When infections are excluded, lung biopsy specimens for diffuse infiltrative pulmonary disease are most likely to be from patients with idiopathic interstitial pulmonary fibrosis or sarcoidosis (1). Most other conditions, such as primary pulmonary hypertension, amyloidosis, vasculitis, and pulmonary Langerhans cell histiocytosis (PLCH), although extremely interesting, are relatively rare and thus are only occasionally encountered in ordinary clinical experience. However, surgical pathologists should have a working familiarity with them.
The development of a systematic method of microscopic examination of lung biopsy material is useful so that all facets of the pulmonary microanatomy are evaluated. The importance of low magnification evaluation cannot be overemphasized. Attention should be focused on the contents of the alveolar space; the condition of the alveolar septa; the conducting airways; and the pulmonary arteries, veins, and lymphatics. In this way, the architectural distribution of pathologic findings (i.e., predominantly intra-alveolar, interstitial, airway, or vascular) is easier to assess, particularly for persons with limited experience.
Various combinations of the pulmonary microanatomy may be affected by certain disease states simultaneously. Some conditions that primarily affect the pulmonary interstitium also cause damage to the small airways and vice versa. For example, usual interstitial pneumonia (UIP) and diffuse alveolar damage (DAD) affect small airways. Conversely, respiratory bronchiolitis affects alveolar spaces. At times, the terminal airways and interstitium may be almost equally involved by diseases, such as hypersensitivity pneumonitis (HP) (2). These overlapping patterns should always be kept in mind, particularly when reviewing small biopsy material. The degree and balance of these morphologic changes form the basis for the histologic diagnosis, but, unfortunately, they often impart little about specific etiology.
Several lung biopsy techniques are in use; each has advantages and drawbacks. Transbronchial lung biopsy is extremely useful and accurate in the diagnosis of pulmonary infections and certain diffuse lung diseases with lymphangitic distributions such as sarcoidosis (3,4). This biopsy method is less accurate, however, for many other diffuse pulmonary conditions because the small biopsy sample may not accurately represent the lung tissue (5) (Table 25.2). However, the usefulness of transbronchial biopsy should not be dismissed out of hand (see the following discussion) (6). On the other hand, open lung biopsy may be as equally unrewarding if the sample is not representative of the disease process or if it is taken from only subpleural lung tissue. Biopsy specimens of the tip of the lingula or other peripheral sites often show advanced fibrosis, which may not necessarily be representative of the deeper pulmonary parenchyma (1). The use of video-assisted transthoracic surgery (VATS) has made obtaining larger tissue samples from multiple areas of the lung easier (7,8). In general, the idea that more tissue is better remains true when one considers the optimal amount of lung biopsy material for diagnosis. However, even with open biopsies, certain histologic artifacts, age-related changes, and nonspecific incidental findings must be recognized (9). Among the most common are mechanical compression or fresh hemorrhage due to the biopsy procedure and rounded clear spaces in the alveoli that can be mistaken for interstitial fibrosis, alveolar hemorrhage, or lipid pneumonia (Fig. 25.1).

Preoperative discussion between pulmonary specialist, pathologist, and surgeon is important to facilitate the appropriate handling of the tissue by the laboratory. A lung biopsy from an acutely ill, immunocompromised patient is treated in a different manner than one from an immunologically normal patient with a slowly progressive diffuse interstitial process.
When an open biopsy is performed, an intraoperative frozen section may be useful so that a judgment can be made about the representation of the disease process in the biopsy. This also ensures that the material arrives in the pathology laboratory fresh and that appropriate bacterial, fungal, or viral cultures and touch preparations can be obtained if necessary. Conditions such as Pneumocystis jiroveci pneumonia, which may require immediate therapy, can thus be rapidly diagnosed. Specialized techniques for mineral analysis, electron microscopy, flow cytometry, or immunofluorescence may also be ordered under the appropriate circumstances (e.g., Goodpasture syndrome, lymphoid lesions, or suspected pneumoconiosis) (10).
In most instances, however, the pathologist’s most important diagnostic tool is well-prepared paraffin section stained with good-quality hematoxylin and eosin (H&E). Oftentimes, trichrome and elastic tissue stains are useful but cannot and should not replace expert correlation with clinical, laboratory, and radiographic information. Judicious use of organism stains, that is, Grocott methenamine silver (GMS) and Ziehl-Neelsen, are often necessary.
Finally, the authors strongly believe that seminal contributions describing puzzling and, oftentimes, rare diseases should still be recognized. Replacing Churg-Strauss syndrome (CSS) with eosinophilic granulomatosis with polyangiitis (EGPA) simply to conform to granulomatosis with polyangiitis (GPA) (Wegener granulomatosis [WG]) and microscopic polyangiitis (MPA) nomenclature slights Drs. Jacob Churg and Lotte Strauss. Drs. Louis Hamman and Arnold Rice Rich’s description of fulminating diffuse interstitial fibrosis, once recognized as Hamman-Rich syndrome but now as acute interstitial pneumonia, should also be acknowledged. Therefore, in appreciation of such pioneering contributions, eponymous disease names are included parenthetically throughout the text.
IDIOPATHIC INTERSTITIAL PNEUMONIAS
Diffuse interstitial inflammation and fibrosis is a common reaction to many injurious agents; therefore, each case presents its own diagnostic riddle. Each of the patterns may also be idiopathic. Microscopic clues about possible etiology are generally lacking, therefore these riddles can only be solved with multidisciplinary discussion (11). Complete details about potential etiologic factors such as drug administration, work or environmental exposure, family history, or other associated diseases must be obtained because they might define an individual case with a specific cause. Radiographic correlation is also required.
The following discussion generally follows the updated American Thoracic Society/European Respiratory Society international multidisciplinary classification scheme (Table 25.3). Although considered a rare idiopathic interstitial lung disease, idiopathic lymphoid interstitial pneumonia (LIP) is best discussed in the “Benign Lymphocytic Infiltrations” section later in this chapter. Idiopathic pleuroparenchymal fibroelastosis along with rare histologic patterns such as acute fibrinous and organizing pneumonia and bronchiolocentric patterns of interstitial pneumonia are not discussed, and the reader is referred to recent publications for complete coverage (12,13).

IDIOPATHIC PULMONARY FIBROSIS
Idiopathic pulmonary fibrosis (IPF) is the clinical term for patients with UIP pattern radiology and histology in the absence of a discernible etiology (11). UIP pattern, although relatively rare, is perhaps the most common condition encountered in biopsies for diffuse interstitial pulmonary fibrosis (9–11).
Although idiopathic, genetic and environmental factors are strongly suspected. Research implicates gene mutations and polymorphisms, smoking, gastroesophageal reflux, drugs, as well as exposure to wood and metal dust (12–14). Rare familial cases are noted, and a common variant in the promoter of the MUCB gene is associated with the development of familial and sporadic IPF (14,15). In many cases, low titers of serum antinuclear antibodies (ANAs) or rheumatoid factor are encountered, which suggests an underlying subclinical collagen disease diathesis (16).
The pathogenesis of UIP is largely unknown, but recent observations have led to the conclusion that inflammation per se may not be as important a factor as was previously thought. In fact, most cases show relatively little inflammation. Emphasis has now switched to the roles of alveolar lining cell damage and fibroblast-myofibroblastic proliferation, that is, abnormal tissue repair, modulated by a growing array of cytokines and growth factors in the production of interstitial fibrosis (17–27).
Clinically, UIP most commonly affects patients in middle to old age. Patients typically present with gradual onset of exertional dyspnea over months or years associated with bibasilar Velcro-type fine end-inspiratory crackles (28). In approximately 40% of cases, the onset is heralded by a viral-like illness, although a viral etiology is not documented. This slowly progressive clinical course is an important feature that immediately separates UIP from more acute causes of diffuse interstitial lung disease. However, periods of rapid decline, termed accelerated phase, are recognized (29).
Physiologically, the earliest change is a reduction in diffusing capacity that occurs at first only with exercise but later also at rest. However, impaired oxygen diffusion across the fibrotic alveolar septa accounts for only a small component of the arterial oxygen desaturation. The major cause is alteration in the ventilation–perfusion relationship that stems from the ventilation of underperfused lung tissue as a result of capillary destruction and perfusion of underventilated alveoli brought about by the profound structural lung alterations. Early in the disease, airway function is normal; however, as interstitial fibrosis progresses, peribronchiolar fibrosis leads to obstructive abnormalities. Lung volumes are normal early in the course, but with progressive fibrosis, the lung volume becomes diminished (30).
Radiologically, diffuse bilateral interstitial abnormalities are most prominent in the lower lung fields and are characteristic but not specific (31). As the condition worsens, progressive fibrosis is associated with shrinkage of the lungs with elevation of the diaphragm and progressive radiographic evidence of “honeycomb lung.” High-resolution computed tomography (HRCT) scans show reticular opacities, traction bronchiectasis, and subpleural honeycomb changes (32). Ground-glass opacities may also be seen and correlate with areas of active alveolitis.
A diagnosis of IPF requires the presence of a UIP pattern on HRCT and/or surgical lung biopsy findings along with the exclusion of other known causes of interstitial lung disease (11) (Table 25.4). Strict adherence to these criteria may spare individuals surgical lung biopsies. However, if biopsies are clinically necessary, then generous samples from more than one lobe should be taken given the possibility of histologic variability in samples from multiple lobes (33–35). Although a recent study suggested that the diagnosis of UIP can be confirmed by transbronchial biopsy in one-third of patients if adequate material is obtained, open or VATS biopsy is the gold standard for the diagnosis (6,36).
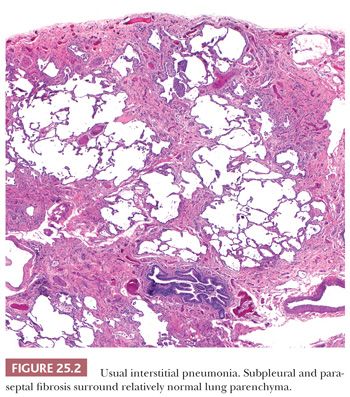
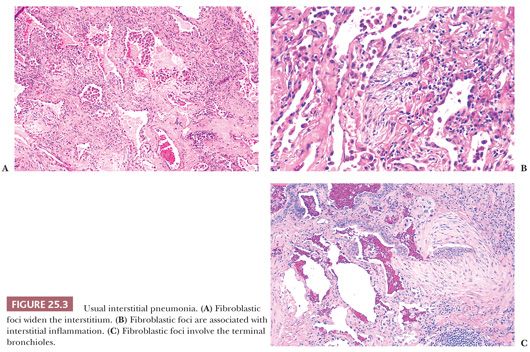
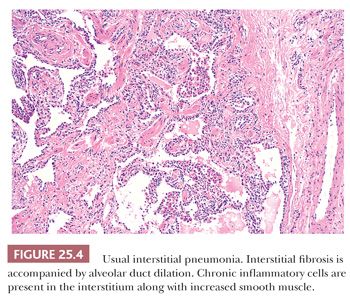
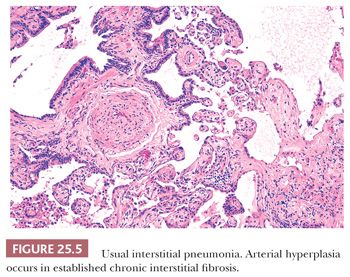
UIP pattern starts with a mainly peripheral lower lobe distribution (Table 25.5). Irregularly distributed areas of interstitial fibrosis with active fibroblastic proliferation alternate with near-normal lung and advanced cystic honeycombing (Fig. 25.2). This pattern is termed temporally heterogeneous given the presence of normal lung, active inflammation, and fibrosis, which has quite cleverly been described as “future,” “present,” and “past” states of disease (37). The fibrosis is predominantly subpleural and paraseptal, although this may be difficult to discern on small surgical lung biopsies. Scattered foci of so-called immature fibrosis, otherwise known as fibroblastic foci, are noted at the interface between fibrotic and relatively normal lung tissue (Fig. 25.3). The fibromyxoid connective tissue proliferations, composed of loose myxoid stroma with spindle cells and occasional inflammatory cells, are interstitial-based and do not fill alveolar spaces. They result from damage to alveolar epithelium and basement membrane (38,39). Of note, the number of fibroblastic foci varies from case to case. A modest interstitial infiltrate of lymphocytes and plasma cells accompanied by intra-alveolar mononuclear histiocytes and lymphocytes is often subtle, whereas alveolar lining cells are often hyperplastic. A pronounced alteration of the pulmonary architecture with dilation of the alveolar ducts and alveoli, diminished pulmonary capillaries, and marked interstitial fibrosis with microscopic honeycomb remodeling develops. These mucin-filled cystic spaces are lined by bronchiolar epithelium (34,40,41). Hyperplastic smooth muscle proliferates around terminal bronchioles (42) (Fig. 25.4). This is thought to be a compensatory mechanism for overcoming increased lung stiffness (decreased compliance). Extreme examples have been termed muscular cirrhosis (43). As fibrosis progresses and the capillary bed is gradually destroyed, pulmonary arteries and arterioles undergo medial hypertrophy (38) (Fig. 25.5). Osseous and squamous metaplasia as well as desquamative interstitial pneumonia–like areas and eosinophil-rich areas may be seen (24). Both squamous and glandular proliferations should not be brushed aside simply as metaplastic without serious consideration of neoplasia given the 14-fold increased risk of lung carcinoma in these patients (44).

