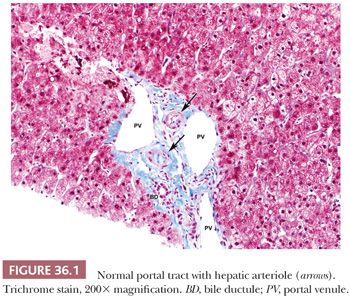
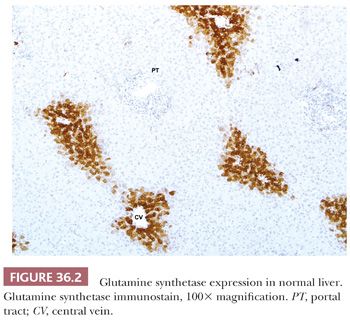
Blood draining from the gastrointestinal (GI) tract flows through the portal veins and eventually into terminal portal venules and then sinusoids, where it mixes with arterial blood (which enters the sinusoids from arteriosinusoidal twigs branching off of the approximately 10 microns in diameter terminal hepatic arteriole) (11). The hepatic arteriole represents a key structure in identification of portal tracts by light microscopy (Fig. 36.1). The complex architecture of the liver lobule leads to structural and functional heterogeneity, such as variable density of fenestration (12) and central ammonium metabolism with associated glutamine synthetase expression (Fig. 36.2) (13). Even the extracellular matrix components of the space of Disse may vary from periportal to pericentral regions (14).
Intrahepatic biliary epithelium is thought to develop from limiting plate hepatoblasts (15) through formation of a ductal plate, which envelops the mesenchyme of early portal tracts. Duct anastomosis progresses from the extrahepatic biliary structures in the porta hepatitis to the ramifying smaller intrahepatic septal ducts, in association with portal vein and hepatic artery branches, which eventually form interlobular bile ducts that drain bile from hepatocytes through the canals of Hering and associated bile ductules. This process is often not complete at birth and contributes to physiologic jaundice (16). Formation of the biliary tree normally involves dismantling of the ductal plate, a physiologic process that may be disrupted/damaged in intrahepatic biliary atresia (17). Liver is the dominant site for hematopoiesis from about 12 weeks’ to 5 months’ gestation (followed by the bone marrow), but hematopoiesis is usually no longer detectable outside the neonatal period.
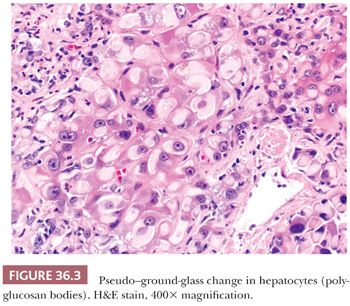
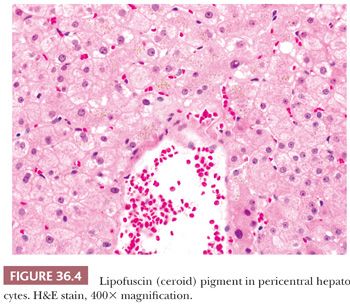
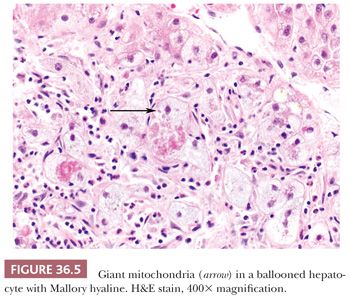
Hepatocytes are not only interconnected through both tight and gap junctions along their lateral membrane (18), but also interfaced with the perisinusoidal space of Disse (without an intervening basement membrane) and a specialized canalicular space. At the limiting plate, the space between hepatocytes and periportal connective tissue is termed the space of Mall, which is in continuity with the space of Disse. The bile canaliculus forms from adjacent hepatocyte canalicular membranes (diameter 1 to 2.5 microns); like the sinusoidal surface, it is covered by microvilli. Synthesis of glycogen represents a key function of the hepatocyte, which also coordinates glycogen breakdown and release as glucose. Smooth endoplasmic reticulum (ER) uses glucoronyltransferase to convert bilirubin to a water-soluble form and uses the p450 system to eliminate toxins in bile (19). Smooth ER may be prominent in some drug-induced liver injury and appear as pseudo–ground-glass change (also termed polyglucosan bodies) (Fig. 36.3), which may be mistaken for hepatitis B cytoplasmic inclusions. A minority of hepatocytes are normally binucleate (20). Mitotic activity is very low and usually no mitotic figures are present on a normal core biopsy. Lipofuscin, iron (i.e., ferritin), and copper (as in cholestasis) may accumulate in lysosomes (Fig. 36.4). Mitochondria account for a substantial portion of hepatocyte cytoplasmic volume and may be visible on light microscopy as giant mitochondria in a variety of settings (e.g., steatohepatitis, Wilson disease, glycogenic hepatopathy) (Fig. 36.5). Hepatocytes, like many other epithelial cells, contain two predominant cytokeratins (CKs), CK8 and CK18, in contrast to biliary epithelial cells, which express CK7 and CK19 (21, 22), although hepatocytes may also express CK7 in the setting of cholestasis (see Section IV). Intermediate filaments are also present and are typically not seen on light microscopy, unless there is network collapse and Mallory hyaline formation.
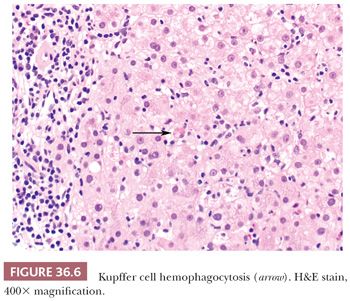
Additional cell types include the hepatic stellate cells (HSCs) (i.e., Ito cells), which contain fat droplets, store vitamin A, and play a role in inflammation, regeneration, and fibrosis (23–27). They are only observed on light microscopy in the setting of Ito cell hyperplasia or lipidosis (e.g., as may be due to hypervitaminosis A). HSC may express glial fibrillary acidic protein (GFAP) and alpha smooth muscle actin when activated (28). Kupffer cells are specialized macrophages with diverse functions that modulate cytokine networks in the liver (29). Kupffer cells are likely derived from both bone marrow–derived monocytes (30) and precursor cells in the yolk sac (31) and have substantial phagocytic activity, which may be readily observed in the setting of hemophagocytosis (Fig. 36.6) (as may be seen due to infection or lymphoma). A substantial normal compliment of lymphocytes (mostly T cells, natural killer [NK] cells, and NK T cells) reside in the liver (32) and likely play a major role in innate and adaptive immunity (33,34).
BASIC PRINCIPLES OF NONNEOPLASTIC LIVER DISEASE
Direct toxic effects, immune response to foreign or self-antigen, ischemia, metabolic disturbance, or bile duct obstruction may all injure the liver. Hepatic response to injury can lead to regeneration, which can return the liver to its normal structure and function, even after extensive injury, or, in the case of ongoing injury, the hepatic response may result in fibrosis, which may progress to cirrhosis with its associated vascular flow changes and systemic effects.
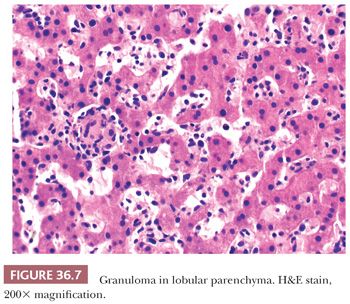
As with inflammation elsewhere in the body, a network of cytokine signaling plays a key role in direction of an immune response within the liver, with some cytokines generally accepted to promote a cell-mediated immune response (e.g., Th1 cytokines such as tumor necrosis factor-α and interferon-γ) and others with a role in amplifying the humoral response (e.g., Th2 cytokines such as interleukin [IL]-10 and IL-4), which may serve to downregulate an overall inflammatory response. Under direction of proinflammatory cytokines, activated macrophages may aggregate to form a granuloma (Fig. 36.7), which is thus a relatively nonspecific finding. A subset of T cells (i.e., regulatory T cells) clearly plays a role in inhibiting cytotoxic T cells and thus protect against autoimmune disease in the liver (35). Recruited lymphocytes may travel to the site of hepatocyte injury in the lobule, but with chronic infection, for example, will also aggregate in portal regions. Neutrophils may extravasate directly through periductal capillaries in response to biliary damage (36–39).
