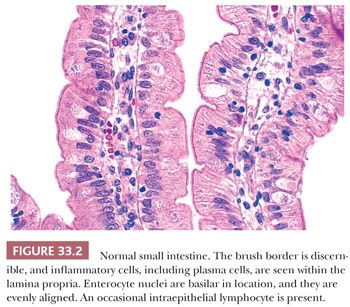
Brunner glands in proximal duodenal specimens have an inconsistent effect on villous architecture (3). Normal-length villi may be encountered overlying the Brunner glands, but the villi usually are distorted and appear short. Similarly, villi are often short and distorted next to or overlying lymphoid aggregates. Such shortened villi should not be misinterpreted as evidence of celiac sprue (CS). The muscularis mucosae tends to hold the mucosa together, resulting in the elegantly normal villi illustrated in Figure 33.1. When muscularis mucosae is not included in the biopsy specimen, the mucosa can spread laterally, giving the impression of shortened villi (3).
In general, the identification of four normal villi in a row indicates that the villous architecture of the whole biopsy specimen is probably normal (3,4). This does not mean that biopsy specimens with less than four aligned normal villi should be considered inadequate for evaluation, because even one normal villous in a proximal small-bowel biopsy specimen rules out fully developed CS. Conversely, finding four normal villi in a row does not necessarily rule out focal lesions, although this is almost always true.
Accumulations of various pigments are frequently seen in the small intestines of apparently normal individuals. The term pseudomelanosis duodeni has been used to describe the accumulation of brown-black pigment in the lamina propria macrophages of the proximal duodenum (3,5). Iron and sulfur are the principal components of these apparently acquired deposits. The condition does not impair the function of the small bowel. Associations include gastrointestinal bleeding, oral iron intake, clinical hypertension, end-stage renal disease, and diabetes mellitus (5). Irregularly distributed, granular, brown-black pigment is commonly encountered in the deep portion of Peyer patches in adults (3,6). The pigment originates in the atmosphere or diet (3,6,7). Accumulating principally within macrophages, this pigment has been shown by x-ray spectroscopy to contain a distinct mineral composition, including silicates, aluminum, and titanium. The pigment is inert and it has no known clinicopathologic significance.
PATTERNS OF ABNORMAL SMALL-BOWEL ARCHITECTURE
The small-bowel mucosal responses to injury are limited, and recognition of a response pattern can be useful in differential diagnosis (Table 33.1). In this chapter, the term severe villous abnormality describes a flat intestinal mucosa in which no villi are seen. Usually, this change is diffuse and is accompanied by epithelial lymphocytosis (at least 30 to 40 intraepithelial lymphocytes per 100 enterocytes) and associated with crypt hyperplasia, which is evidenced by numerous mitotic figures. The terms severe villous abnormality and flat intestinal mucosa are preferred to villous atrophy because the mucosa in the forms associated with crypt hyperplasia is actually of normal thickness. The term variable villous abnormality describes specimens in which the villi either are only focally flat or are less than flat (mild or moderate villous shortening). Many specimens in this category also show increased intraepithelial lymphocytes. These changes may be associated with features that suggest a specific diagnosis (e.g., numerous eosinophils, granulomas, parasites), or they may be nonspecific.

Entities Associated with a Diffuse Severe Villous Abnormality and Crypt Hyperplasia
Celiac Sprue. Celiac sprue, which is also known as gluten-induced enteropathy, gluten-sensitive enteropathy, and nontropical sprue, is a major cause of malabsorption (1). Although the adage “All that flattens is not sprue” is true (8), almost all adult patients in North America with a severe villous abnormality and crypt hyperplasia do, however, have CS. The pathogenesis of CS involves immunologic injury to the enterocyte associated with the ingestion of gluten—the toxic component of cereal grains such as wheat (gliadins), rye (secalins), and barley (hordeins). CS is a human leukocyte antigen (HLA)–associated condition that is primarily associated with the major histocompatibility complex class II alleles DQA1*0501 and DQB1*0201. This HLA-DQ2 allelic combination is found in 98% of patients with CS. HLA-DQ8 (DQA1*03-DQB*0302) is found in the other 2% (1,9). HLA testing is not done for the initial diagnosis of CS because approximately 30% of the apparently healthy white population have it; however, testing negative for both HLA-DQ types makes the diagnosis of CS extremely unlikely (1). Patients with CS usually show a rapid and dramatic clinical, serologic, and histologic improvement with the removal of gluten from the diet, and they quickly relapse following its reintroduction (1,9,10).
The flat mucosa of CS is associated with increased lymphocytes and plasma cells in the lamina propria and increased intraepithelial lymphocytes (Figs. 33.3 and 33.4). About 70% of these intraepithelial lymphocytes express CD8 and about 10%, CD4; the remaining 20% are CD3-positive, CD4-negative, and CD8-negative. Moreover, increased numbers of lymphocytes express the γ/δ T-cell receptor (9). The enterocyte nuclei lose their basilar alignment and become stratified. Neutrophils may be present but they are usually not prominent. The histologic abnormality is most severe in the proximal intestinal mucosa, and it gradually lessens distally. With gluten withdrawal, the abnormalities recede from distal to cephalad in the small-intestinal mucosa. Thus, proximal small-bowel biopsy specimens may remain abnormal for quite some time, even in patients showing marked clinical improvement.