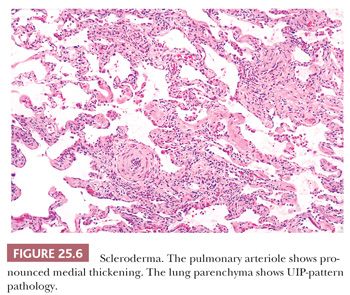
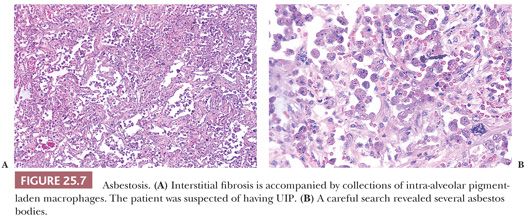
Given the fact that many other diseases may manifest with UIP-like morphology, excluding connective tissue diseases (CTDs), chronic hypersensitivity pneumonitis (cHP), as well as pneumoconiosis, chronic infection, chronic aspiration, and drug effect requires both clinical and radiographic correlation in addition to careful glass slide review. UIP-pattern biopsies from patients with underlying CTD, especially rheumatoid arthritis, usually feature more inflammation including prominent peribronchiolar lymphoid nodules as well as pleural fibrosis (45,46). Scleroderma, besides showing UIP, may also show lymphoid nodules or pulmonary artery medial mucinous changes or laminated fibrosis (47) (Fig. 25.6). These lung manifestations may sometimes precede the overt signs of CTD by several years. cHP often demonstrates advanced fibrosis; however, if the clinical and radiographic features are not enlightening, noting centrilobular (i.e., airway-centered) fibrosis rather than the subpleural and paraseptal pattern noted in true IPF should at least raise the possibility of cHP. Also, given the upper lobe predominance of cHP, biopsies from more than the lower lobe is necessary. Note that cHP may not feature obvious interstitial granulomas (48) (see the following discussion). The possibility of pneumoconiosis should always be considered in any otherwise cryptic case of UIP, and attempts to isolate asbestos and identify other minerals may be necessary (10) (Fig. 25.7). The pathologist should remember, however, that patients exposed to asbestos might also have pulmonary fibrosis that is caused by idiopathic UIP (49).
Adverse drug reaction should also be considered and a thorough drug history should be obtained in all UIP pattern cases (50). One should consult the Drug-Induced Respiratory Disease website, www.pneumotox.com, or standard pulmonary pathology textbooks for lists of drugs known to cause UIP-pattern lung disease (51).
The prognosis of IPF is poor. The disease is slowly progressive and the median survival is 2 to 3 years, although variation is seen among patients. Diminished survival is associated with older age, cigarette smoking, severe shortness of breath, decreased pulmonary function, severity of imaging studies, and greater numbers of fibroblastic foci (28,52,53). Some patients experience acute deterioration that correlates pathologically with the superimposition of acute lung injury qualitatively similar to DAD or organizing pneumonia, the so-called accelerated phase of UIP (29,54). Overall, only approximately 20% of patients have shown objective improvement with steroid treatment, but the lack of controlled pathologic studies makes analysis of retrospective series difficult (28,38,52,54). Cyclophosphamide and other chemotherapeutic drugs have been used, as has colchicine in cases unresponsive to steroids (29). Interferons and newer drugs targeted against growth factors and fibroblasts and that encourage epithelial regeneration as well as several other novel genetic approaches are currently being investigated (22,55). Single lung transplantation may have a 5-year survival rate approaching 44%.
IDIOPATHIC NONSPECIFIC INTERSTITIAL PNEUMONIA
No less complicated a topic than IPF/UIP, nonspecific interstitial pneumonia (NSIP) was first described in HIV-related lung inflammation before being redefined in 1994 as a distinct pattern of interstitial pneumonia separate from IPF/UIP (47). We now recognize NSIP as a distinct idiopathic interstitial pneumonia (IIP) as well as a histologic pattern associated with a variety of clinical disorders including but not limited to cHP, CTD, resolving DAD, HIV, chronic infection, chronic aspiration, and drug reactions (12,56,57). However, our understanding of idiopathic NSIP is incomplete.
Idiopathic NSIP appears to predominantly affect never-smoking women in their 50s. Chronic dyspnea, cough, and fever along with basal crackles are usually noted at presentation. Pulmonary function tests (PFTs) demonstrate a restrictive pattern with reduced gas transfer. Serologic abnormalities, including low serum ANA and rheumatoid factor titers, are seen in a significant minority of patients (58).
HRCT demonstrates bilateral ground-glass opacities along with reticular opacities, traction bronchiectasis, and bronchiolectasis in three-quarters of patients (59). Half of proven cases show diffuse lung involvement, whereas the other half feature predominant bilateral lower lobe disease.
Given its recent elevation to a distinct idiopathic entity, the etiology is unknown. There are likely variable mechanisms at play. It perhaps represents a lung manifestation of an undifferentiated CTD (60). This hypothesis is intriguing given the observation that some patients with CTD develop lung disease years before manifesting systemic symptoms.
Unlike UIP-pattern, NSIP is characterized by a temporally and spatially uniform pattern of interstitial fibrosis with or without inflammation (Table 25.6) (Fig 25.8). Lung architecture is relatively preserved, whereas alveolar walls are expanded by variable degrees of mostly lymphocytes and/or collagen. Neither a bronchocentric nor subpleural/paraseptal distribution is apparent. Cellular and fibrosing patterns are recognized; however, most cases contain fibrosis. The loose fibroblastic or dense collagenous alveolar septal fibrosis should be uniformly distributed. The cellular pattern features diffuse interstitial lymphocytes with rare plasma cells often accompanied by type II pneumocyte hyperplasia. Fibroblastic foci, organizing pneumonia, prominent airway disease, honeycomb fibrosis, and/or muscular cirrhosis are not usual, whereas hyaline membranes, eosinophils, granulomas, inorganic dusts, and Langerhans cells should not be seen.
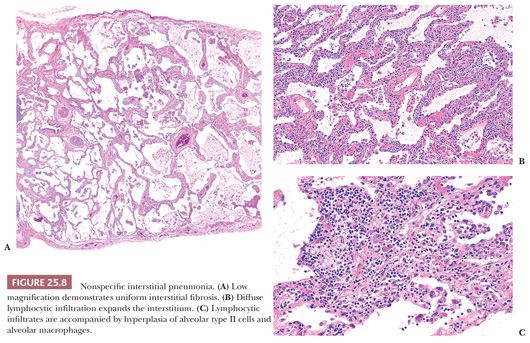

In stark distinction to IPF patients, those with NSIP have a variable but generally favorable prognosis (58,61). Although 20% of patients are dead 5 years following diagnosis, the 10-year survival rate approaches 75% (56). Therapeutic options include corticosteroids and immunosuppressive agents, but supportive studies are lacking (62).
Just as UIP pattern histology is a necessary component of IPF, a diagnosis of idiopathic NSIP requires the exclusion of many etiologies that may present with NSIP morphology. Only in a multidisciplinary setting can one reasonably exclude cHP, CTD, resolving DAD, drug-induced lung disease, occupational exposure, immunodeficiency, and familial pulmonary fibrosis as causes. Morphologically, discerning NSIP pattern from UIP pattern, especially in the setting of underlying emphysema, can be extremely difficult. If fibrosis does not have a subpleural and paraseptal distribution, if honeycomb change is sparse or absent, and if fibroblastic foci are practically impossible to find, then one can favor a diagnosis of NSIP. If one biopsy site demonstrates NSIP histology while another shows UIP pattern, for management purposes, the biopsy should be classified as UIP (33,35). Although CTD lung involvement cannot ever be entirely excluded, HP should be separated given that is a bronchiolocentric inflammatory and fibrosing process with occasional ill-defined granulomas or collections of epithelioid histiocytes (see the following discussion). Diagnosis on a transbronchial biopsy is not possible, especially given the usual presence of alveolar fibrosis in virtually all subbronchial alveolar parenchyma.
CRYPTOGENIC ORGANIZING PNEUMONIA
Cryptogenic organizing pneumonia (COP) is a clinicopathologic entity first described in 1983 (16,63). Although not a typical “interstitial” process, it is included in the IIP classification because it is confused with other IIPs, especially when it progresses to fibrosis (12). Much of the confusion surrounding the fairly easy to recognize morphology relates to its prior designation—bronchiolitis obliterans organizing pneumonia (BOOP) (64). Because “BOOP” is also used with airway diseases including lung transplant rejection lesions, its significance is muddled. In contrast, when used correctly, one cannot help but understand that individuals with COP have organizing pneumonia of unknown etiology. Again, this diagnosis requires clinical and radiographic correlation because the pattern of organizing pneumonia is commonly seen in lung samples from a myriad of pathologic conditions (65) (Table 25.7).

Men and women are equally affected and most are nonsmokers in their sixth decade of life. Patients typically present with a less than 3-month complaint of cough and dyspnea and may be treated with at least one course of antibiotics before being referred to a pulmonologist. Weight loss, fever, and chills as well as myalgias are not uncommon. An elevated erythrocyte sedimentation rate is typically noted (66). Interestingly, PFTs demonstrate a mild to moderate restrictive ventilatory pattern despite the microscopic finding of small airway and airspace obstruction.
HRCT shows patchy subpleural, peribronchial, or bandlike consolidations with air bronchograms. The consolidations migrate in subsequent scans and more than half also demonstrate ground-glass opacities. The reverse halo sign (ground-glass opacities surrounded by consolidations) suggests the diagnosis. Less than 20% of cases present with multiple nodules worrisome for metastatic disease.
The patchy bronchiolocentric histologic pattern is distinctive but not etiologically specific. Chronic inflammatory cells and plugs of loose, matrix-rich connective tissue clog terminal bronchi and bronchiolar lumens and fill alveolar ducts and alveolar spaces (Fig. 25.9). These areas may vary in their stage of organization from case to case but are uniform in any one biopsy (Fig. 25.10). Alveoli adjacent to involved airways contain foamy macrophages, inflammatory cells, and cellular debris sometimes containing cholesterol clefts or fibrin balls. This reaction, which is seen focally within the alveolar space, is a clue indicating that obstruction of small terminal airways has occurred. The alveolar septa show varying amounts of chronic interstitial inflammation and alveolar collapse around involved distal airways. Type II pneumocyte hyperplasia may be seen. Underlying lung architecture is relatively preserved.