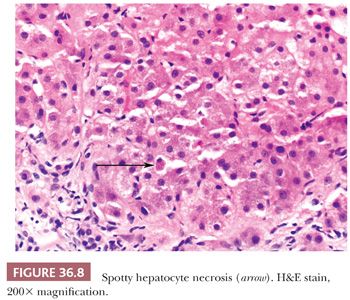
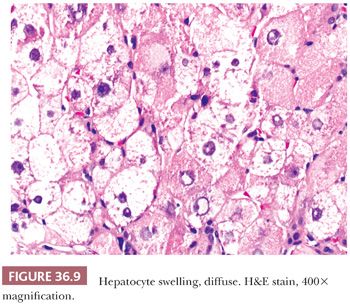
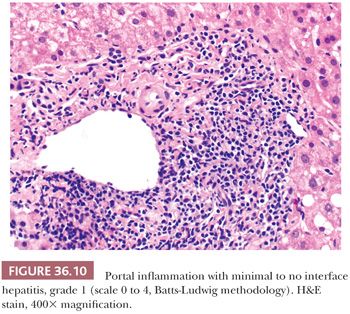
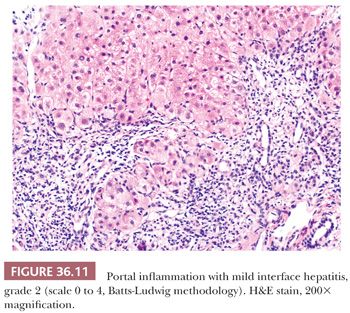
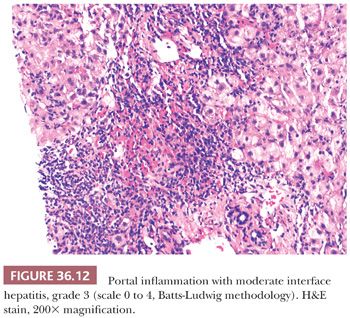
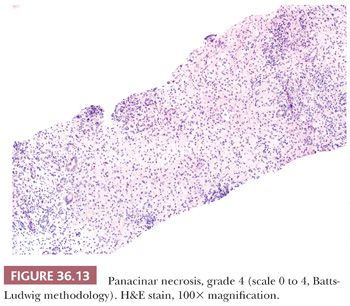
Liver injury most obviously manifests as morphologic evidence of hepatocyte death (Fig. 36.8). Hepatocyte death may occur through multiple pathways (ischemic/toxic, necrosis, apoptosis, autophagy, etc.), but isolated damaged/dying hepatocytes will show overlapping morphologic features and thus related terms, such as apoptosis, spotty necrosis, and acidophil bodies, are used interchangeably. Hepatocyte swelling (Fig. 36.9) may be seen in a variety of settings, but the term ballooning specifically refers to hepatocyte injury in the setting of steatohepatitis and has three morphologic features: (a) large swollen hepatocyte, (b) rarefaction of cytoplasm, and (c) clumping of intermediate filaments into eosinophilic globular areas, which may form Mallory hyaline (also referred to as Mallory-Denk bodies) with loss of CK8/18 (40). A variety of systems are available for grading inflammation/liver injury (Table 36.2), with the Batts-Ludwig system being one of the most commonly used schemes (grade 1, Fig. 36.10; grade 2, Fig. 36.11; grade 3, Fig. 36.12; grade 4, Fig. 36.13).
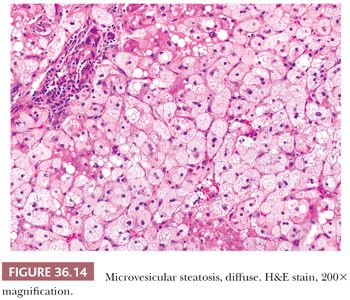
Many metabolic derivatives may accumulate in hepatocytes, but fat droplets, or steatosis, are particularly common in the United States, as this finding is the diagnostic feature of fatty liver disease (discussed in the section “Fatty Liver Disease”). Steatosis may be separated into microvesicular (Fig. 36.14) and macrovesicular forms. Microvesicular steatosis is characterized by very small/fine fat globules and is a result of mitochondrial injury. Macrovesicular steatosis may show multiple fat droplets that do not displace the nucleus (small droplet steatosis) or coalescent fat vacuoles that displace the nucleus (large droplet steatosis). In older descriptions, small droplet steatosis has been classified as microvesicular steatosis; however, based on morphology and pathogenesis, it is best considered as a form of macrovesicular steatosis.
Cholestasis refers to deficient secretion of bile constituents, which may occur from bile duct obstruction or hepatocellular injury and which may manifest with a variety of morphologic changes. Bile may accumulate in hepatocytes, or in canaliculi, and hepatocytes may show swelling and degenerative changes. Bile leaks may form large destructive lakes/infarcts. Mallory hyaline may form in periportal hepatocytes, which along with swelling in this zone has been termed cholate stasis. Portal tracts may show edema and ductular reaction (41). The latter term encompasses both an increase in the number of well-formed duct lumina in a portal tract as well as any apparent metaplastic change of limiting plate hepatocytes into smaller ductular structures. Inspissated bile may be seen, particularly in sepsis-associated bile duct obstruction (i.e., cholangiolar cholestasis).
