82]. The CIC gene encodes the mammalian homolog of Capicua, a transcriptional repressor in Drosophila that is essential for inhibition of RAS/MAPK signaling [83–85]. There is a high level of cross-species homology of the CIC gene, especially in the functionally critical DNA-binding and protein-interacting domains. Somatic CIC mutations in oligodendrogliomas most commonly cluster in the HMG-homology DNA-binding domain in exon 5, and recurrent mutations affecting codons 201, 202, and 215 have been identified (Fig. 8.7). The second most common recurrent mutations are clustered within the exon 20 protein-interacting Gro-L homology domain. A slightly lesser number of CIC mutations are scattered throughout the gene. Strikingly, somatic CIC mutations are very strongly associated with 1p19q co-deleted and IDH mutated OGD, and only found in 2 % of astrocytomas not associated with 1p19q co-deletion. Importantly, the majority of the CIC mutations in this cohort are located outside of exons 5 and 20. This highlights potential selective pressure on the development of CIC mutations in ODG and also the functional significance of these domains. At this time, very little is known about the mechanistic role of CIC in brain tumor development, and hence the impetus to pursue ongoing impetus to pursue ongoing functional investigations into its role functional investigations into its role in the development of this tumor [86]. The close association of CIC mutations with IDH mutations in 1p19q co-deleted ODG suggests that both cooperate in tumorigenesis, which may require a pro-oncogenic environment facilitated by the presence of 2HG. However, it remains unclear whether CIC mutations represent loss-of-function or gain-of-function, and how they contribute mechanistically to the development of ODG. Somatic mutations in FUBP1, located in 1p, also appear to cluster in 1p19q co-deleted ODG [80, 81]. Inactivating mutations of ATRX, which code for a chromatin remodeler, are virtually exclusive to astrocytomas and mixed oligoastrocytomas and rarely found in ODG, whereas CIC and FUBP1 mutations are preferentially associated with ODG and only discovered in 10 % of the former [82]. Therefore, ODG biology and clinical behavior are governed by the complex interactions of the presence and absence of unique genetic alterations. These emerging data highlight the need to develop a molecular classification for these tumors that goes beyond histology and accurately reflects entities with distinct biology and clinical behavior.
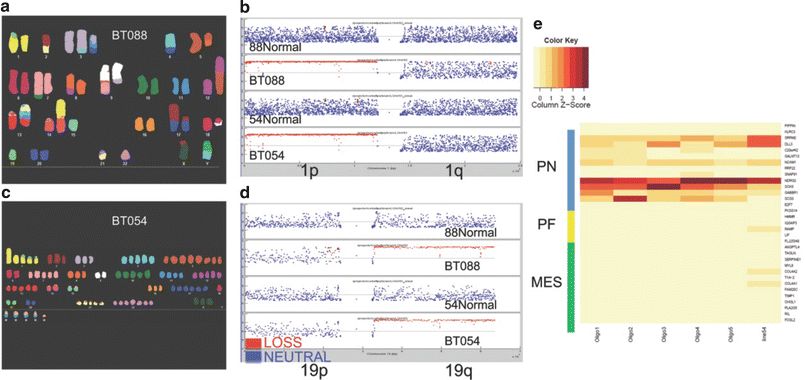
Fig. 8.6




Next generation sequencing of two 1p19q co-deleted brain tumor initiating cell lines derived from ODG patients (a, c) confirmed copy number losses of 1p and 19q (b, d) as well as proneural gene expression signature compatible with ODG (e).
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