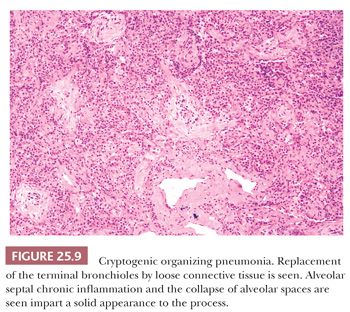
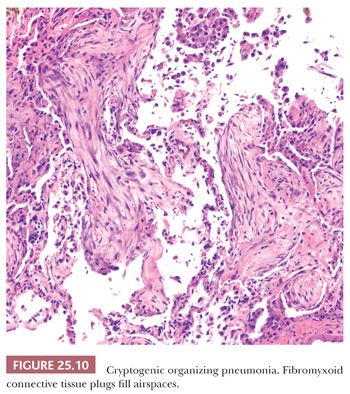
COP cannot be definitively diagnosed on a transbronchial or transthoracic biopsy, but in the proper clinical and radiographic context, the finding of organizing pneumonia in the absence of, for example, granulomas or infectious agents, can confirm the clinical impression (67).
The morphologic differential diagnosis includes DAD; however, this process features edematous thickened inflamed alveolar walls throughout a lobule and usually hyaline membranes. The fibrosing interstitial pneumonias demonstrate prominent alveolar septal fibrosis with varying degrees of parenchymal remodeling. The presence of intra-alveolar neutrophils, necrosis, pronounced acute bronchiolitis, and/or granulomas suggests infection or HP.
The prognosis is usually excellent and, in most patients, steroids are effective in ameliorating the condition, but many relapse within a few months of therapy cessation or tapering below 15 mg/day (64,66,68). Spontaneous recoveries are reported, whereas rare patients are therapy-resistant and develop interstitial fibrosis (69). Approximately three-fourths of patients survive for 5 years (65).
DIFFUSE ALVEOLAR DAMAGE AND ACUTE INTERSTITIAL PNEUMONIA (HAMMAN-RICH SYNDROME)
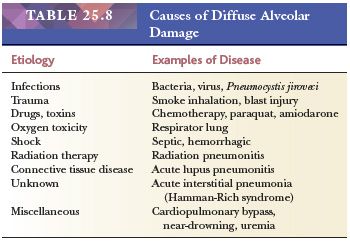
Dad is an acute exudative interstitial pulmonary reaction resulting from many different etiologies (44,70,71) (Table 25.8).
The specific etiology of DAD may be obvious or diagnosable, for example, near-drowning; subsequent to cardiopulmonary bypass; following radiation therapy; or after drug therapy, such as with chemotherapeutic agents or amiodarone, or infections (72–74). In some patients, the cause may be multifactorial, with several factors acting either simultaneously or consecutively—particularly mechanical ventilation with high oxygen concentration (75). However, infection is the most common cause, although idiopathic is almost as common (76). DAD is the pathologic counterpart of the acute respiratory distress syndrome (ARDS). Cases of ARDS showing DAD histology without an identified cause, previously known as Hamman-Rich syndrome, are termed acute interstitial pneumonia (77).
Clinically, disease occurs over a wide age range without sex or smoking associations (70,73,74). Up to 60% of patients are immunocompromised (76). The onset of symptoms is abrupt, generally occurring over 1 or 2 days and consisting of severe dyspnea accompanied by fever and cough, in contrast to the slow, insidious evolution of UIP. Many patients had a prior upper respiratory tract weeks earlier. Auscultation reveals diffuse bilateral crackles. Patients have restrictive physiology along with rapidly progressive hypoxia leading to respiratory failure. Almost all patients fulfill diagnostic clinical criteria for ARDS (78).
Chest radiographs demonstrate bilateral patchy airspace opacifications with air bronchograms that eventually develop into diffuse consolidation. Early CT findings include bilateral patchy ground-glass opacities in dependent areas of the lung yielding a geographic distribution. Organizing disease shows bronchovascular bundle distortion with traction bronchiectasis along with cysts (79,80).
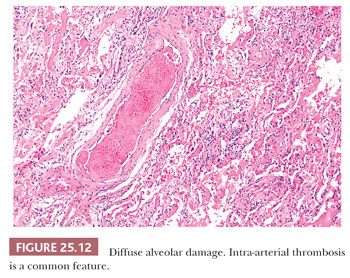
The histologic pattern is characterized by an early exudative phase that develops into an organizing phase. The term diffuse refers to complete involvement of the pulmonary lobule and not necessarily the entire lung sample. The early reaction shows rounded organizing plugs of exudate in the terminal bronchioles and protein-rich edema in the alveolar space, with fibrin-rich eosinophilic hyaline membranes along the surface of the alveolar septa (71) (Fig. 25.11). Type II pneumocytes are reactive with nucleomegaly and atypia. Interstitial edema is usually accompanied by scant numbers of lymphocytes, plasma cells, and a few neutrophils. Intra-alveolar hemorrhage may be present focally but is generally not severe. Thrombi may be seen within pulmonary capillaries and small- to medium-sized pulmonary arterioles (Fig. 25.12). Large numbers of acute inflammatory cells may indicate that DAD is superimposed on another process, such as bacterial pneumonia or alveolar capillaritis (81).
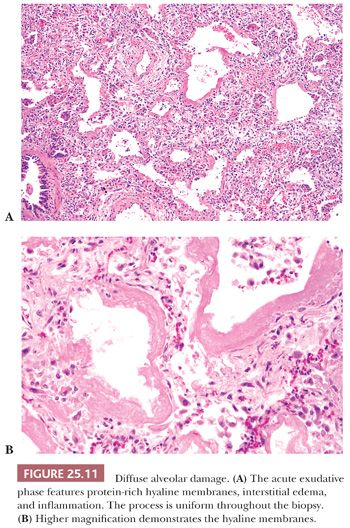
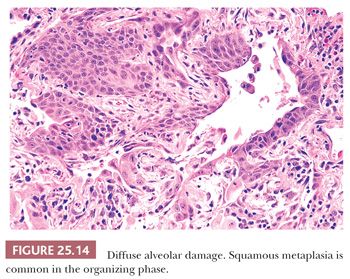
In lesions of one to several weeks’ duration, a phase of organization may ensue that can result in complete resolution of the process or that may progress to interstitial fibrosis (Fig. 25.13). Histologically, the picture is usually uniform, but some biopsy specimens may show a mixed pattern of acute exudation along with areas of organization indicative of repeated bouts of lung injury. Squamous metaplasia due to terminal bronchiolar injury often accompanies the more advanced changes (Fig. 25.14). This sometimes may be mistaken for malignancy.
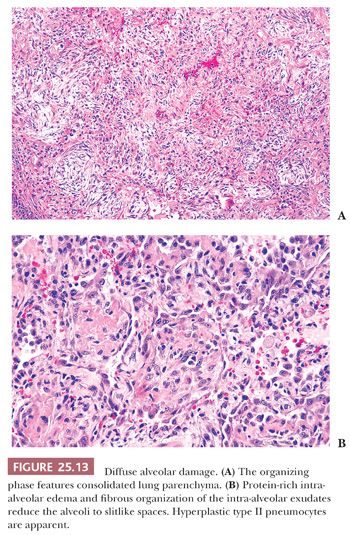
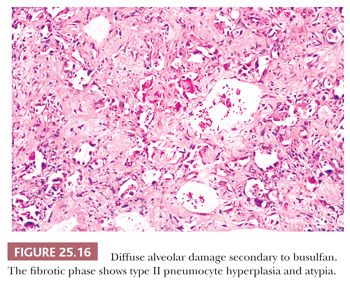
Fibrosis results from the incorporation of proteinaceous exudates by the apposition of newly formed collagen onto the preexisting alveolar framework. Collagen synthesis within the alveolar septal membrane also occurs (Fig. 25.15). In addition, fibrosis may also result from so-called collapse induration caused by nonexpansion of alveolar spaces secondary to the loss of pulmonary surfactant and the direct apposition of damaged alveolar septa without the restitution of normal architecture (82,83). Interstitial fibrosis may occur rapidly over a period of several days, or it may progress slowly over several months. When fibrosis is established, the alveolar septa are densely fibrotic, and the late changes are difficult to differentiate from other causes of diffuse interstitial pulmonary fibrosis. However, residual hyperplasia and atypia of alveolar lining cells and remnants of hyaline membranes may be a clue to preexisting DAD (Fig. 25.16).
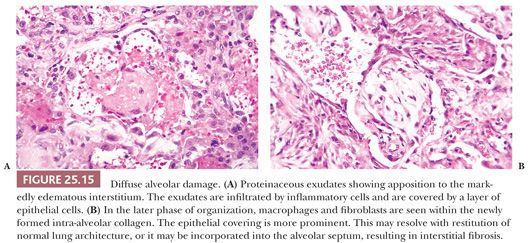
Regardless of the inciting cause, a sequence of events, including the damaging effects of leukocyte enzymes, inactivation of pulmonary repair mechanisms, depletion of surfactant, activation of blood coagulation factors, and fibrinolysis, result in damage to the endothelial cell basement membrane and alveolar type I cells (84). Basement membrane and endothelial injuries are responsible for the protein-rich hyaline membranes and edema. The roles of tumor necrosis factor, interleukins, and other cytokines have been postulated in the pathogenesis of this condition (43,84). The process is reversible but may result in death from impairment of gas exchange through fluid exudates in the early stages or organization and fibrosis.
The presence of hyaline membranes help distinguish DAD from other interstitial pneumonias; however, organizing DAD can be mistaken for organizing pneumonia if one does not appreciate the more diffuse nature of the process and impressive alveolar septal fibrosis. Although fibrosis in DAD may resemble fibrotic NSIP or even honeycomb change in UIP pattern, clinical and radiologic features should discern the entities (85,86).
DAD may fully resolve either in the acute, protein-rich phase or even after biopsy shows organization and fibrosis (84). Death occurs in roughly 40% to 50% of cases, usually after several weeks or months (82,84). Although most patients who recover may show little, if any, residual pulmonary functional disability, some may show persistent radiographic abnormalities with residual functional impairment (84,87,88). Care is supportive. Many cases require assisted ventilation with high concentrations of oxygen to maintain respiratory function, which may itself induce similar histologic changes.
TOBACCO-RELATED INTERSTITIAL LUNG DISEASES
Respiratory bronchiolitis (RB), respiratory bronchiolitis-interstitial lung disease (RB-ILD), desquamative interstitial pneumonia (DIP), and PLCH in addition to UIP/IPF represent tobacco-associated interstitial lung diseases (89,90). Fibrosis associated with emphysema has garnered much attention lately, but its place in the interstitial lung disease classification scheme is not settled (91–95). UIP/IPF was discussed separately in order to distance its clinical course from these other entities. RB, RB-ILD, and DIP can be viewed along a continuum; however, DIP should also be considered separately given its different morphologic differential diagnosis and clinical outcome (96).
RESPIRATORY BRONCHIOLITIS AND RESPIRATORY BRONCHIOLITIS-INTERSTITIAL LUNG DISEASE
RB is simply a morphologic finding present in current smokers and noted incidentally in lung tissue removed for a wide variety of reasons (97,98). Patients are asymptomatic, although PFTs may demonstrate airflow obstruction. Diffuse centrilobular nodules and ground-glass opacities are reported on HRCT. Respiratory bronchioles and surrounding alveolar sacs contain increased numbers of macrophages with finely translucent yellow-brown pigmented cytoplasmic granules, so-called smoker’s macrophages. Mural inflammation and collagen expand bronchiolar walls in almost half of current and ex-smokers (99) (Fig. 25.17). Type II pneumocytes of affected airspaces are often hyperplastic. RB may be an isolated morphologic finding or associated with centrilobular emphysema. Smoker’s macrophages, but not fibrosis or emphysema, eventually clear after smoking cessation (100).