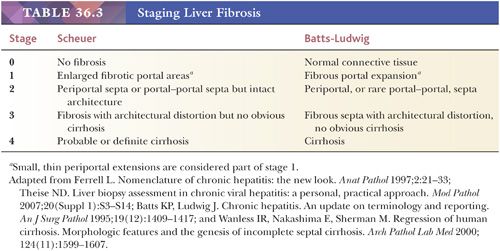
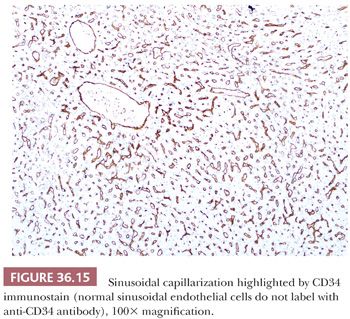
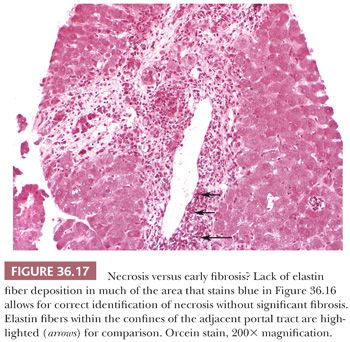
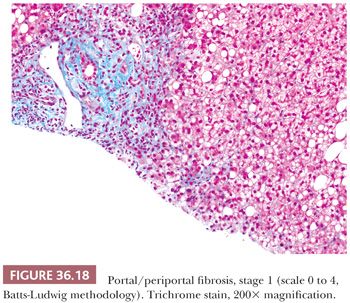
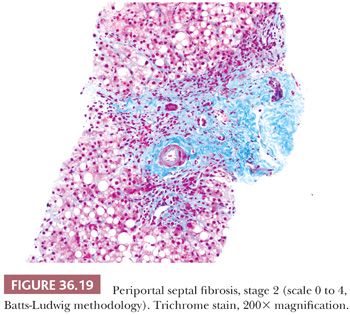
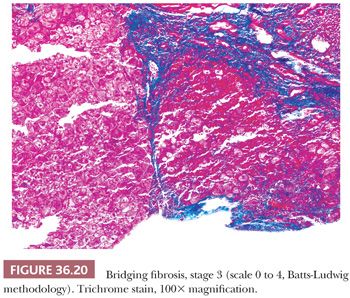
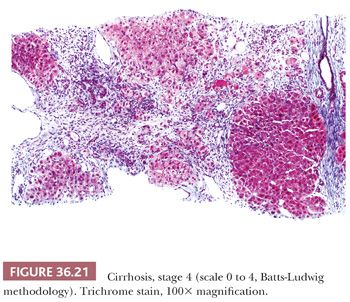
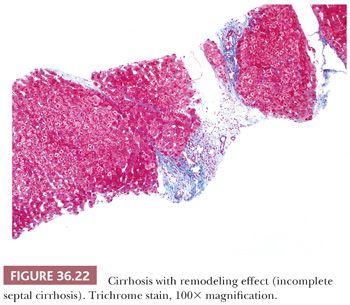
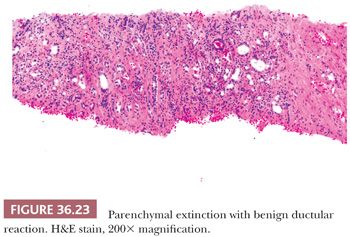
Although some injury may be completely resolved through progenitor cell–driven regeneration, other injuries result in chronic long-lasting changes, namely fibrosis. Collagen fibers (including types I, III, and IV) collect at sites of chronic injury, most likely as a result of activated HSCs (42–44). Fibrosis affects blood flow and leads to sinusoidal capillarization (which can be highlighted on CD34 immunohistochemical staining) (Fig. 36.15) (45,46). Elastin deposition typically precedes collagen fiber deposition (47) and thus can be used as a marker of early fibrosis and aids in distinguishing necrosis from fibrosis on trichrome stain (Figs. 36.16 and 36.17). Several systems for staging liver fibrosis (48) have been developed (Table 36.3); Brunt-Kleiner methodology is most commonly used for steatohepatitis (49) (see later discussion in the section “Fatty Liver Disease”), whereas several systems such as Scheuer, Batts-Ludwig, Metavir, and Ishak methodologies are used for chronic viral hepatitis (50) (portal/periportal fibrosis, stage 1, Fig. 36.18; periportal/incomplete septal fibrosis, stage 2, Fig. 36.19; bridging fibrosis, stage 3, Fig. 36.20; cirrhosis, stage 4, Fig. 36.21). In the commonly used Batts-Ludwig methodology, fibrosis progresses from stage 1 “portal fibrosis” (similar to “enlarged fibrotic portal areas” in Scheuer methodology) to stage 2 “periportal fibrosis” (similar to “periportal or portal–portal septa” formation in Scheuer methodology) and then bridging septal fibrosis (stage 3) and cirrhosis (stage 4). Thin periportal fibrous extensions are considered part of stage 1 fibrosis, whereas better developed incomplete fibrous septa are classified as stage 2 (48,51,52). The result of long-standing chronic injury is cirrhosis, which is defined by diffuse involvement and regenerative nodules isolated by fibrous septa. Fibrous bands may span portal–portal, portal–central, or central–central regions and nodule size may be uniformly small, less than 3 mm (micronodular), or variable (macronodular). Advanced fibrosis, including cirrhosis, can be remodeled and even reversed in some scenarios in which the underlying etiology of hepatocellular injury has been eliminated (so-called incomplete septal cirrhosis) (Fig. 36.22) (53,54). Coincident with the architectural changes of advanced fibrosis, liver vasculature undergoes remodeling and shunting, which is likely to have more impact on liver function than fibrosis itself. Sclerosis around vessels and in the space of Disse leads to vascular shunts, which along with sinusoidal capillarization, allows hepatic arterial blood to bypass parenchyma on its way to the hepatic vein (39). Vascular obstruction in cirrhosis can lead to large zones of parenchymal loss, termed parenchymal extinction, which may form a mass-like lesion on imaging. Parenchymal extinction commonly has extensive ductular reaction, which may mimic a biliary neoplasm (Fig. 36.23).
ACUTE HEPATITIS, AUTOIMMUNE HEPATITIS, AND INFECTIOUS LIVER DISEASE
ACUTE HEPATITIS
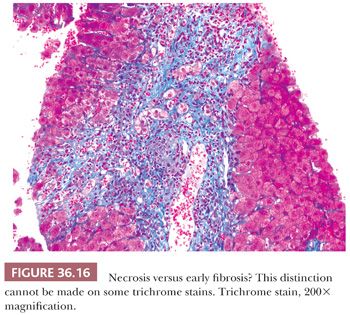
Acute hepatitis is defined clinically as at least a doubling of alanine transaminase (ALT) or aspartate transaminase (AST) (without chronic liver disease history). Alkaline phosphatase (ALP) is either normal or ALT/ALP is increased (≥5). By definition, acute hepatitis is characterized by lobular hepatocellular injury, which manifests as hepatocellular swelling, apoptosis, or dropout. Lobular injury may be random or zonal but in severe cases will form confluent clusters of apoptotic cells or bridge between zones. Bridging necrosis denotes a higher risk of permanent injury in the form of fibrosis. In its most severe presentation, acute hepatitis may lead to acute liver failure manifested by widespread confluent or panacinar necrosis. Massive necrosis refers to loss of almost all hepatocytes (~60% to 70% loss), whereas submassive necrosis (30% to 70%) has more viable hepatocytes, often with periportal sparing. This distinction cannot be made on core liver biopsy, as necrosis is variable throughout the liver, although it is helpful to indicate the extent of injury (e.g., focal, confluent, bridging, panacinar). As the acute phase of liver injury may last up to 6 months, regenerative features are commonly encountered (e.g., numerous binucleate hepatocytes, mitoses, thick plates, prominent Kupffer cells) and active injury may be minimal, especially in a resolving phase. It is important to note that cholestasis (and occasionally ductular reaction) may be present in acute hepatitis (so-called “cholestatic hepatitis”), if bile secretion is inhibited, and thus is not indicative of a chronic biliary obstructive process in this setting. Lobular inflammation is commonly encountered, typically lymphocytic, and may be a predominant feature; bile ducts are usually intact, allowing for exclusion of chronic biliary disease. Portal inflammation is variable. Multinucleated hepatocytes (giant cells or syncytial cells) may be present. By definition, there is no fibrosis; however, distinction between necrosis and fibrosis represents a common diagnostic dilemma. Areas of necrosis show pale staining on trichrome stain as compared to dense coarse collagen in the portal tracts and fibrous septa, but this distinction can be challenging on a poorly stained trichrome stain (Fig. 36.16). Orcein, or other elastic stains, may be used to demonstrate early elastin fiber deposition as a first manifestation of established fibrosis (Fig. 36.17).
