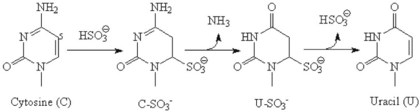
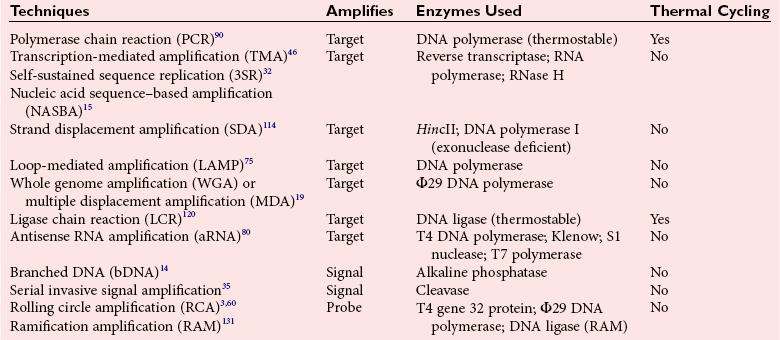
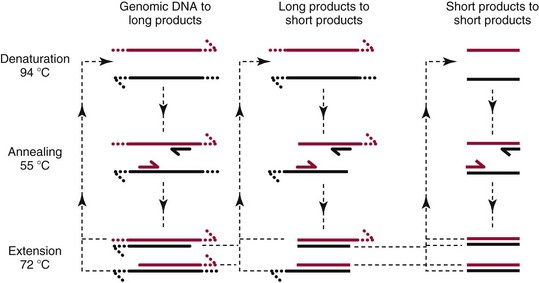
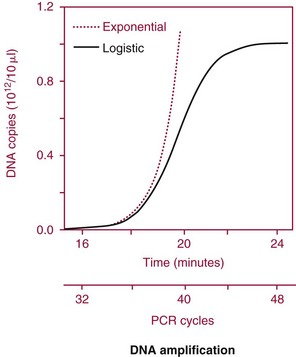
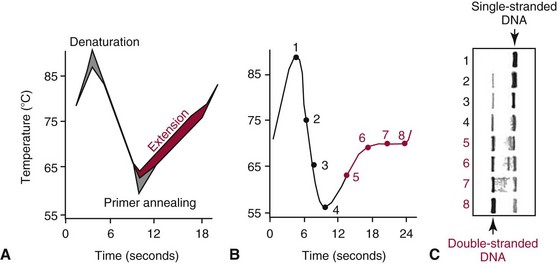
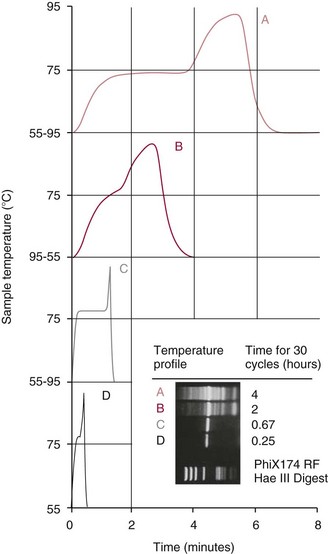
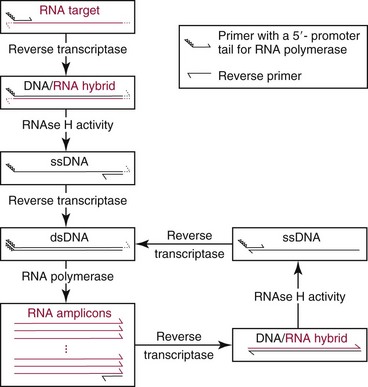
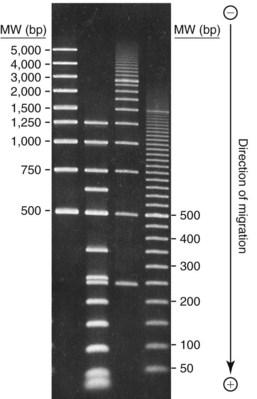
Chapter 17 Molecular diagnostics requires techniques to detect extremely low concentrations of nucleic acids in a background of complex genomic structure. This chapter begins by considering the enzymes that allow us to process nucleic acids into forms suitable for analytical interrogation. We briefly touch on chemical and other processes that aid us in handling nucleic acids (nucleic acid isolation is discussed in detail in Chapter 39). We then discuss amplification techniques that are often necessary to observe or quantify nucleic acid sequences of interest. The tools used to detect or visualize nucleic acids are discussed. Finally, specific methods are described that allow identification, quantification, and/or segregation of individual nucleic acid species. Nucleases are enzymes that hydrolyze one or more phosphodiester bonds in nucleic acid polymers.67 Nucleases may require a free hydroxyl end (exonucleases), with specificity for the 3′ or 5′ end, or may act only on internal bonds (endonucleases). Nucleases can be DNA or RNA specific and may act only on double- or single-stranded polymers. For example, DNAse I digests double-stranded DNA (dsDNA), and S1 nuclease acts only on single-stranded DNA (ssDNA). DNase I can be used to specifically degrade DNA in nucleic acid mixtures when only RNA is of interest. RNAses are very stable enzymes that are common laboratory contaminants. DNA glycosylases are enzymes that remove damaged nitrogenous bases while leaving the sugar-phosphate backbone of DNA intact. Uracil N-glycosylase (UNG) from the bacterium Escherichia coli is commonly used in molecular diagnostics to prevent carryover contamination of DNA from previous amplification reactions (see Contamination Control to Avoid False-Positive Results later in this chapter for details). Restriction endonucleases are found in bacteria; these enzymes prevent replication of foreign DNA.99 Their action is sequence specific, requiring recognition sequences of usually 4 to 10 nucleotides on a dsDNA molecule. Often, these recognition sequences are palindromic (meaning that the same sequence occurs on both of the complementary DNA strands). At each location where these sequences are found, the enzyme cuts both strands in a reproducible manner, resulting in staggered or blunt-end cuts. For example, EcoRI is a restriction enzyme from E. coli that recognizes the six-base sequence GAATTC and cuts between the G and the A on both strands, producing a staggered cut: Ligases catalyze the formation of phosphodiester linkages between two nucleic acid chains.48 DNA ligases are not sequence specific but require the presence of a complementary template. In contrast, RNA ligases used in mRNA processing do not require a template but are sensitive to sequence. Polymerases catalyze the synthesis of complementary nucleic acid polymers using a parent strand as a template.48 In vitro, these enzymes can extend an oligonucleotide primer that is annealed to a template strand. Extension requires that the 3′-OH of the extending end is free, and that nucleotide triphosphates (NTPs) are present in the reaction mixture. Extension stops if you run out of template or NTPs, or if no 3′-OH groups are available at the extending end. DNA polymerases generate DNA strands from a DNA template, whereas RNA polymerases generate RNA strands from a DNA template. Thermostable polymerases, such as Thermus aquaticus (Taq) DNA polymerase, are essential reagents for the automation of many nucleic acid amplification procedures because of their stability at the high temperatures used in these procedures. Some DNA polymerases also have exonuclease activity that can be 3′ to 5′, 5′ to 3′, or both. A polymerase with 3′ to 5′ exonuclease activity can correct mismatched base pairs at the 3′-end of the extending chain. Such proofreading activity increases polymerase fidelity by decreasing the number of errors or misincorporated bases. A polymerase with 5′ to 3′ exonuclease activity can increase polymerase processivity by cleaving any blocking probes or secondary structures, thereby increasing the number of bases incorporated for each extension. Reverse transcriptase is found in retroviruses and catalyzes the synthesis of DNA from an RNA (or DNA) template.2 Retroviruses have RNA genomes, and reverse transcriptase activity is required as part of their replication. Reverse transcriptases do not have proofreading activity and therefore are error prone compared with polymerases. This, however, allows viruses to mutate