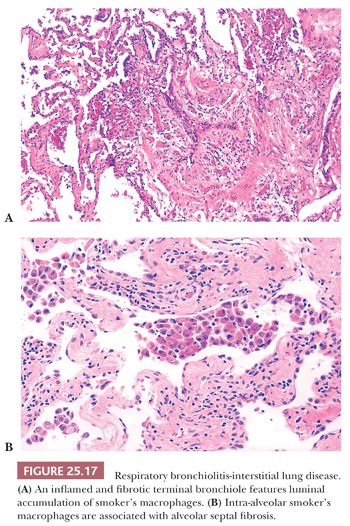
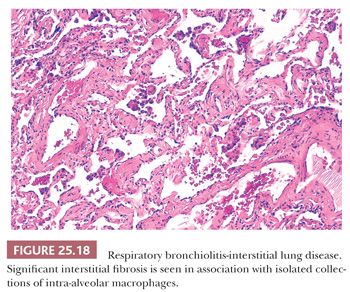
The distinction between RB and RB-ILD is purely clinical. If the clinician establishes the presence of an interstitial lung disease with functional abnormalities (i.e., markedly decreased diffusing capacity and ground-glass opacities/centrilobular nodules along with reticulation at HRCT) and nothing other than RB is noted on the biopsy, then a clinico-radiologic-pathologic diagnosis of RB-ILD can be made (101) (Fig. 25.18).
RB-ILD patients may complain of developing dyspnea or either a new or changed cough. Men are affected twice as often as women, with 30 pack years of cigarette smoking, usually in their fourth to fifth decades of life. Affected individuals usually improve with smoking cessation.
DESQUAMATIVE INTERSTITIAL PNEUMONIA
DIP is confusing for several nonmorphologic reasons. The term desquamative interstitial pneumonia is a misnomer as the lesional cells are alveolar macrophages and not desquamated pneumocytes, as originally believed. In addition, DIP in children is a different disease representing a surfactant-related process in many cases with recognized genetic mutations (102). Controversy existed regarding whether DIP was an early phase of UIP or an entirely separate entity (68,103,104). This debate has cooled off, as most authorities believe the diseases are separate; however, the relationship between DIP and NSIP is uncertain (12,97,100,105).
DIP mostly affects male smokers in their fourth or fifth decades of life. However, approximately 10% of patients are nonsmokers and the disease can affect patients following occupational exposures, drugs, and in the context of viral and autoimmune illnesses (106) (see the following discussion). Patients develop dyspnea and dry cough slowly over weeks to months and may rarely progress to respiratory failure. Diffusion capacity is decreased, whereas lung volumes are normal or mildly restrictive. Chest radiographs are often normal, whereas CT demonstrates lower lung zone and sometimes peripheral ground-glass opacifications (107).
Pathologic changes are generally diffuse. Alveolar spaces are filled with mononuclear histiocytes with eosinophilic cytoplasm and finely dispersed dusty brown pigment (Fig. 25.19). The cytoplasm of these cells may stain positively with periodic acid-Schiff (PAS) after diastase, and the brown pigment stains with Prussian blue (104). Alveolar walls are expanded by plasma cells, a few eosinophils, and scattered lymphoid nodules. Diffuse septal fibrosis may be seen as well. Type II pneumocytes are often hyperplastic and may be especially prominent. Fibroblastic foci are not observed, but peripheral honeycomb lung may be seen.
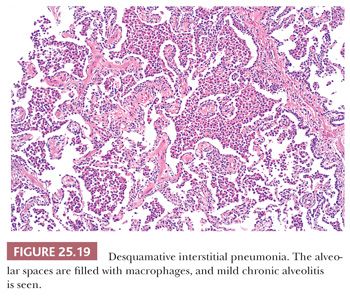
DIP is discerned radiographically and morphologically from RB-ILD by its diffuse lung involvement rather than the bronchiolocentricity noted in latter. Lymphoid aggregates are also only found in DIP. Temporal homogeneity along with absence of fibroblastic foci discerns DIP from UIP. Prominent intra-alveolar macrophages are not a typical finding in active NSIP; however, chronic DIP with fibrosis may resemble fibrotic NSIP. Clinicopathologic correlation, again, is extremely important.
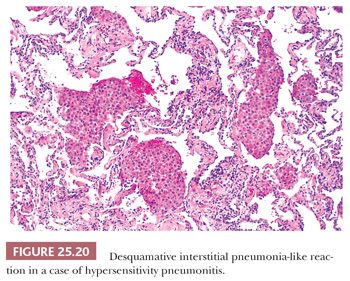
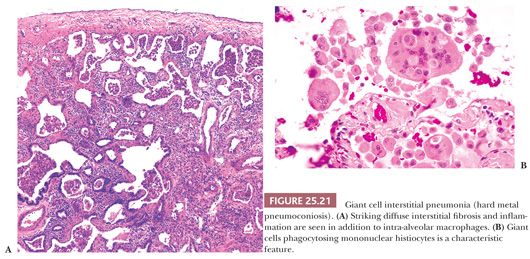
It is important to recognize that DIP can also be a nonspecific reaction pattern in other conditions and that a DIP-like pattern can be seen adjacent to areas that are otherwise diagnostic of such conditions as PLCH, HP, eosinophilic pneumonia, or pulmonary hemorrhage (108) (Table 25.9) (Fig. 25.20). Foci that are histologically indistinguishable from DIP may also be seen in patients with asbestosis, in aluminum workers, in those with drug reactions, or in patients with cobalt pneumoconiosis (109–111). In patients with industrial exposure to hard metals, diffuse interstitial lung disease characterized by large numbers of intra-alveolar macrophages, similar to those seen in DIP, may develop, but it is accompanied by multinucleated giant cells that phagocytose other histiocytes. This giant cell interstitial pneumonia is extremely rare, and some consider it a specific response to hard metal exposure (112) (Fig. 25.21).

The prognosis of DIP is much better than that for UIP and NSIP. Most patients recover after smoking cessation, and steroids are effective for treatment. Only a few patients experience progression to significant interstitial fibrosis. In recent studies, the survival of patients without honeycombing was 100% (38,52,113).
PULMONARY LANGERHANS CELL HISTIOCYTOSIS
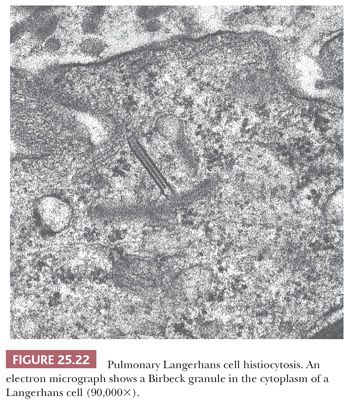
Alternative terms are eosinophilic granuloma, Langerhans cell granulomatosis, and histiocytosis X (137). PLCH is a disease of unknown etiology characterized histologically by tissue infiltration with localized collections or sheets of eosinophilic histiocytes (Langerhans cells). Ultrastructurally, the histiocytes contain specific pentilaminar intracytoplasmic structures (Birbeck granules) that impart specificity to the Langerhans cells (114) (Fig. 25.22). The disease may be localized to a single site, or it may involve several different organs simultaneously or progressively. Pulmonary involvement is seen in approximately 20% of cases with multicentric disease, which may also involve bone, skin, lymph nodes, spleen, pituitary gland, and other areas. The lung is the only organ of involvement in more than 50% of cases (115,116).
PLCH affects young adults between the ages of 20 and 40 years and accounts for up to 5% of all diffuse lung disease biopsies (1). The etiology is unknown, but an association with cigarette smoking is significant. Almost all patients are current or past smokers (117). In fact, a diagnosis of PLCH should be suspect in a nonsmoker. Langerhans cells are a subtype of dendritic cells. The method by which they coordinate airway immune responses, particularly in response to tobacco smoke, however, remains unexplained (118). Molecular studies have shown the clonal nature of the Langerhans cells in extrapulmonary sites (119–121). Approximately half of pulmonary cases may be clonal by human androgen receptor (HUMARA) assay, although doubt has been expressed as to whether this reflects a neoplastic process (122).
Two-thirds of patients have clinical disease, which varies in severity from relatively mild dyspnea to severe shortness of breath, hemoptysis, and pleuritic chest pain associated with spontaneous pneumothorax; the latter is noted in about 15% of patients. The remainder are asymptomatic despite an abnormal chest radiograph (115,117). Ten percent to 15% of patients may also have extrapulmonary disease, such as polyuria and polydipsia due to diabetes insipidus, pain from skeletal involvement, or skin rashes due to cutaneous involvement. Blood eosinophilia is not seen. Low diffusing capacity to carbon monoxide is noted in approximately 70% of patients and related to pulmonary vascular dysfunction. A restrictive pattern gives way to an obstructive pattern as the disease progresses.
Radiologically, the disease usually shows diffuse bilateral upper and middle lung interstitial or reticulonodular infiltrates that are more pronounced in the peripheral lung fields. Rare cases may show single or multiple nodules that are suggestive of malignancy (116,123). In chronic cases, cysts predominate in the subpleural regions and are accompanied by interstitial fibrosis. HRCT scans show characteristic and highly specific centrilobular 0.1 to 1.0 cm nodules and up to 2.0 cm thick or thin-walled cysts sparing the lower lung fields (123). Early nodular lesions may be fluorodeoxyglucose-positron emission tomography (FDG-PET) avid.
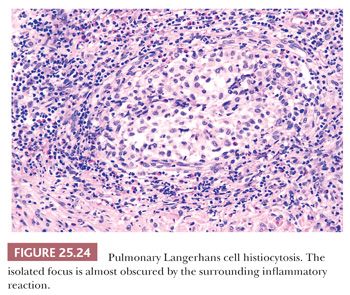
Pathologically, a diagnosis rests on the identification of Langerhans cells with appropriate architectural and cytologic findings. The disease is bronchiolocentric with stellate or medusa-like extensions into surrounding pulmonary interstitium, including small arteries and veins (Fig. 25.23). The cellular nidus is often nodular (Fig. 25.24). Langerhans cells aggregate and vaguely resemble granulomas. Obliterated small arteries and veins are commonly seen. Eosinophils along with macrophages and lymphocytes are always present, but their numbers vary from lesion to lesion in a single biopsy.
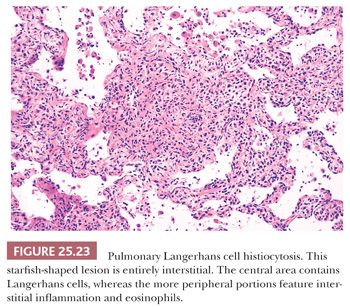
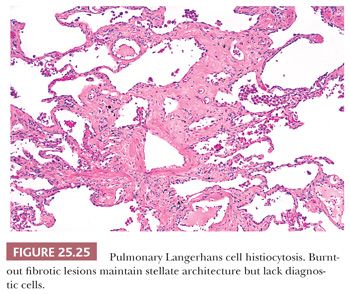
As the disease progresses, Langerhans cells and other inflammatory cells diminish, and fibrosis replaces the cellular phase (Fig. 25.25). Pulmonary hypertensive changes may develop. Organizing pneumonia, identical to that seen in other fibrosing lung diseases, may also be present. Older lesions often show subpleural acellular stellate scars. These may show the remnants of burned-out foci containing foamy histiocytes, smoker’s pigment, or hemosiderin-laden macrophages. In the appropriate clinical and radiographic setting, these stellate areas of fibrosis are characteristic of the healed phase of the disease. A careful search may disclose Langerhans cells.