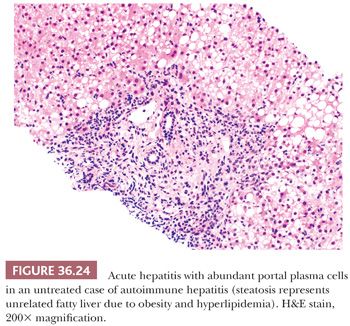
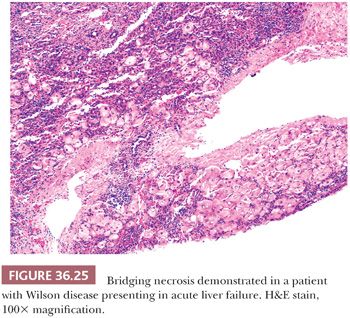
Acute hepatitis can be divided into six morphologic subpatterns, each with its distinct morphologic features and differential diagnoses (Table 36.4). The inflammation-predominant pattern is the most common and shows hepatocellular injury accompanied by a prominent inflammatory infiltrate. The differential diagnosis includes acute viral hepatitis (AVH), drug-induced liver injury (DILI), autoimmune hepatitis (AIH), Wilson disease (WD), and celiac disease (CD). Serology is not reliable in excluding AVH; instead, nucleic acid testing (typically with polymerase chain reaction [PCR]–based assays) could be considered to allow for window period detection. DILI has variable histologic manifestations but may have prominent eosinophils, granulomas, centrizonal necrosis, and cholestasis out of proportion to lobular activity. Careful clinical evaluation of drug/herbal history and correlation with temporal profile of disease onset is necessary. Transaminitis typically subsides with cessation of the offending agent but may persist for months. Histologic and clinical findings for specific drugs are summarized and updated on a website maintained by the National Institutes of Health (NIH) (www.livertox.nih.gov). Most cases of AIH will have fibrosis, but up to 10% lack fibrosis at initial presentation. High necroinflammatory activity, interface inflammation, hepatocyte rosette formation, and abundant plasma cells (Fig. 36.24) are the most suggestive histologic features, but these findings may be less prominent at various phases of disease or with therapy. Elevated serum immunoglobulin G (IgG) and demonstration of autoantibodies (antinuclear, smooth muscle, liver-kidney microsomal, soluble liver antigen) are helpful. Definitive diagnosis is based on a scoring system proposed by the International Autoimmune Hepatitis Group (55). In addition to steatosis, chronic hepatitis, and cirrhosis, WD can present with acute hepatitis and liver failure (Fig. 36.25) (described in more detail in the section “Metabolic, Genetic, and Developmental Disorders”). CD commonly is associated with a mild transaminitis, which may normalize after a gluten-free diet and shows mild nonspecific reactive hepatitis on biopsy. Rare instances of acute hepatitis have been reported in CD, and liver involvement can also manifest as chronic hepatitis or nodular regenerative hyperplasia (NRH). Serologic testing for CD should be considered in all cases of unexplained liver dysfunction. Inflammation-predominant hepatitis can show zonation and, when injury is confined to the centrizonal region, special consideration should be given to AIH and DILI, specifically drugs such as minocycline, nitrofurantoin, and methyldopa. Idiosyncratic reactions to isoniazid, monoamine oxidase (MAO) inhibitors, phenytoin, valproate, and various antibacterial (e.g., sulfonamides) and antifungal agents (e.g., ketoconazole) may result in inflammation-predominant acute hepatitis that progresses to liver failure. Herbal agents should be carefully evaluated for known hepatotoxins, which include chaparral leaf, germander, pennyroyal oil, Jin Bu Huan, and kava kava among others (56).

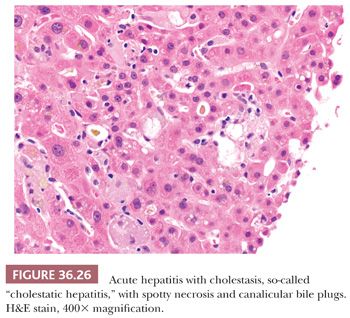
In cholestatic hepatitis (Fig. 36.26), the ALP is typically elevated but is less than five times normal and ALT/ALP is less than five. The morphologic features are similar to inflammation-predominant acute hepatitis and are accompanied by cholestasis. The differential diagnosis includes all entities described earlier but in particular, viral hepatitis A, E, and DILI. Commonly implicated drugs include chlorpromazine and macrolide antibiotics (e.g., erythromycin) (56).
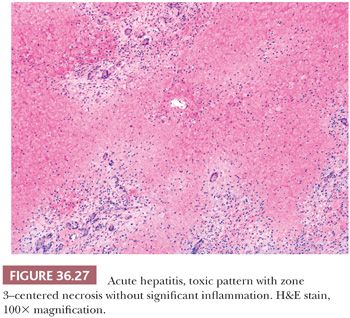
The toxic pattern manifests as necrosis with essentially no inflammation (Fig. 36.27), which indicates a direct toxic effect rather than an immune-mediated event. The differential diagnosis is therefore limited to exposures with known liver toxicity (e.g., halothane, acetaminophen, cocaine, 3,4-methylenedioxy-N-methylamphetamine [MDMA or “ecstasy”], organic solvents, toxic mushrooms [e.g., Amanita phalloides], some herbal medications, herpes simplex virus [HSV] 1/2, and adenovirus). Acute vascular injury, as in ischemia, and acute-onset vascular outflow obstruction, as in Budd-Chiari syndrome, can also show this pattern of injury.
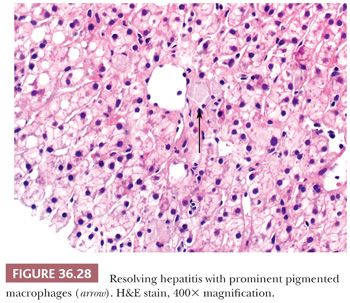
The resolving hepatitis pattern of injury is characterized by mild portal and lobular inflammation with scattered pigmented macrophages in sinusoids (Fig. 36.28), often clustered around central veins. There is usually only mild hepatocellular necrosis manifested by rare/spotty acidophil bodies. The differential diagnosis includes a resolving phase of any acute hepatitis, but it is especially common in DILI, as the offending agent has often been stopped prior to biopsy. A similar mild hepatitis pattern of injury, with or without prominent sinusoidal macrophages, is seen as a nonspecific response in systemic autoimmune and inflammatory disorders (e.g., systemic lupus erythematous, rheumatoid arthritis, and CD) and systemic or abdominal infections such as appendicitis and cholecystitis.
Giant cell hepatitis (i.e., postinfantile giant cell hepatitis, syncytial giant cell hepatitis) is not only similar to inflammation-predominant acute hepatitis but also shows multinucleate syncytial hepatocytes (defined as numerous hepatocytes with four or more nuclei [57]). Although not specific, this pattern in adults is most commonly seen in AIH. Syncytial giant cell hepatitis is common in neonatal hepatitis (see the section “Metabolic, Genetic, and Developmental Disorders”).
In mild acute hepatitis, it may be difficult to reliably distinguish hepatitis versus biliary pattern of injury. Correlation with serologic findings, liver enzymes, and autoantibodies is important in establishing an underlying etiology. Demonstration of periportal hepatocyte copper, by copper stains, and staining of periportal hepatocytes with CK7 is helpful in establishing a biliary etiology.
ACUTE LIVER FAILURE
Acute liver failure manifests as onset of hepatic encephalopathy and coagulopathy within 8 to 26 weeks of symptom onset (in patients without prior chronic liver disease). Drugs, such as acetaminophen overdose, are the most frequent etiology, but many of the etiologies of acute hepatitis can progress to acute liver failure. The histologic pattern can help in determining the etiology: inflammation dominant (e.g., DILI, AIH, WD), necrosis dominant (e.g., acetaminophen overdose, other toxins, HSV, vascular causes), and microvesicular steatosis (e.g., drugs such as valproate and tetracycline, Reye syndrome). Extensive sinusoidal involvement by amyloidosis or metastatic disease can rarely present as acute liver failure.
Liver biopsy–associated bleeding is more common in the setting of severe coagulopathy and so transjugular biopsy may be performed (although biopsy may also be entirely contraindicated). The etiology may remain undetermined in at least 15% of adult acute hepatitis cases that progress to liver failure (58).