quickly and develop drug resistance. In vitro, reverse transcriptase is used to make complementary DNA (cDNA) copies of RNA in samples and may be used for cloning, probe preparation, and nucleic acid assays. Sonication, shearing, or chemical cleavage can be used to cut DNA or RNA into smaller fragments so as to make the analytical processes that follow more efficient. Sonication generates nucleic acid fragments of 100 to 1000 bases in length, depending on the conditions used. Shearing can be performed by a compressed air device known as a nebulizer. For chemical cleavage, several metal-ion–catalyzed chemistries can cleave single-stranded and double-stranded nucleic acids. Some alkylating compounds can cleave and label nucleic acids at the same time. One example is 5-(bromomethyl)-fluorescein, which is catalyzed by metal ions to fragment RNA or DNA, and then labels those fragments with fluorescein.10 This can be used in nucleic acid preparation for microarray analysis. Bisulfite treatment of DNA is used to analyze the methylation status of cytosine (C) residues in DNA. Sodium bisulfite (NaHSO3),36 optionally used together with ammonium bisulfite,97 converts C into uracil (U), but does not affect 5-methylcytosine. The chemical process of bisulfite treatment is shown in Figure 17-1. The process works effectively only on single-stranded DNA, and so the sample needs to be denatured by heat, alkali, or chaotropic agents such as urea or formamide. Analysis of the bisulfite-treated DNA is usually performed after nucleic acid amplification. DNA polymerases used for amplification will treat 5-methylcytosine as C (no change in sequence), but they will treat unmethylated C that is converted to U as a T (thus a sequence change of C to T). Many methods can be used to detect and quantify the altered sequences that result from bisulfite treatment of methylated DNA, including preferential amplification, detection with probes, melting techniques, and sequencing.49 One limitation of bisulfite treatment is that it significantly degrades DNA. Depending on the protocol, as much as 90% of the DNA can be lost. Affinity enrichment of methylated DNA is an alternative to bisulfite treatment that may provide higher yields. One example is to enrich methylated DNA by immunoprecipitation with an antibody raised against DNA containing 5-methylcytosine.117 Up to 90-fold enrichment in methylated DNA can be achieved by immunoprecipitation. Another method is to use methylated DNA binding protein to capture double-stranded methylated DNA on an affinity column or other solid support.83 Reversal of formalin (formaldehyde) cross-linking is required before DNA or RNA can be analyzed from tissues prepared in formalin-fixed paraffin-embedded (FFPE) blocks. Formalin modifies nucleic acids with monomethylol groups, which cause nucleic acids to cross-link by forming methylene bridges to amino groups.63 These cross-links interfere with many processes, including reverse transcription and polymerase reactions. Heating the DNA or RNA reverses cross-linking, and most protocols use alkali (pH 9 to 12) conditions to reverse formalin cross-linking in DNA and acid (pH 3 to 6) conditions for RNA. Depending on the age of the FFPE specimen, nucleic acid can be degraded into short fragments. Achieving adequate detection limits is a central concern for clinical applications of nucleic acid analysis. Techniques that increase the amount of the nucleic acid target, the detection signal, or the probe are referred to as amplification methods. Examples of amplification methods are listed in Table 17-1. In target amplification, a well-defined segment of the nucleic acid region of interest (the target sequence) is copied many times by in vitro methods. Areas outside the target are not amplified. In signal amplification, the amount of target stays the same, but the signal is increased by one of several methods, including sequential hybridization of branching nucleic acid structures and continuous enzyme action on substrate that may be recycled. Finally, in probe amplification, the probe (or a product of the probe) is amplified only in the presence of the target. Amplification techniques often can achieve more than a million-fold amplification in less than an hour. When the amount of target nucleic acid is increased by synthetic in vitro methods, target amplification occurs. PCR90 is the best known and most widely applied of the target amplification methods. Because of the commercial availability of thermostable DNA polymerases, kits, and instrumentation, this method has been widely adopted in research and is routinely used in the clinical laboratory. PCR requires a thermostable DNA polymerase, (deoxy)nucleotides of each base (collectively referred to as dNTPs), the target sequence, and a pair of oligonucleotides (referred to as