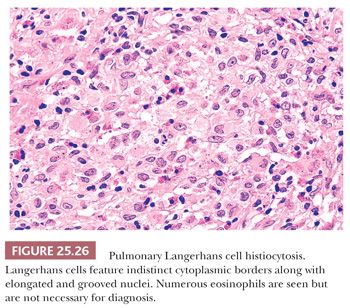
Diagnostic Langerhans cells have somewhat glassy pink cytoplasm with indistinct cell borders, lending a syncytial quality to the infiltrate. The nuclei are distinctive and show longitudinal coffee bean grooves and an undulating or indented nuclear membrane. These Langerhans cells are necessary for the diagnosis (Fig. 25.26). Immunohistochemically, the nucleus and cytoplasm stain positively with antibodies to S-100 protein, which is sometimes a useful tool in differentiating Langerhans cells from pulmonary macrophages. Langerin and CD1a are also positive (114,124).
Because a variety of inflammatory conditions, including chronic obstructive pulmonary disease and lung carcinomas, may contain reactive Langerhans cells, caution should be exercised in diagnosing PLCH, particularly in transbronchial biopsies (125,126). Yet a suggestive transbronchial biopsy in the proper clinical and radiographic setting may obviate the need for an open lung biopsy (127).
The differential diagnosis includes other tobacco-related diffuse lung diseases. Surrounding lung tissue commonly shows smoking-related changes characteristic of RB-ILD or, in exceptional cases, DIP. In RB-ILD, interstitial collections of Langerhans cells are not identified; however, alveoli contain varying numbers of pulmonary macrophages containing pigmented smoker’s granules and eosinophils that are sometimes accompanied by hemosiderin. Compared with Langerhans cells, these macrophages have darker cytoplasm and sharper cell borders, lack the characteristic grooved nuclei, and do not stain with S-100 protein. They do, however, react with macrophage markers such as CD68. Confusion with DIP may be a problem, particularly in transbronchial or poorly representative open lung biopsies. Considering the possibility of PLCH is wise under such circumstances when one is confronted with DIP-like changes. This problem may be compounded because both DIP and PLCH patients may clinically be asymptomatic, and the chest radiographs in the two conditions may at times be similar. Eosinophilic pneumonia is also a differential consideration but usually features intra-alveolar macrophages, reactive type II pneumocytes, and intra-alveolar fibrin (115).
Eosinophilic pleuritis is a nonspecific pleural reaction of sometimes abundant eosinophils mixed with mesothelial cells that is seen in specimens that have usually been resected for the treatment of pneumothorax of any cause. It has been confused with PLCH, and care should be taken in such clinical situations (128). Eosinophilic vasculitis has been described in subpleural blood vessels in this condition (129).
Although the course of disease in adults is variable and unpredictable, the prognosis of PLCH is generally favorable (119). Only the presence of pulmonary hypertension reliably predicts a poor outcome (130). Despite tobacco cessation, up to 20% of patients worsen (115,123). Steroids or chemotherapy may be effective in these individuals, whereas transplantation is the final option. PLCH patients also have an increased incidence of secondary malignancies, including lymphoma (117).
ACUTE EXACERBATIONS OF FIBROTIC INTERSTITIAL LUNG DISEASE
An abrupt development of clinical and radiologic acute lung injury in patients with known or perhaps undiagnosed fibrosing interstitial pneumonia is considered an acute exacerbation (29,131). First reported in patients with IPF/UIP, the spectrum has expanded to include patients with CTD-associated UIP, NSIP, cHP, as well as asbestosis and DIP (132–134).
Patients, usually male, present with unexplained worsening of dyspnea, usually within 30 days, along with new bilateral ground-glass opacities or consolidation superimposed on a reticular or honeycomb pattern on CT (54,135). Patients usually require intubation and mechanical ventilation. In some instances, this rapid clinical and radiographic presentation is the patient’s first presentation with the chronic disease (136). The etiology is not known, but possibilities include undiagnosed infection, stress of surgery including anesthesia and oxygen, and gastroesophageal reflux (137).
Pathology demonstrates chronic fibrosing interstitial pneumonia with superimposed DAD or organizing pneumonia (138). The challenge for the pathologist is to recognize underlying UIP, NSIP, or cHP. Fibroblastic foci of UIP are parallel arranged tufts of fibromyxoid connective tissue attached to areas of fibrosis or alveolar walls with overlying regenerating epithelium, whereas organizing pneumonia features tissue plugs in alveolar ducts and respiratory bronchiolar lumens unassociated with fibrous tissue. Extremely consolidated lung may mask underlying disease; radiologic correlation can be helpful in such cases.
High-dose steroids are typically administered, but mortality averages 70% (135).
HONEYCOMB LUNG
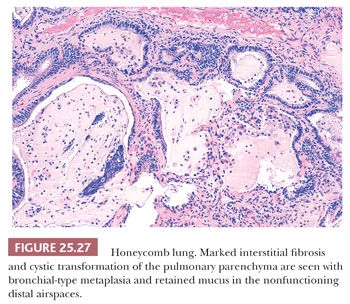
Honeycomb lung is the final common pathway of the many different diseases that progress to chronic interstitial fibrosis and is preferable to the term end-stage lung. The lungs are spongy, stiff, and fibrotic, with readily discernible cystic changes apparent to the naked eye (139). Microscopically, dense interstitial and peribronchial scarring is seen along with diminished or absent alveolar capillaries, marked medial hyperplasia of pulmonary arteries and arterioles, and increased peribronchial and interstitial smooth muscle. Cystic dilation of alveolar ducts and alveoli decrease surface area for gas exchange (Fig. 25.27). This profound restructuring results in diffuse cystic transformation and metaplasia of the alveolar lining to mucinous or columnar epithelium. This epithelium often features cytologic atypia. The terminal bronchi and dilated alveoli may contain abundant mucus and acute inflammatory cells. Such patients are at increased risk of developing lung cancer (44). Precise characterization of the underlying lung disease may be impossible, unless previous biopsy material is available or less advanced histologic changes are present in other areas of the biopsy.
ADDITIONAL INTERSTITIAL LUNG DISEASES
Although most surgical pathologists evaluating a wedge lung biopsy showing any type of fibrosis immediately wonder about IPF/UIP or NSIP, the differential diagnosis should stretch beyond the IIPs.
HYPERSENSITIVITY PNEUMONITIS (EXTRINSIC ALLERGIC ALVEOLITIS)
HP, also known as extrinsic allergic alveolitis, develops from repeated inhalational exposure to occupational, recreational, or domestic environmental antigens (Table 25.10). The disease can mimic other lung conditions given its varying presentations as diffuse acute, subacute, or chronic interstitial disease (140). Multidisciplinary discussions are necessary to render a correct diagnosis given the rarity of the disease. No more than 13% of interstitial lung disease is attributable to HP, an antigen is not identified in up to 30% of patients, and radiographic and histopathologic overlap with other chronic fibrosing interstitial pneumonias are common (141).

HP is the pulmonary manifestation of hypersensitivity to several categories of inhaled antigens from various plant sources, such as moldy hay, grain, sugar cane, bark, cheese, or even cork dust. Animal proteins from the feces of pigeons or parakeets or from the feathers of turkeys or ducks have also been implicated, and certain drugs may also produce this reaction (142). The varying types of exposure are reflected in the numerous names given to patients with HP, including bird fancier’s lung, farmer’s lung, maple bark stripper’s lung, humidifier lung, and others. Hot tub lung is a peculiar form of HP (see the following discussion).
HP is immunologically mediated by the combined effects of circulating immune complexes and cell-mediated immunity to the inhaled antigens. Genetic factors, including linkage to the major histocompatibility complex, also play a role. The host response is thought to be a combination of type III and type IV immunologic reactions, whereas antigen size, solubility, and concentration are relevant. Most of the lymphocytes in the tissue reaction are CD3+, CD8+ T-suppressor cells derived from both the lung and the peripheral blood (142,143). The pathogenesis is not fully understood but appears to be related to repeated antigenic exposure, which stimulates chemotaxis and sensitizes T lymphocytes and macrophages, resulting in granuloma formation. Immune complexes result in the fixation of complement to the tissues and the subsequent inflammation. The end result is the elaboration of growth factors that stimulate fibroblasts to produce collagen (142–144). This mechanism of fibrosis is thought to differ from that of UIP (145).
Because antigen factors, host factors, and circumstances of exposure determine the scope of the disease, epidemiologic data is sparse (146). In general, there is no gender bias; however, in occupational forms of HP, pigeon keepers and farmers are typically male. Although usually an adult disease, the pediatric population is not spared. Peak seasonal variations are noted in farmer’s lung (late winter when stored hay is dispersed for cattle feed) and bird fancier’s lung (during the sporting season), whereas rainy springs lead to increased incidence rates for farmers mowing and baling hay. Interestingly, tobacco smoking appears protective (141).
Acute HP is the most common clinical presentation. In sensitized individuals exposed to a large challenge, an acute febrile illness is marked by the abrupt onset of dyspnea and cough; chest pain develops within 4 to 6 hours. These symptoms peak at 6 and 24 hours and last for hours to days. Bibasilar crackles may be heard and PFTs demonstrate a restrictive defect and hypoxemia. Radiographs mimic pulmonary edema. Because this presentation usually resolves within days, lung biopsies are almost never performed. Reports describe airspace edema, fibrinous exudates, and interstitial neutrophils (i.e., organizing acute lung injury) in addition to characteristics of subacute HP (134) (see the following discussion).
Subacute disease is the form most familiar to surgical pathologists. Patients present with cough and dyspnea over days to weeks. HRCT features patchy or diffuse symmetric bilateral ground-glass opacities, numerous 3- to 5-mm centrilobular nodules, and mosaic attenuation on inspiration with lobular air trapping on expiration, all with an upper lobe distribution (147). Thin-walled 3- to 25-mm cysts may be seen.
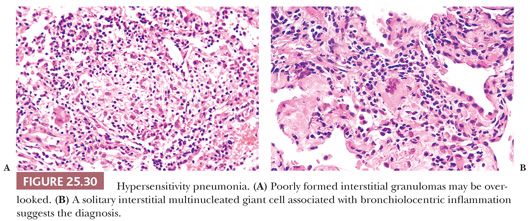
Although transbronchial biopsy may raise the possibility of HP, excisional lung biopsies are strongly favored and demonstrate the classic histopathology triad of (a) cellular bronchiolitis; (b) cellular interstitial pneumonia; and (c) small, poorly formed nonnecrotizing granulomas and/or single multinucleated histiocytic giant cells in up to 80% of patients with clinical diagnoses of HP (148).
The inflammatory process has an uneven bronchiolocentric distribution and also affects the interstitium and alveolar spaces (2,148,149). Thus, focal areas may show patchy interstitial pneumonia along with areas of more confluent, solid inflammatory infiltration that alternate with relatively spared areas (Fig. 25.28). Scattered bronchiolar walls and surrounding alveolar septa are expanded with lymphocytes and variable numbers of plasma cells. Peribronchiolar lymphoid aggregates are commonly seen. Nodules of organizing fibroblasts, histiocytes, and inflammatory cells within the lumen of distal airways result in luminal obliteration. This may dominate the histologic pattern and obscure the other features in some cases (Fig. 25.29). Interstitial inflammation is densest in the peribronchiolar regions and may contain occasional neutrophils or eosinophils. Histiocytes with pink or foamy clear cytoplasm may be seen in small clusters within the interstitium, the presence of which is sometimes an important diagnostic clue (Fig. 25.30). These infiltrates are also associated with at least mild focal interstitial fibrosis and correlate with the CT-noted centrilobular nodules. The granulomas or single multinucleated histiocytes, identified in up to 70% of cases, are typically located in the bronchiolar walls or peribronchiolar interstitium but may extend into alveolar walls or alveolar spaces. These granulomas lack surrounding concentric fibrosis, as seen in sarcoidosis. Cholesterol clefts, Schaumann bodies, asteroid bodies, and/or oxalate crystals may be seen within the histiocyte cytoplasm but have no diagnostic importance.