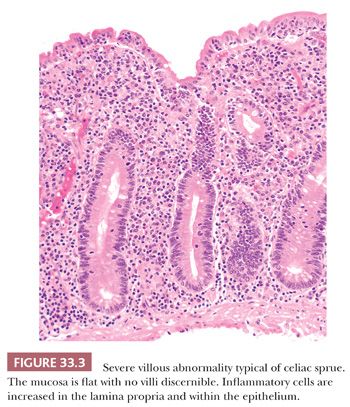
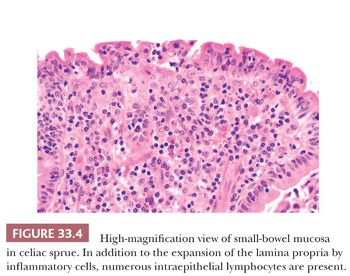
A pathologist does not make the diagnosis of CS. All that can be said is that the specimens contain a severe villous abnormality that is consistent with CS. Definitive diagnosis depends on the demonstration of a suitable clinical presentation, compatible serologic tests (e.g., immunoglobulin [Ig] A antiendomysial antibodies, IgA anti-tissue transglutaminase antibodies), appropriate small-bowel histology, and clinical and serologic response to gluten withdrawal (1,9). Gluten challenge and repeat small-bowel biopsy are no longer recommended (1).
Mucosal lesions of CS can be classified into five types using the Marsh-Oberhuber scheme (1,9,11,12). Type 0, the preinfiltrative lesion, is essentially normal. Type 1, the infiltrative lesion, is characterized by intraepithelial lymphocytosis (at least 40 per 100 enterocytes). The type 2 lesion, which is also known as the hyperplastic lesion, shows a mild variable villous abnormality with epithelial lymphocytosis. The type 3 destructive lesion represents the classic CS lesion described earlier. Some investigators subdivide the type 3 lesion into a type 3a with partial villous shortening, a type 3b with a more severe villous change, and a type 3c that is flat (1,9,12). The hypoplastic type 4 lesion is considered an atrophic end-stage lesion that is seen in a minority of patients unresponsive to gluten withdrawal; it includes the lesion of collagenous sprue (discussed in the section “Collagenous Sprue”).
The histologic differential diagnosis includes all entities that may cause at least a focal severe villous abnormality, including the following: common variable immunodeficiency, protein allergies other than gluten, some cases of infectious gastroenteritis (13), rare cases of tropical sprue (14), stasis (15), Zollinger-Ellison syndrome (4), chronic ischemia, inflammatory bowel disease (IBD) including Crohn disease (CD) (16), and nonspecific duodenitis. Clinicopathologic correlation is essential for proper diagnosis. All biopsy specimens should be carefully evaluated for plasma cells because their absence in selective IgA immunodeficiency and common variable immunodeficiency syndrome (CVID) is easy to overlook. Numerous neutrophils, cryptitis, and crypt abscess formation are usually not part of CS; in such cases, entities such as infectious gastroenteritis, Zollinger-Ellison syndrome, IBD including CD, nonspecific duodenitis, and stasis syndromes should therefore be considered.
The most common cause of unresponsiveness after implementing a gluten-free diet is that the diet is not really gluten-free (1,9). Furthermore, wheat is commonly used as an extender in processed foods, and it occasionally is present in seemingly noncereal grain products such as ice cream, cocoa mixes, instant coffee, and salad dressings. Medications, vitamins, and mineral supplements may also contain gluten (17). If dietary indiscretions are ruled out, patients may have refractory or unclassified sprue, which may respond to the administration of azathioprine, mesalamine, or corticosteroids (1,18). Refractory sprue can also be associated with cavitation of mesenteric lymph nodes and hyposplenism (19). Persistent symptoms despite gluten withdrawal with small-bowel histologic improvement should be a clue to search for comorbidities that may cause persistent diarrhea, such as pancreatic insufficiency, secondary lactase deficiency, bacterial overgrowth, coexisting IBD, or collagenous or lymphocytic colitis (see the section “Lymphocytic Enterocolitis”) (20). Lymphoma must also be considered in nonresponsive patients, and this should prompt a re-review of biopsy specimens or rebiopsy. Furthermore, abnormal intraepithelial T-cell immunophenotypes and T-cell receptor gene rearrangements in some patients with refractory sprue suggest that many refractory patients could, in fact, have low-grade T-cell lymphomas (18,21) (see the section “Enteropathy-Associated T-Cell Lymphoma, or So-Called Ulcerative Jejunoileitis”).
OTHER PROTEIN ALLERGIES
Patients with allergic reactions to chicken, soy protein, milk, eggs, and tuna fish have been reported to show a flat small-bowel mucosa similar to that seen in CS (22–25). The definitive diagnosis depends on identifying the offending protein, showing a response to its withdrawal from the diet, and demonstrating recrudescence of symptoms and pathology with its reintroduction.