primers) complementary to opposite strands flanking the sequence to be amplified (the target sequence). In the first step, target duplexes are denatured into single strands by heat (Figure 17-2). When the mixture is cooled, primers provided in great excess (usually over a million times the concentration of the initial target) specifically anneal to complementary sequences on the target. Once the primers are annealed, the action of the polymerase synthesizes two new DNA strands by extending each of the two primers at their 3′ end. The primers are designed to recognize sequences of the target that are close enough together that the polymerase extends each strand far enough to include the priming site of the other primer. Usually, the optimal temperature for polymerization (or DNA synthesis) is an intermediate temperature between the annealing temperature (at which primer hybridizes to its target) and the denaturation temperature (at which the newly generated DNA strand dissociates from its template). The second cycle also begins with denaturation, but now twice as many strands (the original genomic DNA and the extension products from the first cycle) are available for primer annealing and subsequent extension. Temperature cycling (typically) employs three temperatures: a high temperature sufficient to denature the target sequence, a low temperature that allows annealing of the primers to the target, and a third temperature optimal for polymerase extension. The instrument that takes samples through the multiple steps of changing temperature is known as a thermocycler. Repetitive thermocycling results in the exponential accumulation of the short product (consisting of primers and all intervening sequences). If the efficiency of each cycle is optimal, the number of target sequences doubles each cycle (the efficiency is 100% or 1.0). PCR efficiency depends on the primers and the temperature-cycling conditions, along with the presence or absence of polymerase inhibitors. Amplified products accumulate exponentially in the beginning cycles of PCR. At some point, however, the efficiency of amplification falls and eventually the amount of product plateaus (Figure 17-3) as the result of exhaustion of components or competition between primer and product annealing (i.e., the single strands of product are at such high concentrations that they anneal to each other rather than to the primers). The S-curve shape is similar to the logistical model for population growth. In a typical PCR reaction using 0.5 µmol/L of each primer, the maximum DNA concentration achievable is about 100 billion copies/µL. The kinetics of PCR suggests that rapid transitions between temperatures with minimal or no pauses (temperature plateaus) provide a better paradigm of PCR amplification (Figure 17-4). Denaturation, annealing, and extension are very rapid reactions as shown by experiments in capillaries.125 The use of temperature “spikes” at denaturation and annealing, instead of extended temperature plateaus, allows for rapid cycling with the appropriate instrumentation.129 The actual time required for PCR depends on the size of the product, but when it is less than 500 base pairs (bp), 30 cycles are easily completed in 15 minutes. Furthermore, rapid amplification improves specificity. Figure 17-5 shows PCR amplification of the same product amplified at different cycling speeds. With conventional slow cycling, many nonspecific products are generated (cycling profile A). These products disappear as the cycling time is decreased (profiles B, C, and D). In fact, amplification yield and product specificity are optimal when denaturation and annealing times are minimal. Requirements for denaturation, annealing, and extension in PCR have been reviewed.128 Initial denaturation of genomic DNA may be required before PCR cycling, depending on how the DNA sample was prepared. Boiling of the sample or an initial denaturation step of 5 to 10 seconds before PCR cycling may be necessary. During PCR, however, denaturation occurs very rapidly. Even for long PCR products, denaturation is complete in less than 1 second after the denaturation temperature is reached. Anything greater than a denaturation time of “0” serves only to degrade the polymerase. If longer denaturation times are required, either the sample is not reaching temperature or heat-activated polymerases are being used. Product specificity is optimal when annealing times are less than 1 second. Longer annealing times may be required if the primer concentrations are low. The required extension time for each cycle depends on the length of the PCR product. Extension is not instantaneous, although it is much faster than common practice would suggest. Extension rates of Taq polymerase under optimal conditions are 50 to 100 bases per second. Despite its common use, a 5 to 10 minute final extension is not necessary to complete extension. Products can be as small as 40 bp to about 40 kilobases (kb). To amplify products longer than 2 kb, mixtures of polymerases that include some 3′-exonuclease activity to edit mismatched nucleotides are usually used. Instead of separate annealing and extension temperatures, both processes can be carried out at the same temperature, resulting in two-temperature, instead of three-temperature, cycling. Although this simplifies the demand on instrumentation and programming, it limits the choice of primers and requires a longer extension time at suboptimal temperatures. 