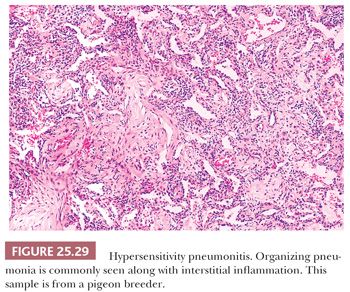
Secondary findings include airway mucostasis, bronchiolar epithelial squamous metaplasia, and scattered foci of organizing pneumonia. Trapped cellular debris is often transformed into collections of foam cells and cholesterol clefts in distal airways and alveoli secondary to bronchiolar obstruction. Intra-alveolar edema and proteinaceous exudates are generally inconspicuous, giving the overall histologic pattern a dry, cellular appearance rather than an exudative one.
Chronic insidious exposure to low levels of sensitizing antigen may lead to diffuse interstitial lung disease, which, in some cases, may progress to interstitial fibrosis and honeycomb lung. The histologic pattern and variability of HP is important to recognize because clinical suspicion may be low in some patients. The correct interpretation of lung biopsy material can lead to the identification of the inciting antigenic agent and the prevention of progressive lung disease.
Radiographically, chronic HP demonstrates reticular opacities attenuated by honeycomb remodeling and volume loss. Thus, the entity overlaps significantly with UIP/IPF and NSIP. The absence of lower lung zone predominance along with lobular areas of decreased attenuation and presence of centrilobular nodules may aid in distinguishing the diseases (150).
Morphologic features seen in typical subacute HP are seen in a large percentage of cases of cHP; however, cHP may also demonstrate UIP-pattern fibrosis, relatively homogeneous linear fibrosis approximating NSIP, or irregular predominantly peribronchiolar fibrosis. A mixture of UIP-patterns with peribronchiolar fibrosis is also reported (48,151). The differential diagnosis is difficult due to secondary changes. One cannot be certain that inflammation or organizing pneumonia is HP-related or a secondary result of underlying parenchymal architectural distortion. Furthermore, most experts agree that granulomatous inflammation dissipates with chronicity; at least 30% of subacute cases do not have demonstrable granulomas (152). Recognizing a single granuloma is important, as it at least suggests the possibility of HP and will remind the clinical team to consider this diagnosis. Chronic vascular changes, including intimal and medial hyperplasia, are seen in cHP as well as virtually all chronic fibrosing interstitial pneumonias.
Together with the history of exposure and pathologic findings, identifying precipitating serum immunoglobulin G (IgG) antibodies to an offending agent are extremely useful in diagnosis (74,84). However, precipitating antibodies by themselves are not diagnostic of HP because many people are sensitized to a variety of agents yet they never develop lung disease (153,154). Thus, the presence of the antibodies cannot be used as an absolute criterion for diagnosis without other accompanying features. On the other hand, the absence of precipitating antibodies may suggest a different disease process.
The morphologic differential diagnosis for both subacute and chronic HP includes many entities depending on the dominant microscopic feature. NSIP, LIP, organizing pneumonia, and COP are considerations when interstitial inflammation overwhelms the bronchiolitis and granulomatous findings. UIP-pattern and NSIP are considerations when fibrosis reigns supreme, whereas bronchiolocentric interstitial pneumonias are difficult to separate from HP when bronchiolitis is prominent (155,156). Granulomatous entities including sarcoidosis, drug-related disease, aspiration pneumonia, and even Pneumocystis pneumonia should be considered. One should arrive at a descriptive diagnosis, mindful of the classic triad of HP features, and discuss the findings with both clinicians and radiologists in order to best serve the patient.
Patient outcomes are varied depending on the antigen and host factors. The prognosis is generally good and the disease may respond well to steroid treatment (142,148,157). Some patients, particularly bird fanciers, have repeated bouts of antigenic exposure that result in chronic pulmonary impairment and honeycomb lung (2). Patients with fibrosis fare poorly (101,158). The best treatment is the identification of the offending antigen and the removal of the agent from the patient’s environment before fibrosis develops. The role of steroid therapy in those with cHP is not defined.
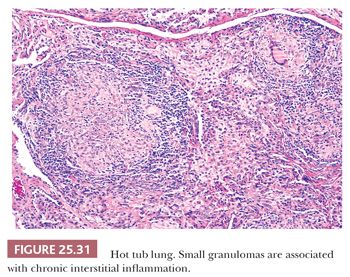
Hot tub lung should be considered in the differential diagnosis of HP. It is associated with Mycobacterium avium-intracellulare complex (MAC), but it is uncertain whether the disease is a hypersensitivity reaction or infection (159,160). Middle-aged individuals with at least a 1-month exposure to a hot tub present with an acute flulike illness with cough and dyspnea. Unlike HP, PFTs are obstructive, whereas CT reveals diffuse centrilobular micronodules, diffuse ground-glass opacities, and lobular air trapping with a lower lobe predominance. A tree-in-bud pattern, indicative of cellular bronchiolitis, may be seen. Tissue samples feature exuberant well-formed nonnecrotizing granulomas with a pronounced bronchiolocentric distribution. Cellular bronchiolitis and interstitial pneumonia are also present. Although the granulomas can be seen in airspaces and in the interstitium, their presence in bronchiolar walls and lumens is particular (Fig. 25.31). Necrotizing granulomas are noted in up to 10% of cases, and up to 25% reveal acid-fast organisms on Ziehl-Neelsen stains (161). Cultures are positive in almost all cases. Hot tub avoidance is usually curative, but some patients are treated with steroids or even antimicrobial drugs (162).
Another condition caused by the toxic effects of large doses of inhaled organic dusts, which is usually seen after the unloading of grain silos, is pulmonary mycotoxicosis (or organic dust syndrome) (163,164). Unlike HP, this condition is not mediated by hypersensitivity but presumably by a direct effect of biologic dusts on the lungs, causing acute bronchopneumonia, DIP, or DAD, which may progress to organization (163,164). Numerous fungal organisms can be seen histologically, and cultures have shown a mixed variety of fungi, including Fusarium and Penicillium. Complete recovery is the rule. The disease is separate from silo-filler’s disease, which is seen in patients after they have filled grain silos; it is caused by the toxic effect of inhaled nitrogen dioxide and produces acute lung injury (165,166).
SARCOIDOSIS
Sarcoidosis is a common multisystemic granulomatous disease of unknown etiology that, in more than 90% of cases, affects the lungs. It is one of the most common conditions for which a lung biopsy is performed (167). An infectious cause has long been sought, and familial and environmental case clusters have been reported (168). Using molecular probes for mycobacteria and human herpesvirus-8 (HHV8), recent studies have shown a positive association in some cases (169–172). One provocative theory suggests that foreign environmental particles, too small to see with ordinary microscopy, are causal in genetically prone individuals (173).
Because of the nonspecific nature of the granulomatous reaction, the diagnosis of sarcoidosis rests with careful assessment of the clinical, radiographic, and pathologic aspects of each case. Clinically, most patients are young, ranging from 20 to 40 years of age, but the disease can occur at any age. It is more common in African Americans. Approximately half of patients are asymptomatic, and the diagnosis is made after a biopsy for an abnormal chest radiograph. Other patients may have an active febrile illness that is marked by severe dyspnea, or they may have uveitis or erythema nodosum. Dyspnea and cough are the most common symptoms of lung disease. Approximately 2% of cases may slowly progress to a fibrotic process that may lead to interstitial fibrosis, honeycomb lung, and death (174).
Radiographically, the pattern is variable. The most common abnormality is hilar adenopathy, which is seen in 50% of cases; 30% show both hilar adenopathy and parenchymal infiltrates and 12% parenchymal infiltrates only. A normal chest radiograph is found in 8% of cases (174). HRCT reveals small nodules in almost all cases (175). Cases with established fibrosis may have evidence of honeycombing with cysts and bullae.
Laboratory tests are sometimes helpful in diagnosis. Approximately 80% of patients with active sarcoidosis have elevated angiotensin-converting enzyme in the serum, but this is not specific because it can be seen in a small percentage of other conditions as well (176). The bronchial lavage fluid contains increased numbers of CD4+ T-helper lymphocytes and reflects increased number of these cells in affected tissue (177). Cutaneous anergy is commonly seen, and the B-cell hyperactivity is reflected by the associated serum polyclonal hypergammaglobulinemia.
Pathologically, sarcoidosis is characterized by lymphangitic nonnecrotizing epithelioid cell granulomas, which, in the lungs, involve primarily the pleura, the connective tissue septa, and the interstitium around pulmonary vessels and bronchi (178) (Fig. 25.32). The granulomas may occur singly or may form nodular collections (Fig. 25.33). Granulomatous angiitis is common (179).
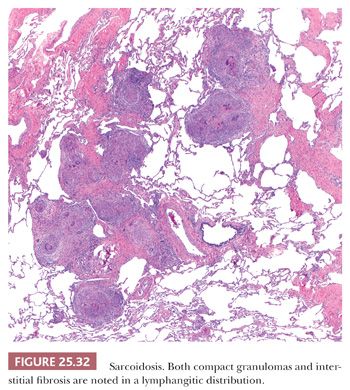
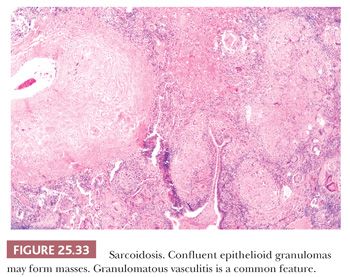
The granulomas are small and are composed of epithelioid histiocytes and multinucleated giant cells that sometimes contain nonspecific birefringent crystalline, asteroid, or Schaumann bodies. The histiocytes are tightly packed and the granulomas discretely marginated, often with a few lymphocytes mixed in (Fig. 25.34). The outer borders often show concentric laminated collagen, giving them an onionskin appearance. Focal necrosis may occur within the center. Sometimes necrosis and, more rarely, cavitation may occur within the larger confluent areas, making differentiation from infection difficult. These cases, termed necrotizing sarcoidal granulomatosis (NSG), are considered a variant form of nodular sarcoidosis by most observers (167,180,181) (see the following discussion). A mild lymphocytic interstitial alveolitis in the alveolar septa may accompany the granulomas in about two-thirds of patients; this is thought to precede the formation of the epithelioid granulomas, and it decreases over time (182). The granulomas themselves may resolve completely or they may form nodular hyaline scars. Severe interstitial fibrosis with cystic blebs may be a result of progressive sarcoidosis, and, in advanced cases, clinical and radiographic correlation is necessary to arrive at a correct diagnosis. The finding of hyalinized granulomas in such cases is extremely helpful.

Unlike its sensitivity for other chronic diffuse interstitial lung disease, transbronchial lung biopsy is highly accurate in making the diagnosis of pulmonary sarcoidosis. However, endobronchial ultrasound-guided transbronchial needle aspiration may preempt the need for endobronchial or transbronchial biopsies (2,183,184). Although the diagnosis is one of exclusion and special stains for infectious agents should be performed in all cases and the tissue should be cultured, finding a characteristic granuloma in the appropriate clinical setting is diagnostic, whereas a nonspecific fibrotic or inflammatory sample should not dissuade the clinician from the diagnosis if the clinical suspicion for sarcoidosis is high. In such cases, a repeat biopsy is indicated.
Nonnecrotizing sarcoid-like granulomas may also be seen in such diverse conditions as lymphoid pulmonary infiltrates, lung tissue adjacent to carcinoma, mycobacterial or fungal infections, vasculitides, and HP. In HP, the granulomas are often composed of more loosely arranged epithelioid cells, and they have a tendency to be associated more with airways than with blood vessels, fibrous septa, or pleura, in contrast to sarcoidosis. Berylliosis is histologically indistinguishable from sarcoidosis, and a history of exposure and mineral analysis of biopsied tissue are required for diagnosis. Difficulties in diagnosis may also occur from transbronchial lung biopsies taken from areas adjacent to tumors because the biopsy may misleadingly sample a granulomatous reaction and not the neoplasm.