CHRONIC HEPATITIS
Chronic hepatitis is a form of liver injury that persists for at least 6 months after initial presentation. Common etiologies include AIH, WD, α1-antitrypsin deficiency (A1ATD) (both WD and A1ATD are described in more detail in the section “Metabolic, Genetic, and Developmental Disorders”), and some hepatotropic viruses. Drug-related chronic hepatitis is uncommon but is well described with certain drugs.
AUTOIMMUNE HEPATITIS
AIH may present as acute hepatitis, including a fulminant form, chronic hepatitis, and cirrhosis. AIH is more common in women (peak incidence at 35 to 40 years) and often shows hypergammaglobulinemia (IgG >1.5× normal) and/or autoantibodies (antinuclear antibodies [ANAs], smooth muscle antibodies [SMAs], and liver-kidney microsome antibody [LKM]). AIH patients commonly express human leukocyte antigen (HLA) DR3 or DR4. Bilirubin levels may be elevated, ALP is usually not significantly elevated, and transaminitis may be mild to severe. The clinical course may “wax and wane,” but patients will progress to cirrhosis if untreated. AIH usually responds to immune suppression, but a prolonged period of treatment may be needed and relapse is possible. The predominant histologic finding is interface and lobular hepatitis, often with prominent plasma cells. Hepatocyte rosette formation may be seen. The bile ducts are largely intact, but focal lymphocytic infiltration and mild epithelial injury can be seen in areas of intense portal inflammation. Definitive diagnosis of AIH requires correlation with clinical parameters such as IgG level and autoantibodies: ANA (type 1 AIH), SMA (type 1 AIH), LKM (type 2 AIH, younger patients), soluble liver antigen/liver-pancreas antibodies (SLA/LP) (type 3 AIH), perinuclear antineutrophil cytoplasmic antibodies (p-ANCAs), and liver enzyme findings, (59) and as scored in the International Autoimmune Hepatitis Group (55). Histopathologic findings remain important in establishing a diagnosis of AIH, as serologic autoimmune markers are notoriously nonspecific, being noted in hepatitis C virus (HCV) infection (60,61) and in fatty liver disease (62). Overlap syndromes represent AIH overlap with primary biliary cirrhosis (PBC) or primary sclerosing cholangitis (PSC) and may be concurrent or sequential (see the section “Chronic Biliary Obstructive Disease”).
Autoimmune Hepatitis—Primary Biliary Cirrhosis Overlap Syndrome
PBC is characterized by bile duct destruction (nonsuppurative), cholate stasis in a periportal/periseptal distribution, absence of canalicular bile, florid duct lesions, and less prominent lobular and/or interface activity. Antimitochondrial antibodies (AMAs) are usually positive, and there is evidence of cholestasis on copper and CK7 stains. ANA may be positive in PBC or autoimmune cholangiopathy (AMA-negative PBC) and does not suggest overlap syndrome.
By definition, clinical, biochemical, and/or histologic features of both PBC and AIH are present simultaneously or consecutively in the same patient. The diagnosis should be considered if two of three typical features of AIH (ALT elevation five times above normal, IgG elevation twice normal [or SMA], or moderate to severe lymphocytic interface or lobular activity) and PBC (ALP elevation two times above normal [or gamma-glutamyl transpeptidase (GGT) elevation five times above normal], AMA, or florid duct lesion) are present (63,64). The diagnosis of AIH-PBC overlap syndrome has significant impact on treatment. Immunosuppressive agents such as steroids are not indicated in PBC as they are ineffective and can induce or worsen osteoporosis, whereas failure to treat with steroids can lead to rapid progression in AIH-PBC overlap syndrome (65–67). The pathologist’s primary role is to highlight significant interface and lobular activity in a case that otherwise resembles PBC and highlight bile duct injury in a case that otherwise resembles AIH. This will enable the hepatologist to integrate the overall features for a diagnosis of overlap syndrome.
Autoimmune Hepatitis—Primary Sclerosing Cholangitis Overlap Syndrome
AIH/PSC overlap is most often diagnosed in the pediatric setting and is more likely to require transplantation than AIH (68). Definitive diagnosis of AIH/PSC requires histologic evidence of AIH and cholangiographic evidence of PSC (69). Periductal sclerosis is unlikely to be present, but other evidence of biliary obstructive disease (e.g., bile duct injury, duct loss, periportal cholestatic effect) should prompt cholangiography to exclude a PSC component.
HEPATOTROPIC VIRUSES
Hepatitis A (HAV), B (HBV), and E (HEV) viruses can lead to acute hepatitis, whereas acute presentation of hepatitis C is extremely rare and is subclinical in nearly all the cases. Hepatitis E may persist in immunocompromised patients.
Hepatitis A
Hepatitis A clinical disease results from fecal–oral contamination and is generally mild and easily diagnosed by serology. Sewage contamination can result in concentration of virus in shellfish such as oysters and is a source of sporadic infection in the United States. Newly infected patients are positive for immunoglobulin M (IgM) anti-HAV antibodies; IgG antibodies signify past exposure and do not indicate acute hepatitis A if IgM antibodies are absent. Hepatitis A typically follows a short benign course and does not cause chronic hepatitis. Rare cases of hepatitis A can pursue a variant course and are more likely to be biopsied:
1. Fulminant hepatitis: Rare cases of acute hepatitis A can be associated with massive or submassive necrosis and clinically present with acute hepatic failure.
2. Hepatitis A, cholestatic variant: These patients present with cholestatic features that may persist for several months. The cholestatic features include hyperbilirubinemia, pruritus, and a cholestatic liver enzyme pattern (i.e., elevated ALP with relatively mild elevation of ALT and AST). Acute hepatitis A should be considered in all cases of unexplained hepatitis with cholestasis.
3. Hepatitis A, relapsing variant: Although hepatitis A does not lead to chronic hepatitis, rare patients can present with recurrence of symptoms after complete recovery. Relapse usually occurs within 3 weeks, is milder than the initial illness, and often shows cholestatic features. Immune manifestations such as purpura, nephritis, and arthralgia are common. Uneventful recovery occurs in nearly all cases.
Hepatitis B and D
Hepatitis B presents as acute hepatitis in 20% to 25% of the patients exposed to the virus, which can occur through blood or body fluid exposure. In Asia and Africa, where HBV infection is endemic, vertical transmission is common. A majority of the patients (~65%) remain asymptomatic after acquisition of HBV infection. Chronic disease develops in less than 5% of infected individuals. The histologic features are similar to other causes of acute hepatitis, and diagnosis is based on serologic findings. Hepatitis B surface antigen (HBsAg) appears in the serum 4 to 26 weeks (average 8 weeks) after the infection. In self-limited cases, HBsAg may disappear before onset of symptoms or persist during the symptomatic phase. The disappearance of HBsAg is followed by anti-HBs antibody after a window period of a few weeks. The presence of IgM anti–hepatitis B core (HBc) antibody (or nucleic acid testing) can be useful for diagnosis during this window period. Conversion of IgM (anti-HBc) to IgG occurs in a few months and is the hallmark of prior hepatitis B exposure. In chronic infections, HBsAg persists and anti-HBs antibodies do not appear. Hepatitis D virus (HDV) relies on HBV for effective infection and can be present as part of initial blood-borne co-infection with HBV or as a secondary infection with new exposure to HBV/HDV, which is more common in Africa and the Middle East. Anti-HDV IgM is an indicator of recent infection, and anti-HDV IgG will be present in chronic HBV/HDV co-infection. Hepatitis D superinfection of a patient with HBV may result in fulminant hepatitis (70).