1. Avoid primers that anneal to themselves or to other primers. Particularly avoid complementation at the 3′-end of primers, and especially do not use a primer that has reciprocal 3′-end complementation. 2. Choose primers that are specific to the target. Avoid simple sequence repeats and common repeated sequences, such as Alu repeats. If your target has close relatives, design your primers so that they will anneal only to your intended target. Targets that need to be avoided include pseudogenes (for genomic DNA) and related bacterial or viral strains (for microorganisms). 3. One should avoid primers that have sequence complementary to internal sequences of the intended product, especially at the 3′-ends of the primers. 4. Primers should have between 18 and 25 bases that are matched in melting temperature (Tm) to each other. A primer greater than 17 bases long has a good chance of being unique in the human genome. 5. Unless there is a reason to amplify longer targets, product length should be less than 500 bps. Even shorter products (<200 bps) amplify with the highest efficiency. 6. It is important to search for sequences similar to the candidate primers that are present in the background DNA likely to be present in your assay (http://www.ncbi.nlm.nihgov/BLAST/). Many primer selection programs are available commercially and others can be freely obtained over the Internet. However, very few if any of the selection rules have been empirically tested. Taq DNA polymerase and other polymerases have a terminal transferase activity that may add a single unpaired nucleotide on the 3′-end of PCR product strands. In the presence of all four dNTPs, dATP is preferentially added. This means that some percentage of the double-stranded products generated by PCR will have a protruding A at one or both ends. Although this does not influence most detection protocols, it can complicate some systems with high size resolution. On the other hand, this feature is useful for high-efficiency cloning and ligation of PCR products.134 Because PCR can detect a single molecule of target sequence, a small amount of contamination in a sample can easily produce a false-positive result. The greatest potential for contamination comes from the product of the amplification reaction, referred to as the amplicon (used interchangeably with PCR product). After amplification, each reaction mixture may contain as many as one trillion copies of the amplicon. Thus, minute aerosol droplets contain more than enough target for robust amplification. Amplicon can contaminate reagents, pipettes, and glassware. It is easy to turn a laboratory into a Dr. Seuss fiasco.95 Experience has dictated the use of laboratory procedures that minimize contamination by amplicons.52 These include physically separated areas for preamplification and postamplification steps, positive-displacement or filter barrier pipettes to minimize aerosol contamination, and the use of prealiquoted reagents. The most effective way of all is to not let the product out of the tube. Methods that perform amplification, detection, and characterization in a closed tube virtually eliminate the risk of product contamination. Even with these precautions, a negative control or blank (all reactants minus target DNA) is one of the most important controls for PCR. Another variant method called allele-specific PCR enables preferential amplification of one genetic allele over another. The 3′-end of one primer is placed at the polymorphic site and is extended readily only if it is completely complementary to the target. This strategy is used for distinguishing a gene from its pseudogenes and for genotyping of single-nucleotide polymorphisms (SNPs). Allele-specific PCR can also be used to determine haplotypes.66 Conventional PCR averages the amplification results of the many individual template molecules in a reaction. Because of this averaging, certain questions are difficult to answer. For example, quantification of copy number variants and minor allele fractions in the background of wild-type DNA, although possible, is difficult by bulk PCR. It is also difficult to determine whether multiple variants are on the same or different chromosomes. Single-molecule