The clinical course varies (168). In half of cases, disease resolves spontaneously within 2 years and also does so in many other patients within 5 years. Remission is less likely after 5 years of disease. Twenty percent of patients have permanent clinical symptoms, mainly due to irreversible pulmonary fibrosis. Mortality approaches 8%, mostly due to pulmonary involvement rather than cardiac, central nervous system, or hepatic disease (185,186). There is no known cure, and most drugs target tumor necrosis factor alpha. There are no accepted guidelines for treatment. Steroid treatment for symptomatic disease has short-term benefits, but there is no evidence for long-term effect (168). Cytotoxic agents, along with cytokine modulators and antimicrobial drugs, may target particular organ system disease (187).
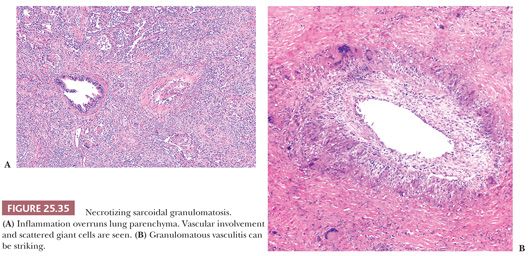
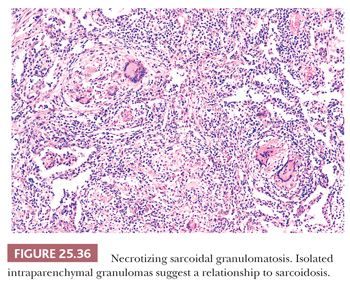
Necrotizing Sarcoidal Granulomatosis
Most regard NSG entity as a form of sarcoidosis. Patients usually present with mild symptoms of low-grade fever, cough, and rarely hemoptysis. The chest radiograph may show a single nodule or multiple bilateral nodules, sometimes with cavitation. Hilar lymph node enlargement is variable (180,181,188).
This variant of sarcoidosis features compact sarcoidal granulomas, which often coalesce and form a large nodular collection of granulomatous inflammation. Central necrosis can be striking. Pulmonary arteries characteristically show giant cell vasculitis (Fig. 25.35). This can take the form of discrete, sarcoid-like granulomas within vessel walls, or it may be similar to the vasculitis seen in giant cell arteritis with radially arranged giant cells without discrete granuloma formation. Both types of vascular lesions may be associated with chronic lymphocytic infiltration, mural fibrosis, and luminal obliteration. The similarity to sarcoidosis is illustrated by some cases that show typical sarcoid-like granulomas in the adjacent interstitial tissue, within the alveoli, or next to the interlobular connective tissue septa and blood vessels of the lung (180) (Fig. 25.36). Other cases that have been described as NSG have a more necrotizing granulomatous appearance, similar to GPA (WG) or infection. The involvement of the hilar lymph nodes by sarcoid-like granulomas is unusual but it has been observed, as have rare examples of uveitis or other forms of extrapulmonary involvement (189–192). NSG, like sarcoidosis, is a diagnosis of exclusion. The pathologist must take care to exclude tuberculosis or fungal infection in each case (180,193). The prognosis is generally excellent and the condition responds well to steroid therapy, or it may be cured by surgery.
PULMONARY LYMPHANGIOLEIOMYOMATOSIS
Lymphangioleiomyomatosis (LAM) is a rare chronic, progressive, and fatal disease considered by some to be a low-grade, destructive, metastasizing neoplasm (194). This condition is characterized by a peculiar and histologically unique pattern of interstitial smooth muscle proliferation in the lung that, qualitatively, bears close similarity to that seen in angiomyolipoma of the kidney. It may be associated with the full expression of tuberous sclerosis or with similar smooth muscle proliferations of the abdominal or thoracic lymphatic ducts and lymph nodes in combination with renal angiomyolipomas, or it may occur only in the lung (195–198). Molecular genetic studies demonstrate similar allelic losses, most often in the TSC1 and TSC2 genes in patients with tuberous sclerosis complex (TSC) and isolated pulmonary LAM. This evidence confirms the relationship between these conditions (199–201). Mutations in the TSC2 tumor suppressor gene result in dysregulation of the mammalian target of rapamycin (mTOR) pathway. Only 1% to 3% of patients with TSC have full-blown pulmonary LAM, but HRCT scans have shown that more than 20% of TSC patients have some evidence of subclinical lung disease (202,203). Unlike tuberous sclerosis, pulmonary LAM occurs almost exclusively in women who are usually in their childbearing years, although examples in both children and men, all with TSC, have also been reported (204,205).
Shortness of breath, dyspnea on exertion, cough, and repeated pneumothorax are the most common presentations as well as the predominant cause of disability. Hemoptysis and chylous pleural effusions also occur. Characteristically, PFTs disclose increased total lung capacity and increased residual volume due to air trapping also with reduced diffusion capacity and flow rates.
Radiographically, the chest film may be normal. More commonly, a pattern of interstitial fibrosis or reticulonodular densities is seen, sometimes with cysts and blebs. Unlike the more common interstitial processes, however, the lungs may appear expanded or normal in size rather than shrunken. This results from the smooth muscle proliferation around the bronchioles, which leads to air trapping. HRCT scans show numerous thin-walled cysts throughout the lungs. These can be seen even in patients with otherwise normal chest radiographs (206).
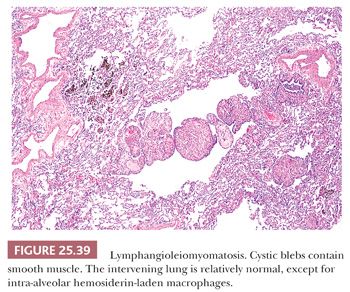
Pathologically, a patchy and haphazard lacy pattern of round and spindled smooth muscle proliferation within the interstitium and around the bronchi, bronchioles, veins, and lymphatics is seen. These appear to spin off the muscle coat of these structures and extend into the walls of adjacent alveolar ducts and alveoli in a whorled pattern that gives this process its unique histologic appearance (Fig. 25.37). The muscle cells themselves often contain optically clear cytoplasm and abundant intracytoplasmic glycogen (Fig. 25.38). Air trapping and overdistention occurs as the result of bronchiolar obstruction, which may lead to cystic blebs and pneumothorax (Fig. 25.39). Localized pulmonary hemorrhage and hemoptysis may develop secondary to venous obstruction, whereas lymphatic obstruction may lead to lymphatic cysts, which may rupture into the pleural space, causing chylous pleural effusion.
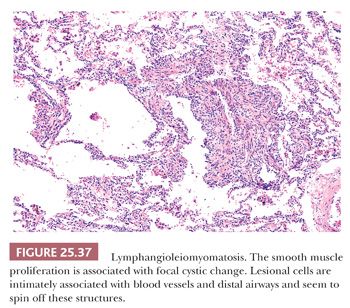
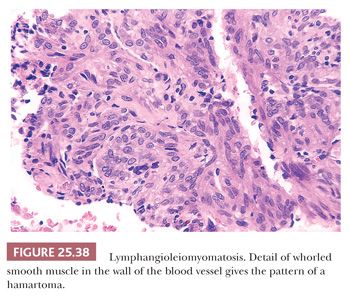
The smooth muscle stains positively with melanoma antigen HMB45, a feature shared with both angiomyolipoma and clear cell tumor of the lung and also TSC-associated lesions (207,208). Larger lesional cells are more likely to be estrogen and progesterone receptor–positive than the smaller cells. A diagnosis can be made on transbronchial samples if the clinicoradiographic findings support the light microscopic impression including HMB45 positivity of lesional cells.
LAM must be distinguished from benign metastasizing leiomyomas of lung. This condition is also seen predominantly in female patients and is also hormonally dependent; however, this disease has a much better prognosis. Histologically, the smooth muscle proliferation is composed of single or multiple nodules that are localized rather than diffuse. Cysts and blebs are usually not a feature in this disorder. Most authors regard the entity as well-differentiated leiomyosarcoma that has metastasized to the lung, usually from a “benign” tumor of the uterus resected long ago (209,210). Similar to LAM, estrogen receptors have been demonstrated (211).
Rare cases of metastatic endometrial stromal sarcoma may become cystic, causing pneumothorax, and these must be differentiated from LAM (212). In contrast to LAM, this is HMB45-negative and often positive with CD10.
Chronic PLCH, emphysema, and UIP pattern may also focally resemble LAM. The former features predominantly upper lobe stellate nodular scars and may feature Langerhans cell–rich lesions. Emphysema cysts are typically thin and acellular. UIP pattern fibrosis differs in that the cystic structures and smooth muscle hyperplasia are associated with other features of early honeycomb lung and are generally confined to the subpleural lung regions.
Finally, LAM must be differentiated from diffuse pulmonary lymphangiomatosis (213). This condition can be seen in men, women, and children. The symptoms most commonly are wheezing and shortness of breath. Pathologically, the lesion is characterized by prominent lymphatic vessels in the pleura and connective tissue septa accompanied by variable degrees of oftentimes kaposiform smooth muscle proliferation. Chylothorax may occur, but cysts, pneumothorax, and HMB45 positivity are not seen. The disease may be progressive and may lead to death.
LAM is progressive. Death may occur within 1 year or the disease may be more protracted, with greater than 10-year survival. Ten-year survival rates range from 30% to 90% (214,215). Progressive airflow restriction leads to respiratory failure and cor pulmonale. LAM may contain estrogen and progesterone receptors, and some patients respond to antiestrogen therapy (216). A LAM histology score grades the extent of replacement of normal lung tissue by both cystic lesions and lesional cells (214). Semiquantitative grading suggests differences in survival and time to transplantation. Serum vascular endothelial growth factor D (VEGF-D) concentration correlates with disease severity and treatment response to mTOR inhibitor sirolimus (217–219). Heart-lung transplantation is performed (220). Interestingly, disease recurrence in lung allografts originates from the transplant recipient rather than from the donor lung (221).
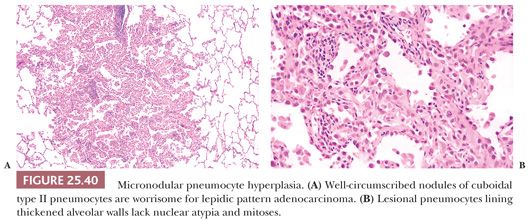
Micronodular pneumocyte hyperplasia (MPH) is another lung lesion seen in up to half of TSC patients with or without LAM. They are most common in premenopausal women. These discrete usually less than 0.5-cm well-demarcated nodules featured crowded alveolar pneumocytes lining slightly thickened alveolar walls. The pneumocytes have abundant eosinophilic cytoplasm and vesicular nuclei with smooth nuclear contours and rare eosinophilic inclusions. Intra-alveolar macrophages impart a solid appearance to this lesion at low magnification. (222–225) (Fig. 25.40). Unlike LAM, hormone receptors and HMB45 immunohistochemical stains are negative. Most examples of MPH do not appear to be progressive, although the clinical course is often dominated by coexisting LAM. Rare cases of MPH may lead to respiratory failure (226).
AMYLOIDOSIS
Pulmonary amyloidosis can take the form of localized primary tracheobronchial involvement, nodular parenchymal masses, or diffuse interstitial septal amyloidosis that accompanies primary amyloidosis or paraproteinemia secondary to plasma cell myeloma or Waldenström macroglobulinemia (light chain–derived [AL] amyloid). It is also seen as part of secondary amyloidosis (protein A–derived [AA] amyloid) (227–230) (Table 25.11).