Hepatitis C
Among hepatotropic viruses, hepatitis C is the most commonly encountered etiology of chronic hepatitis in the United States, with the majority of infections progressing to a chronic stage. Like HBV, HCV is a blood-borne pathogen, but risk of transmission with other body fluids is lower than with HBV. HCV RNA appears in the blood within a few weeks, and nucleic acid testing is the earliest option for detection and is also performed to determine viral load. HCV antibody does not appear in the serum until 2 to 26 weeks (average 6 to 12 weeks), and because the clinical course is mild, it is unusual for acute HCV to manifest clinically. HCV genotyping is clinically useful (71), and genotype 3a is notably associated with significant steatosis.
Hepatitis E
Hepatitis E is a pathogen transmitted via the fecal–oral route that is endemic in Asia, India, Africa, and Mexico. In the United States, there are reports of infection following exposure to infected pigs. In healthy people, the disease is often self-limited and can lead to acute liver failure in pregnant women. Immunosuppressed patients, specifically transplant patients, may develop chronic liver disease (72). Anti-HEV IgM signifies recent infection, whereas anti-HEV IgG signifies past exposure. Viral RNA can also be detected in blood or stool with nucleic acid testing.
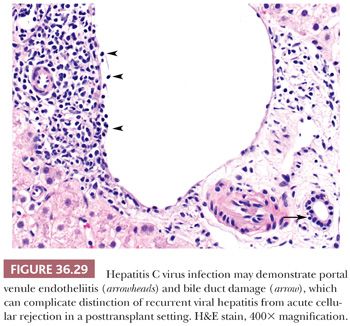
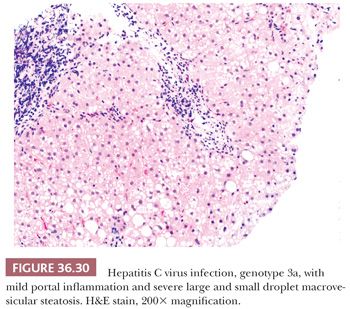
Liver biopsy is needed to assess for stage of fibrosis and need for antiviral therapy in chronic viral hepatitis. Plasma-based testing for markers of fibrosis has so far proven imprecise in defining fibrosis severity (73–76), but advanced imaging techniques (e.g., transient elastography) have more accuracy (77), especially in advanced fibrosis. Biopsy remains the gold standard for staging and is particularly important when there are competing etiologies for liver injury (e.g., HCV and steatohepatitis). Chronic viral hepatitis, in native liver tissue, most commonly has a predominant portal lymphocytic infiltrate often with germinal centers and variable interface and lobular activity (Figs. 36.10 to 36.13). Endotheliitis and bile duct damage may also be seen (Fig. 36.29), which can complicate distinction between recurrent viral hepatitis and acute cellular rejection in the posttransplant setting (see the section “Transplant Pathology”). The inflammatory activity is graded using one of several commonly used schemes (Table 36.2). Ideally, a core biopsy for grading and staging of chronic viral hepatitis will be taken from the right lobe, away from the subcapsular region, and will be of sufficient length and thickness to obtain greater than 10 portal tracts (78–81). Mild fatty change may occur entirely due to viral hepatitis and not in relation to metabolic risk factors for fatty liver (most typically with HCV genotype 3a) (82) (Fig. 36.30).
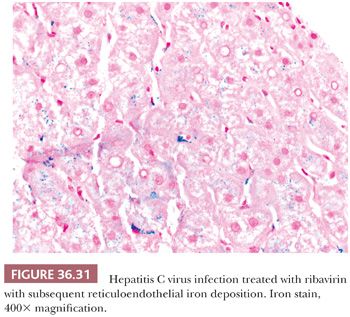
Alcoholic liver disease often coexists with HCV infection, and there may be an increased risk of progression to cirrhosis (83). On iron stain, hepatocellular iron raises suspicion for hereditary hemochromatosis, which may have variable penetrance (see the following texts). Reticuloendothelial iron, on the other hand, may be seen after transfusion or perhaps in relation to antiviral treatment (e.g., ribavirin) (84) (Fig. 36.31). Patients with chronic viral hepatitis are at risk for dysplasia and carcinoma. Large cell change (common in HBV) and small cell change should be identified and included in the report, as they portend an increased risk (or in the case of small cell change, a precursor) of carcinoma (see Chapter 37).
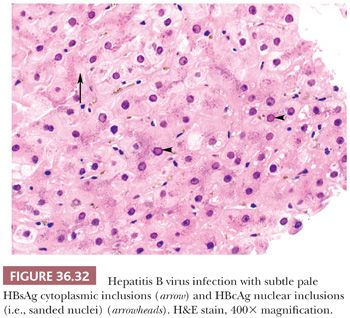
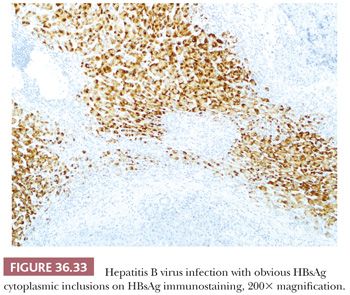
Viral inclusions can be seen only in HBV (and HDV), which manifest as ground-glass cytoplasmic inclusions (HBsAg) and sanded nuclear change (HBcAg or HδAg) (Fig. 36.32). These inclusions can be highlighted on immunostaining (Fig. 36.33), which can allow distinction from mimics (e.g., cyanamide toxicity, Lafora disease, fibrinogen storage disease, drugs such as barbiturates, glycogen inclusions, and polyglucosan bodies) (Fig. 36.3). Absence of nuclear HBcAg could be due to sampling or suggest suppression of HBV replication by co-infection with HCV or HDV, which could be excluded by serologic testing (85,86).
It is relatively common for HCV (less often in the setting of HBV infection) patients to have low-titer autoantibodies, and it can be challenging to distinguish AIH with false-positive HCV serology from true HCV infection (87). Some studies have shown that autoantibodies have no influence on the natural history of hepatitis C, whereas others have shown that these patients may have more aggressive disease with higher necroinflammatory activity and fibrosis (88,89). Recent reports indicate that there is no difference in treatment response and disease progression in hepatitis C with or without antibodies (90,91). If the biopsy shows high necroinflammatory activity, viral nucleic acid testing and serologic testing for additional antibodies, such as p-ANCA, that are rare in HCV infection can be considered (92,93).