PCR (sometimes also called digital PCR) is a technique that uses a dilute solution of template that is distributed across many reaction compartments such that each of those compartments will have no template or a single molecule of template. Instead of conventional tubes, these compartments may be minute aqueous droplets in a water-in-oil emulsion (emulsion PCR),102 PCR colonies (polonies) on a thin film of acrylamide gel,68 or clusters on the surface of a planar flowcell generated by bridge amplification,4 or they may be found on beads with clonally amplified template attached to their surface.62,96 When amplification is observed in one of the massively parallel reactions, this means that clonal amplification occurred from a single template molecule. By analyzing each of these clonal reactions, it is possible (1) to determine whether multiple sequence variants are carried on one chromosome or split among two chromosomes, (2) to detect hemizygous gene or exon level deletions, and (3) to count minority events such as a mutation in a gene that is present in a very small fraction of cells in the specimen. Single-molecule PCR is used as the first step for many of the high-throughput sequencing methods reviewed later in this chapter. Several other methods for target amplification have been developed and are described briefly here. Transcription-based amplification methods are modeled after the replication of retroviruses. These methods are known by various names, including transcription-mediated amplification (TMA),30,43 nucleic acid sequence–based amplification (NASBA),15 and self-sustained sequence replication (3SR) assays.32 They amplify their target without temperature cycling (isothermally) and use the collective activities of reverse transcriptase, RNase H, and RNA polymerase. The most widely used is TMA, illustrated in Figure 17-6. Two primers, a reverse transcriptase, and an RNA polymerase are used. The primer complementary to the RNA target has a 5′-tail that includes a promoter sequence for RNA polymerase. This primer anneals to the target RNA and is extended by the reverse transcriptase, creating an RNA/DNA duplex. The RNA strand is degraded by the RNAse H activity of the reverse transcriptase, allowing the second primer to anneal. The reverse transcriptase then extends the second primer to create double-stranded DNA that includes the promoter. RNA polymerase recognizes the promoter and initiates transcription, producing 100 to 1000 copies of RNA. Each strand of RNA then binds and extends the second primer, forming a RNA/DNA hybrid; the RNA in the hybrid is degraded, the promoter primer binds and extends to produce double-stranded DNA that can be transcribed, and the cycle repeats. As in PCR, all reagents are included and amplification is exponential with completion in less than an hour. Unlike PCR, these methods do not require temperature cycling (except for an initial heat denaturation if a DNA template is used). They are particularly advantageous when the target is RNA (e.g., human immunodeficiency virus [HIV] and hepatitis C virus [HCV] in blood bank nucleic acid testing). Another isothermal amplification technique is strand displacement amplification (SDA).114 After heat denaturation of DNA in the presence of four primers—dCTP, dGTP, dUTP, and a modified deoxynucleotide (dATPαS)—two enzymes are added: an exonuclease-deficient polymerase and a restriction enzyme. The two flanking primers that enter into exponential amplification have a restriction site added to their 5′-end and get nicked by the restriction enzyme, allowing displacement of strands that in turn can be primed, extended, and nicked. Deoxy-ATPαS is used so that restriction sites include a hemiphosphorothioate linkage to allow single-strand nicking, instead of cutting through the double strands. The target amplification methods presented so far produce a defined length of RNA or DNA according to primer placement. In contrast, loop-mediated methods produce a wide range of different length concatemers of the target sequence. Primer design includes tails at the 5′-end of some of the primers that are complementary to the target sequence. After replication of complementary strands, these ends become free 3′-ends capable of extension. By forming a loop, these 3′-ends prime and extend the hairpin (self-priming) initiating concatemer formation. After initial DNA denaturation, the process can be isothermal if strand displacement primers are included. In the basic version, two strand displacement primers and two loop-forming primers recognize six segments in the target.75 In another version, allele-specific amplification with five primers and one competitive probe recognizes seven segments in the target.1 In both versions, a variety of product concatemers are formed, and the reactions can be