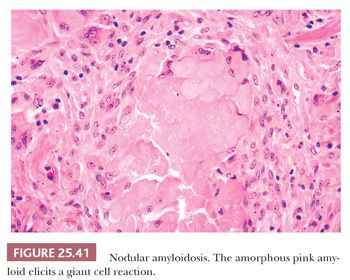
The most common form is nodular intraparenchymal amyloidosis. Such patients are usually elderly and asymptomatic, and they have no evidence of paraproteinemia or systemic disease. They come to medical attention because of an abnormal chest radiograph that shows single or multiple tumorlike masses (231). Microscopically, glassy pink collections of amyloid form nodular masses of 1 to several centimeters. Foreign body giant cells may appear to phagocytose the amyloid material (Fig. 25.41). Ossification and calcification of the amyloid may occasionally be seen. The amyloid exhibits the tinctorial quality of apple green, Congo red birefringence with polarized microscopy and has the ultrastructural qualities of amyloid fibrils. In some cases, plasma cells may show clonal restriction of light chains, suggesting low-grade immunoproliferation (231).
Tracheobronchial amyloidosis affects the submucosa of the airways as diffuse narrowing or nodular single or multiple tumor like collections (230). The patients may present with hemoptysis, obstructive pneumonitis, atelectasis, or asthma-like symptoms that are related to bronchial obstruction. The diagnosis is usually made from bronchus biopsy material. Death may result from massive bleeding (232,233).
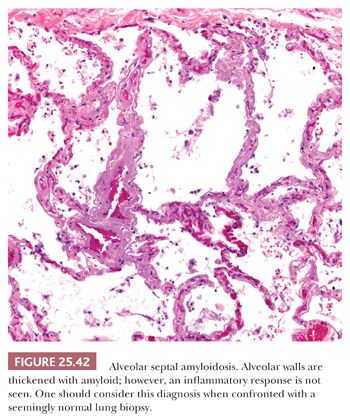
Diffuse septal amyloidosis predominantly involves the alveolar septa as diffuse interstitial deposits that may lead to physiologic abnormalities identical to those seen in advanced UIP or other forms of diffuse lung fibrosis (Fig. 25.42). The pulmonary arteries are also infiltrated, and, rarely, pulmonary hypertension may result. This condition is most commonly associated with generalized systemic amyloidosis. Cardiac involvement with congestive heart failure and pulmonary edema may confuse the clinical picture, which may mimic primary heart disease. The survival rate is poor and no effective treatment exists. Histologic differentiation of diffuse septal amyloidosis from either fibrin or interstitial fibrosis is usually not a problem. Rare cases of light chain deposition disease have shown Congo red–negative granular deposits with the light microscope appearance of amyloid. Such cases may sometimes be associated with true amyloid (234,235).
PULMONARY ALVEOLAR PROTEINOSIS
Since its first description, the pathogenesis of pulmonary alveolar proteinosis (PAP) has remained unknown. However, multiple associated factors have been described (236–238). Some cases have been observed following exposure to inorganic dusts such as silica, aluminum, and fiberglass, among others, and are regarded as a secondary phenomenon (239–241). Areas identical to idiopathic PAP are seen around tuberculous lesions or in patients with immunosuppression or leukemia (242–244). The pathologic changes likely reflect an imbalance between the production of surfactant by alveolar type II cells and the inability of defective lung macrophages to remove this material, which results from the presence of antibodies to granulocyte-macrophage colony stimulating factor (GM-CSF) (245). GM-CSF is found in both serum and lavage fluid of PAP patients, which inhibits macrophage function (246).
PAP is seen in adults of all ages, but infants and children may also be affected (247). The majority of patients are cigarette smokers (248). Clinically, shortness of breath, cough, sputum production, and fever are observed. Radiographically, a predominantly bilateral perihilar and bibasilar infiltration that often resembles pulmonary edema is seen on the chest radiograph. Unilateral disease has, however, also been seen. Occasional patients may have a normal chest radiograph. HRCT reveals bilateral ground-glass and abnormal interlobar septal thickening (238).
Pathologically, the alveoli are filled with homogeneous pink-red, PAS-positive, granular material with cholesterol clefts as well as with the ghost outlines of degenerated macrophages and alveolar type II cells (Fig. 25.43). The alveolar septa are normal except for occasional areas of mild lymphocytic infiltration. Alveolar type II cells may be prominent in areas and sometimes show foamy cytoplasm (Fig. 25.44). Ultrastructurally, the intra-alveolar exudate is seen to contain lamellar bodies in various stages of dissolution, along with cellular debris and lipid particles. The intra-alveolar exudate has been demonstrated to be rich in surfactant (249).
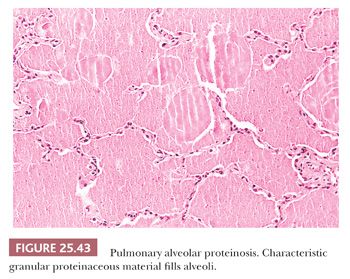
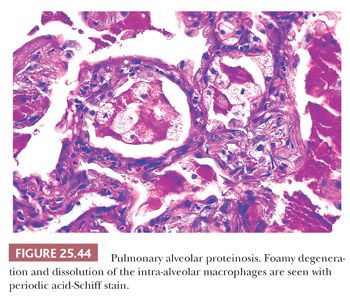
This condition represents yet another type of pulmonary reaction pattern that is not actually a specific disease. In most clinical cases, the etiology is unknown. The proteinaceous material is an excellent culture medium for microorganisms, and infection with such organisms as Nocardia, mycobacteria, Aspergillus, or others may complicate the clinical course. The frothy intra-alveolar exudates of P. jiroveci may closely resemble PAP, and special stains should be conducted in all cases. Since the introduction of bronchoalveolar lavage as treatment, few cases progress to honeycomb lung (250). Lavage is effective and it may be repeated if relapse occurs. Steroids should not be administered (236,250).
PULMONARY EOSINOPHILIC REACTIONS
The pulmonary eosinophilias are presumed hypersensitivity reactions characterized by tissue and usually blood eosinophilia. The inciting agents are most commonly fungi, parasites, or drugs, or they may be unknown (251). Clinical syndromes include asthma, tropical pulmonary eosinophilia syndrome, Löffler syndrome, acute eosinophilic pneumonia, chronic eosinophilic pneumonia (CEP), and allergic bronchopulmonary fungal disease (252). EGPA (CSS) is considered a systemic vasculitis and is discussed in that section later in this chapter.
ASTHMA
Asthma is a very common lung disease characterized by acute, episodic paroxysmal and usually reversible airway narrowing. More than 300 million humans are affected, but they may not all have the same disease (253). Atopic and nonatopic forms, although not mutually exclusive, are recognized (254,255). Genetic and environmental factors are implicated in both types. Chest radiographs are usually normal, whereas HRCT may demonstrate bronchial wall thickening, mucoid impaction, centrilobular opacities, and/or bronchial dilation (256). Morphologic features of asthma, first described in autopsy series, are recognizable in bronchoscopic and surgical specimens.
Atelectasis, hyperinflation, bronchiectasis, and mucus plugs are classic macroscopic findings, but airway abnormalities can also be appreciated, even in small biopsy samples. Bronchial and bronchiolar lumens contain mucin admixed with sloughed bronchial epithelium, inflammatory cells, and elongated bipyramidal needle-shaped (Charcot-Leyden) crystals. The eosinophils are scattered throughout the mucin and not layered, and small airways may contain mucous casts (Curschmann spirals). Basement membrane may be exposed, although it is not known whether the sloughed epithelium is actual or artifactual. Goblet cell metaplasia and enlarged seromucinous glands in larger airways are seen. The basement membrane of larger airways is appreciably thickened and bronchial and bronchiolar smooth muscle is hypertrophied (257) (Fig. 25.45). Airway walls, especially the subepithelial compartment, are expanded by eosinophils, plasma cells, lymphocytes (particularly CD8+ T cells), mast cells, occasional neutrophils, and dilated blood vessels. Nonatopic asthma does not feature as many eosinophils (254). Regardless of the types of inflammatory cells, submucosal thickening leads to airway narrowing and increased airway resistance.
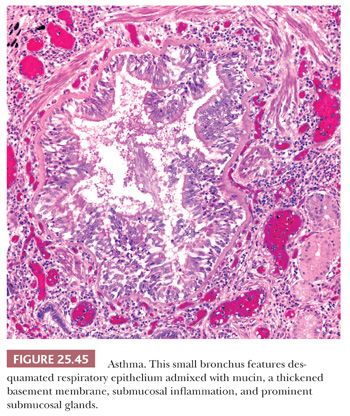
The morphologic differential diagnosis is essentially limited to chronic bronchitis and RB. However, prominent airway eosinophils and marked smooth muscle hyperplasia, although not absolute, are more often seen in asthma. RB may feature bronchiolectasis and intraluminal mucin, but thickened basement membranes and eosinophils are not conspicuous. Treatment relies on bronchodilators and long-term suppression of airway inflammation.
TROPICAL PULMONARY EOSINOPHILIA
Tropical pulmonary eosinophilia syndrome is caused by nematodes that inhabit the lymphatics and subcutaneous tissues. Wuchereria bancrofti and Brugia malayi are most often implicated. Adult worms release larval microfilariae into the host’s bloodstream at night and cause an immediate type of eosinophil-rich hypersensitivity reaction. Patients are awakened by coughing attacks, wheezing, fever, and fatigue. Blood eosinophil counts may be greater than 50,000/μL, and total blood IgE concentrations are high. The disease is rarely seen outside of tropical regions. Lung biopsy demonstrates nodular eosinophilic bronchopneumonia that develops into a fibrosing granulomatous infiltrate. One may identify rare degenerating microfilaria within the pulmonary capillaries (258). In the absence of microfilariae, the differential diagnosis includes other eosinophilic syndromes and vasculitis including EGPA (CSS) (see the following discussion).
LÖFFLER SYNDROME (SIMPLE PULMONARY EOSINOPHILIA)
Löffler syndrome is a transient febrile illness marked by mild respiratory symptoms and fever along with peripheral blood eosinophilia and pulmonary infiltrates on chest x-ray. It is thought due to Ascaris infection with larva passing through the lungs (259). Transitory peripheral airspace consolidation and ground-glass attenuation are described on CT. Lung biopsy usually is not necessary for diagnosis but shows interstitial and intra-alveolar eosinophils (Fig. 25.46). The process responds to corticosteroids; however, treatment is usually not necessary due to its self-limiting course (260).
ACUTE EOSINOPHILIC PNEUMONIA
Acute eosinophilic pneumonia, first described in 1986, is rare (261,262). In addition to the idiopathic form, numerous associations and causes are recognized, including but not limited to working with mulch, firework smoke exposure, tobacco smoke, drug exposures, and World Trade Center dust exposure. Nonasthmatic noninfected patients, both young and old, present with acute hypoxic respiratory failure along with rapid onset of fever, chills, and myalgias of less than 1-month duration. Paradoxically, peripheral eosinophilia is usually lacking at presentation but generally develops during the illness. Bronchoalveolar lavage cell counts contain greater than 25% eosinophils. CT shows diffuse bilateral irregular airspace consolidations and bilateral pleural effusions. The histologic picture features acute and organizing DAD with interstitial and lesser numbers of intra-alveolar eosinophils. Hyaline membranes may be a bit more eosinophilic than those seen in typical DAD and eosinophilic vasculitis is absent (Fig. 25.47). Although typical DAD may feature scattered eosinophils, any case with interstitial or intra-alveolar groups of 5 to 10 eosinophils should raise the possibility of acute eosinophilic pneumonia. Hyaline membranes exclude CEP from consideration. Transbronchial biopsy samples may not be representative; however, clinical history should help separate the entities. Acute eosinophilic pneumonia responds well to steroids, whereas DAD does not, and a relapse-free course is typical (263,264).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