NONHEPATOTROPIC VIRUSES
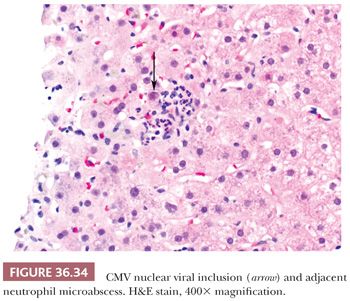
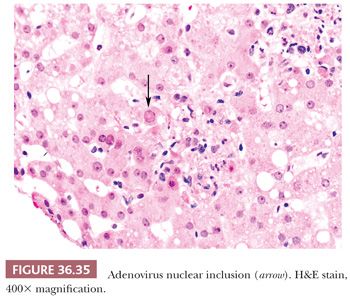
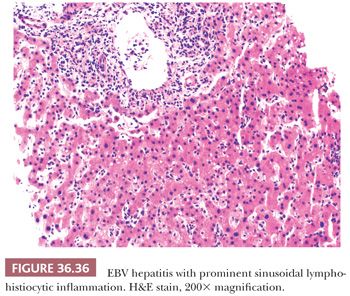
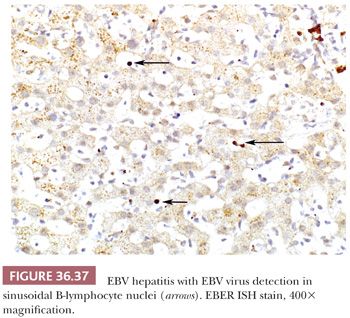
Several nonhepatotropic viruses may also cause significant liver disease (e.g., cytomegalovirus [CMV], adenovirus, HSV, Epstein-Barr virus [EBV]) and should be considered in the appropriate clinical context. Viral inclusions may be seen with CMV (Fig. 36.34), HSV, and adenovirus (Fig. 36.35) infection, but viropathic change is relatively insensitive, especially if the patient has received antiviral therapy. HSV often affects immunocompromised individuals, but herpetic hepatitis can occur in immunocompetent hosts, and both scenarios may present with prominent hepatocellular necrosis. EBV causes infectious mononucleosis that usually occurs in adolescence in Western countries. Liver involvement is mild but occurs frequently. Liver transaminases can be elevated, but jaundice is rare. The typical histologic feature is a diffuse infiltrate of lymphocytes in the sinusoids (Fig. 36.36), often with nonnecrotizing granuloma formation. Hepatocellular damage is usually not prominent, and apoptotic bodies are rare. Adenovirus, as with HSV, may demonstrate large zones of hepatocellular necrosis. CMV hepatitis in the immunocompetent host is typically mild; as with EBV, the liver shows sinusoidal lymphocytosis and hepatocellular injury is mild or absent. Severe involvement with necrosis can occur in the immunocompromised settings and rarely in immunocompetent individuals. CMV hepatitis is well described in allograft liver and often is associated with neutrophil microabscesses in the lobule (Fig. 36.34). Hemophagocytosis (Fig. 36.6) may also be noted with EBV infection. Immunohistochemical or in situ hybridization (ISH) (e.g., EBV-encoded RNA [EBER] ISH [Fig. 36.37]) staining for the respective viruses can be used to confirm the diagnosis.
Other less common viral infections that can cause acute hepatitis with widespread necrosis include yellow fever, dengue fever, and Ebola fever. Parvovirus B19 can cause fulminant hepatitis in children. Mild nonspecific liver inflammation can be seen in many infectious processes, such as human herpesvirus 6 (HHV-6), Coxsackie virus, rubella, and measles virus infection.
OTHER INFECTIONS
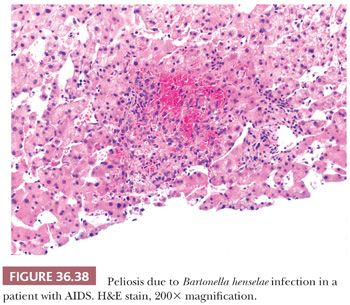
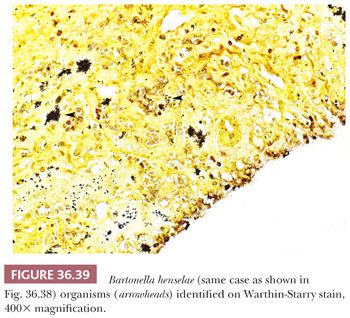
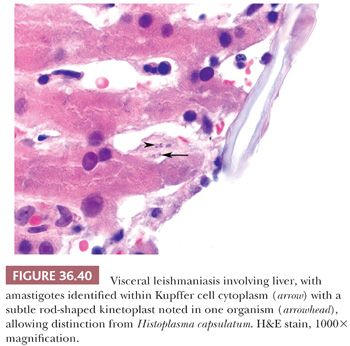
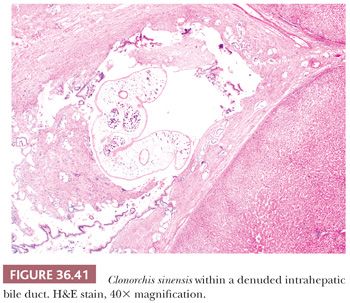
Many systemic bacterial and fungal infections can involve the liver, which often demonstrates a nonspecific reactive hepatitis; other manifestations include abscess (e.g., with bacteria, fungal organisms, or ameba infections), granulomas (e.g., with bacteria, fungal organisms, or parasite infections), fibrin ring granulomas (e.g., with Q fever infection), peliosis (Fig. 36.38) (e.g., Bartonella sp. infection, typically in the setting of severe immune deficiency, as in HIV infection/AIDS) (94), cysts (e.g., with Echinococcus granulosus infection), intracellular organisms (e.g., with visceral leishmaniasis or histoplasmosis), or bile duct involvement by parasites (e.g., with Clonorchis sinensis infection). Culture is usually most helpful in definitive organism identification. Special stains may be useful for Mycobacteria sp. (Ziehl-Neelsen stain), Bartonella sp. (Warthin-Starry stain) (Fig. 36.39), fungal infections (Grocott methenamine silver [GMS] or periodic acid-Schiff with diastase [PASD] stains), and Fusobacterium sp. (Warthin-Starry stain), which can lead to liver abscess and may be missed on routine culture (95). A number of parasitic organisms may also present with abscess, granulomas, or fibrosis. Common parasitic diseases of the liver include schistosomiasis, amoebiasis, and visceral leishmaniasis (Fig. 36.40), as well as infection with E. granulosus (see Chapter 37) or C. sinensis (Fig. 36.41). Hepatic schistosomiasis is caused by Schistosoma mansoni (Africa, Middle East, and Latin America) or Schistosoma japonicum (East Asia) and is contracted by exposure to cercaria released by infected fresh water snails. Schistosome eggs may be identified within hepatic granulomas, and the host immune response may lead to portal fibrosis without regenerative changes (i.e., “pipe-stem fibrosis”) and portal hypertension.
FATTY LIVER DISEASE
Nonalcoholic fatty liver disease (NAFLD) is defined as evidence of fat in the liver in the absence of a significant alcohol consumption history or other reasons to have secondary fat accumulation (Table 36.5) (96). NAFLD may be subclassified as nonalcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH). NAFL represents steatosis without histologic liver injury, whereas NASH represents steatosis with histologic evidence of liver injury (as defined in the following texts). Progression to cirrhosis and/or development of hepatocellular carcinoma (HCC) may occur in NASH (97) but is not expected in NAFL. NASH cirrhosis is defined as cirrhosis with current or previous evidence of NAFLD. NAFLD and NASH prevalence are estimated at 20% and 3% to 5% in the United States, respectively (96,97). Risk factors for NASH include metabolic syndrome, dyslipidemia, diabetes mellitus type 2, and obesity (96) (Table 36.6). Metabolic syndrome manifests with at least three of the following: blood pressure greater than 130/85 mm Hg, increased waist circumference (>102 cm in men and >88 cm in women), fasting blood sugar greater than110 mg/dL, triglycerides greater than 150 mg/dL, and low high-density lipoprotein (HDL) (i.e., <40 mg/dL in men, <50 mg/dL in women). Serum transaminases often correlate poorly with histologic findings, and biopsy is required to diagnose steatohepatitis (96,98).