completed in less than 1 hour. Instead of specific amplification of one target to improve sensitivity, methods that amplify all genomic DNA or mRNAs are useful when the target is in short supply. For example, multiple-displacement amplification uses exonuclease-resistant random hexamers and a highly processive polymerase to amplify DNA nonspecifically.19 Initial DNA denaturation is not necessary, and the reaction proceeds isothermally. Similarly, messenger RNA can be generically amplified with a poly(T) primer modified with an RNA polymerase promoter.80 After reverse transcription, second-strand DNA synthesis, and transcription, antisense RNA is produced. Both whole genome and antisense RNA amplification are also useful as nucleic acid purification methods before amplification or detection. Instead of increasing the concentration of target, signal amplification techniques use nucleic acids to magnify the detection signal. The branched-chain DNA (bDNA) method is one of these techniques in common use. The bDNA approach hybridizes the target nucleic acid to multiple capture probes affixed to a microtiter well.108 This is followed by hybridization to a series of “extender,” “preamplifier,” and amplifier probes. The final, highly branched amplifier probe includes multiple copies of signal-generating enzymes that act on a chemiluminescent substrate to produce light. Nucleotide analogs isoC and isoG (isomers of C and G that are complementary to each other but not to other nucleotides) are often used to increase the specificity of the signaling cascade. When two probes overlap on one target, an “invasive” cleavage reaction can be catalyzed by certain structure-specific nucleases. The cleaved fragment, in turn, can cause invasive cleavage of a secondary probe in the shape of a hairpin. The hairpin probe can be designed as a fluorogenic indicator by using a reporter/quencher pair of dyes that are separated by cleavage. This serial sequence of events (primary invasion and cleavage, followed by secondary invasion and cleavage of an indicator probe) is known as the serial invasive signal amplification reaction.35 After DNA denaturation, cooling, and the addition of enzymes, the reaction is run at a temperature at which both the primary and secondary reactions recycle. If a primer is annealed to a closed circle of DNA in the presence of a processive, displacing polymerase, the complement of the circle will be synthesized over and over again with displacement of the tandem repeats.60 If two primers are used in opposite orientation, progressively more complex branches will be formed in an exponential reaction. The rolling circle can be formed by ligation of the two ends of a linear probe on template DNA. Ligation may happen directly, after polymerization through a gap, or after annealing of an additional, allele-specific oligonucleotide. Quantitative analysis at the end point of amplification is usually carried out with the use of calibrators with known amounts of target or a target mimic. Sample nucleic acid may be quantified by comparison with an internal standard of known amount that is added at the time of sample processing to control for efficiency of nucleic acid purification. These internal standards can be DNA fragments, plasmids, or RNA packaged into synthetic phage or virus particles to mimic real viruses (so-called armored RNA115). One such strategy uses a competitor template, which is amplified by the target primers but generates an amplicon with sequence or size different from that of the expected amplicon. This competitor template is added in varying amounts to replicates of the sample before amplification. When the target and the competitor are present in equal concentrations, the amounts of the two different products will also be the same. The competitor is present in the same tube as the sample, so any variation in enzyme activity affects both products identically. This method is not used frequently in clinical applications because of the requirement for multiple assays on each patient sample. Nucleic acid molecules absorb ultraviolet light maximally at 260 nm owing almost entirely to the constituent bases. This property can be used to measure the nucleic acid content of a solution. DNA double helices have lower UV absorbance than an equivalent number of nucleotide monomers, and when DNA is denatured into single strands (ssDNA) (e.g., by heat or pH), UV absorbance increases.6 If a double-stranded DNA preparation is pure, a 50 mg/L solution has an absorbance of 1.0 at 260 nm. RNA has a greater absorbance, so only about 30 mg/L gives an absorbance of 1.0. More precise estimates for oligonucleotides are based on dinucleotide contributions.8 It is common to assess the purity of a nucleic acid preparation by its ratio of absorbances at 260 nm and 280 nm (260 : 280 ratio). In contrast to nucleic acids, proteins absorb maximally at 280 nm. A pure preparation of nucleic acid should have a 260 : 280 ratio between 1.7 and 2.0 that depends on base content. Lower values suggest significant protein contamination. Although absorbance measurements are simple and precise, they are not sensitive. Fluorescent stains that bind to nucleic acid are 1000 to 10,000 times more sensitive than absorbance measurements. The best known example of a nucleic acid dye is ethidium bromide, a positively charged, intercalating dye for double-stranded DNA and to a lesser extent, single-stranded DNA and RNA. Cyanine dyes are also popular stains for nucleic acids; they do not fluoresce unless they are bound to nucleic acids, thus providing very low background. With the appropriate optics, single molecules of DNA can be visualized with cyanine-based nucleic acid stains.79 Nucleic acid dyes can detect DNA and RNA in gels or in solution (such as in real-time PCR). The first practical example of nonradioactive probes used a biotin-labeled analog of dUTP.53 Despite the altered steric configuration, this nucleotide is incorporated by both DNA polymerase and terminal transferase. Other functional groups, such as digoxigenin, may also be used as affinity labels through chemical linkage to a dUTP and incorporation into polynucleotides. Alternatively, oligonucleotide probes can be labeled during synthesis with biotin or amino linkers for subsequent attachment to indicator molecules. Labels at the 5′- or 3′-end of the molecule are usually preferred because central modifications may interfere with hybridization. Electronic detection of nucleic acids is attractive for its simplicity. Hybridization events can be detected by redox indicators that recognize the DNA duplex, or by other hybridization-induced changes in electrochemical parameters, such as conductivity or capacitance.78,107 However, it is difficult to make electronic methods sensitive and specific enough that nucleic acid amplification is not required. Advances in oligonucleotide synthesis and fluorescence detection have made fluorescently labeled probes the preferred reporter for nucleic acid analysis. Many fluorescent labels are now available, allowing color multiplexing for applications such as DNA sequencing, fragment length analysis, DNA arrays, and real-time PCR as reviewed later in this chapter. Techniques such as fluorescence polarization, fluorescence resonance energy transfer (FRET), and fluorescence quenching can provide additional detection specificity. Fluorescence polarization can be used to distinguish free from bound label, if the molecular rotation of the probe changes upon binding.51 Molecular rotation primarily depends on the size of the molecule, so binding of a small probe onto a large target results in a polarization increase that can be measured. FRET techniques depend on the distance between two spectrally distinct fluorescent labels. Two labeled probes may be brought closer together by adjacent hybridization. Alternatively, two labels on the same probe may end up farther apart by hydrolysis or hybridization. Fluorescence quenching or augmentation does not always require FRET. For example, fluorescence may change merely by hybridization of a fluorescent oligonucleotide to its target. The effect depends on the dye and on inherent quenching from G residues in the target and/or probe.16,72 Alternatively, quenching moieties can be purposely incorporated into probes.55,106 Nucleic acid discrimination techniques are divided into three categories: 1. Electrophoretic methods that physically separate nucleic acids based on molecular size and/or shape. 2. Alternatives to electrophoresis that determine the size, base content, and/or sequence of nucleic acids without electrophoresis. Examples include high-performance liquid chromatography (HPLC), mass spectrometry, and high-throughput sequencing. 3. Hybridization assays that identify specific nucleic acids by annealing or melting of complementary nucleic acids. Some techniques use both electrophoresis and hybridization. Electrophoresis is the most commonly used method for DNA and RNA analysis. Both DNA and RNA are negatively charged and will migrate toward the positively charged electrode when an electrical field is present within an appropriately buffered solution. Separation of different nucleic acids occurs when mixtures are allowed to travel through a neutral sieving polymer under the electrical field. Separation is based primarily on molecular size, with smaller molecules traveling faster through the polymer than larger ones (Figure 17-7). When very large molecules (≥50 kb) have to be separated, pulsed electrical fields are used to help move these molecules through the polymer matrix.64 Separation also occurs based on the physical conformation, or