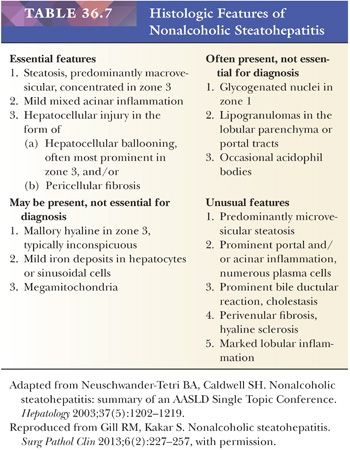
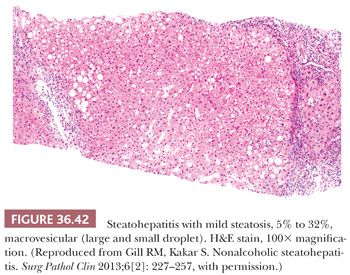
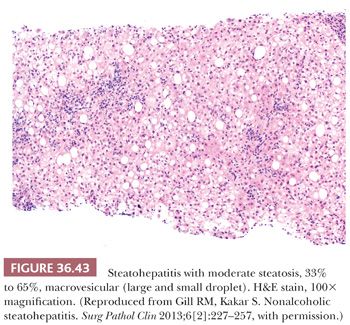
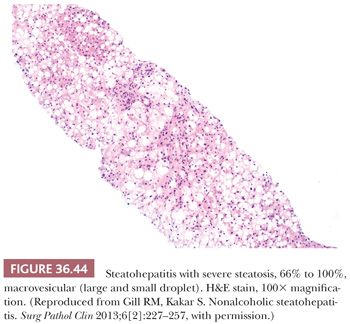
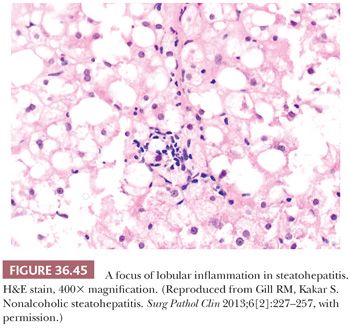
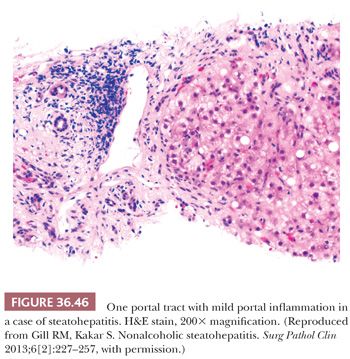
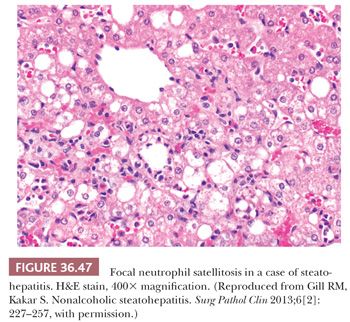
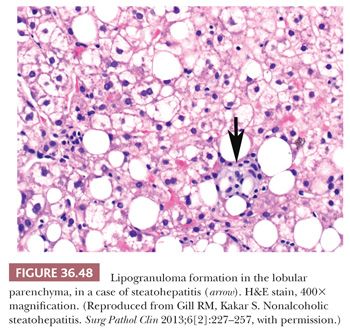
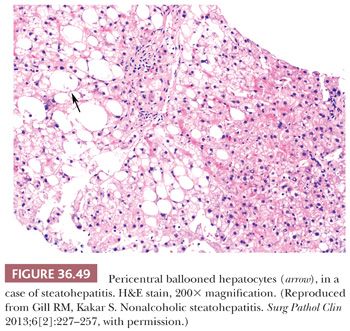
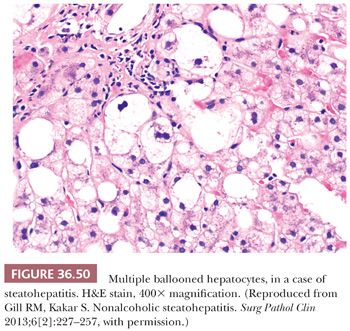
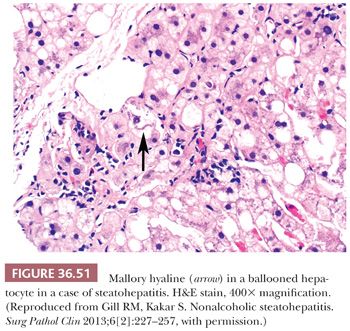
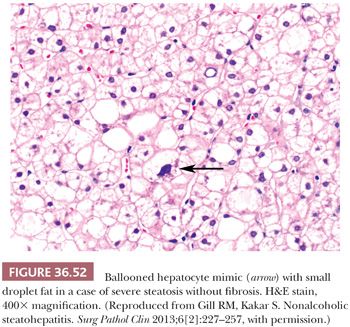
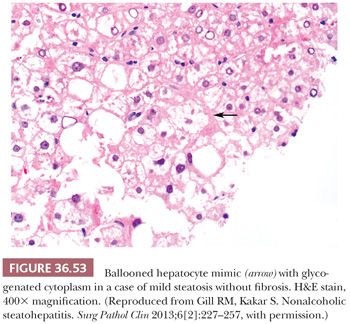
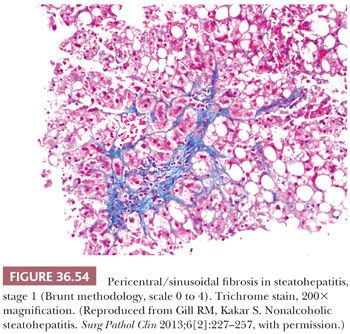
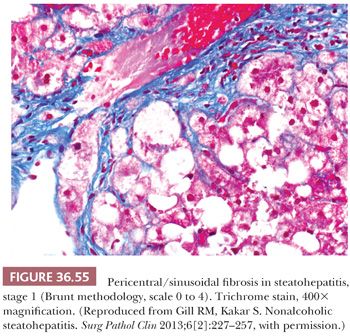
Essential microscopic features for the diagnosis of steatohepatitis (99,100) (Table 36.7) are steatosis, inflammation, and hepatocellular injury in the form of hepatocyte ballooning and/or pericentral fibrosis. Significant steatosis is defined as greater than 5% macrovesicular steatosis (includes both large and small droplet fat), usually with pericentral accentuation (Figs. 36.42 to 36.44). Inflammation is usually mild and more prominent in the lobule (Fig. 36.45), with or without mild portal inflammation (Fig. 36.46). Neutrophils are typically present usually in small numbers and can surround ballooned hepatocytes (neutrophil satellitosis) (Fig. 36.47). This finding is more common in alcoholic and amiodarone-related steatohepatitis. Lymphocytes, histiocytes, and lipogranulomas are also common (Fig. 36.48). Occasional eosinophils, pigmented macrophages, and microgranulomas may be present. Steatohepatitic hepatocellular injury manifests as ballooned hepatocytes or, in the chronic phase, as pericellular fibrosis, typically starting around central veins. Hepatocellular ballooning is characterized by an increase in cell size, rarefaction of cytoplasm, and condensation of cytoplasm into eosinophilic globular areas (Figs. 36.49 and 36.50) (99). When conspicuous, the globular structures are referred to as Mallory hyaline (Fig. 36.51). All three characteristics must be present for definite interpretation as hepatocellular ballooning. Hepatocytes with small droplet steatosis (Fig. 36.52) or with excessive glycogen (Fig. 36.53) can be enlarged or clear but do not have all three characteristics and should not be interpreted as ballooned hepatocytes. Spotty hepatocyte necrosis is also common. Pericellular fibrosis involving sinusoids in zone 3 results in a “chicken wire” or “spider web” pattern of fibrosis (Figs. 36.54 and 36.55) (101).
NONALCOHOLIC STEATOHEPATITIS GRADING AND STAGING
Evaluation of fibrosis stage is important in NASH and can influence treatment decisions; advanced fibrosis (stages 3 and 4) will prompt consideration of future transplantation. Steatohepatitis fibrosis usually starts around the central vein and thus should not be staged using the Batts-Ludwig methodology (52). Two staging schemas are available for NASH (100,102) (Table 36.8), but in practice, the Brunt methodology is sufficient for most clinical reports (Figs. 36.56 to 36.58 ). Although hepatologists are often interested in the grade of steatosis, there is no need to present a formal evaluation of the NASH activity score (NAS) (Table 36.9), which is primarily a research tool (100).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