shape, of the molecule. For instance, (1) single-stranded molecules may fold into secondary structures or they may stay as flexible linear structures; (2) linear double-stranded molecules may form heteroduplexes with mismatched bases or they may stay in their original homoduplex forms; and (3) circular double-stranded nucleic acids may be nicked and take a relaxed open-circular structure or they can be in a more compact superhelical structure. Under nondenaturing conditions, each of these shapes may influence the way nucleic acid molecules travel through the electrophoretic matrix. Separation based on shape can provide useful information, but it can also confuse size-based analysis. Electrophoresis of DNA is performed under nondenaturing or denaturing conditions depending on the application. RNA electrophoresis is commonly performed as a quality control check before transcript quantification or microarray expression analysis. RNA can be degraded easily by tissue or environmental RNAses, so it is important to assess the quality of the RNA used in these methods. Because RNA often has secondary structure, electrophoresis is usually performed under denaturing conditions to abolish these structures. Microfluidic chips with integrated microelectrophoresis are commercially available to rapidly assess RNA integrity by inspection of ribosomal RNA peaks (Figure 17-8). Although specific transcripts are not detected by this method, only small amounts of starting RNA are needed. Table 17-2 lists common electrophoresis-based techniques described further in this section. TABLE 17-2 Commonly Used Electrophoresis-Based Techniques Southern blot analysis (Figure 17-9) separates restriction fragments by agarose gel electrophoresis and transfers (blots) them to a membrane for easier manipulation in the following steps and for selective visualization by labeled probes. It reveals polymorphisms in the DNA sequence based on the RFLP profile. After electrophoresis, acid treatment fragments the DNA to make it easier to blot, and base treatment denatures the DNA to single strands for better binding to the membrane and probe hybridization. After neutralization, the DNA is transferred to a nitrocellulose or nylon membrane and immobilized. The membrane is then incubated with one or more probes that hybridize to the fragments of interest. The hybridized probes then are visualized by autoradiography or chemiluminescence. Southern blotting detects RFLPs, including large structural alterations such as deletions, duplications, insertions, and rearrangements. The procedure was named after its inventor, E. M. Southern,100 and was the first discrimination method with adequate sensitivity and specificity for DNA analysis of single-copy genes in complex genomes. However, it requires large amounts of DNA (10 to 50 µg/lane) and is very labor intensive and time consuming. Today, it has largely been replaced by PCR.
Nucleic Acid Techniques
Enzymes That Act On Nucleic Acids
Nucleic Acid Treatments that Do Not Use Enzymes
Amplification Techniques
Polymerase Chain Reaction (PCR)—Target Amplification
Details of the PCR Process
PCR Kinetics and Rapid Cycling
PCR Optimization and Primer Design
3′-End of PCR Products
Contamination Control to Avoid False-Positive Results
Asymmetric PCR and Allele-Specific PCR
Single-Molecule PCR or Digital PCR
Other Forms of Target Amplification
Transcription-Based Amplification Methods
Strand Displacement Amplification
Loop-Mediated Amplification Methods
Whole Genome and Whole Transcriptome Amplification
Other Approaches to Amplification
Branched-Chain DNA: Signal Amplification
Serial Invasive Amplification: Signal Amplification
Rolling Circle Amplification: Probe Amplification
End Point Quantification in Amplification Assays
Detection Techniques
Generic Measurement and Visualization of Nucleic Acids
UV Absorbance
Fluorescent Staining of Nucleic Acids
Reporter Molecules and Labeled Probes
Indirect Probe Detection
Fluorescent Labels
Discrimination Techniques
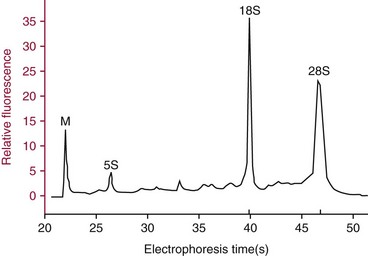
Electrophoresis
Techniques Using Electrophoresis
Abbreviation
Primary Application
PCR/Restriction fragment length polymorphism
RFLP
Detection
PCR (RT-PCR) fragment analysis
Detection
Southern blotting
Detection
Northern blotting
Detection
Dideoxy-termination sequencing
Detection
Single-nucleotide extension assay
SNE
Detection
Oligo ligation assay
OLA
Detection
Multiplex ligation-dependent probe amplification
MLPA
Quantification
Heteroduplex migration assay
CSGE
Scanning
Single-strand conformation polymorphism analysis
SSCP, SSCA
Scanning
Denaturing gradient gel electrophoresis
DGGE
Scanning
Temperature-gradient electrophoresis
TGGE, TGCE
Scanning
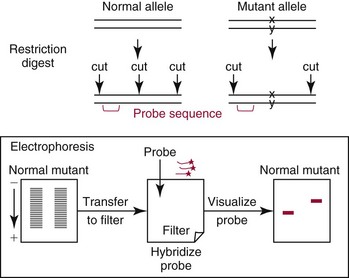
Southern and Northern Blotting
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Nucleic Acid Techniques