LYMPHOCYTIC ENTEROCOLITIS
CS and other sprue-like lesions may be associated with a colonic epithelial lymphocytosis (26,27), with or without gastric epithelial lymphocytosis (28). Approximately 25% of patients with CS who have also had colonic biopsy have demonstrated this form of lymphocytic colitis. Similarly, about 15% of patients with lymphocytic colitis who also had small-bowel biopsy showed sprue-like histology (29). Colonic microscopic abnormalities in patients with CS occur after experimental exposure to wheat or gliadin enemas (30), suggesting that the entire intestinal tract may be susceptible to gluten-induced injury. In some patients with true CS (responsive to gluten withdrawal), one possibility is that occult dietary gluten actually reaches the colon and induces the histologic changes of lymphocytic colitis. However, approximately half of the patients with sprue-like small-bowel lesions and lymphocytic colitis have not responded to gluten withdrawal. The term lymphocytic enterocolitis has been coined to describe this refractory sprue–like condition associated with colonic mucosal abnormalities (26).
Entities Associated with a Variable Villous Abnormality and Crypt Hypoplasia
Marasmus and/or Kwashiorkor. Biopsy specimens from malnourished patients with marasmus (i.e., severe calorie and protein deficiency) may be normal, but patchy areas of decreased villous height that are associated with markedly decreased mitotic activity have been reported (31). In kwashiorkor (i.e., low protein but adequate caloric intake), 6 of 10 patients reported by Brunser et al. (32) showed lesions indistinguishable from those of CS. Some cases have shown a variable villous abnormality associated with increased intraepithelial lymphocytes (33). The mitotic rates in crypts are decreased in kwashiorkor, but this is not as profound as in marasmus.
Megaloblastic Anemia. Nutritional deficiency of folate and vitamin B12 may result in impaired epithelial cell replacement because of decreased DNA synthesis. Consequently, a variable villous abnormality with or without megaloblastic epithelial changes can be seen (34). Evaluation of mucosal biopsy specimens usually allows differentiation from CS because, in folate and vitamin B12 deficiency, (a) the villous abnormality is not severe and it is associated with decreased mitoses in the crypts; and (b) increased inflammatory cells are not present (35,36).
Radiation and Chemotherapy Effect. Because radiation therapy and chemotherapeutic agents inhibit DNA synthesis, the intestinal mucosal changes are similar to those in folate and vitamin B12 deficiency, and they are associated with decreased mitotic activity in the crypts. Chemotherapy and irradiation may also cause focal necrosis of epithelial cells (apoptosis) and increased numbers of chronic inflammatory cells within the mucosa and submucosa (37–39).
Microvillous Inclusion Disease. Microvillous inclusion disease, which also includes cases classified as microvillous dystrophy or atrophy, is an inherited autosomal recessive condition associated with mutations of MY05B gene that codes for myosin Vb, a protein involved in polarized epithelial cell trafficking (40). First reported under the designation familial enteropathy, it causes intractable diarrhea with steatorrhea in infants (41). Diarrhea persists despite total parenteral nutrition, and the patients rarely survive beyond the age of 2 years without small-bowel transplantation (40). The entity should be recognized so that genetic counseling can be offered. Small-bowel biopsy specimens show a severe villous abnormality with crypt hypoplasia. In general, the mucosal specimen may resemble CS, but inflammatory cells including intraepithelial lymphocytes are usually not increased. Transmission electron microscopy establishes the diagnosis by identifying abnormal microvillous structures at the luminal border of the enterocyte and apical intracytoplasmic inclusions lined by microvilli in the same cells (42). The intracytoplasmic vacuoles can also be detected with PAS stain or with carcinoembryonic antigen (CEA) immunostaining (43). Prominent surface enterocyte cytoplasmic CD10 and Rab11 immunoreactivity may also be used to detect microvillous inclusion disease (44,45).
Entities Associated with a Nonspecific Variable Villous Abnormality
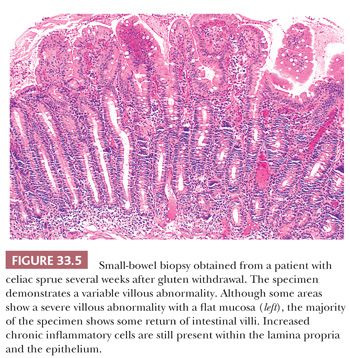
Many diseases are associated with nonspecific variable villous abnormalities that are usually not flat. Some patients show intraepithelial lymphocytosis with essentially normal small-bowel mucosal architecture (46,47). Only about 10% of these biopsy specimens are from patients with clinically latent or partially treated CS (Fig. 33.5) (46); other conditions also enter the differential diagnosis (Table 33.1).
DERMATITIS HERPETIFORMIS
The similarities between dermatitis herpetiformis and CS are numerous, including response to a gluten-free diet and predisposition to malignant lymphoma (48). Approximately 60% to 80% of patients with dermatitis herpetiformis show a villous abnormality of the proximal small intestine (49–51). However, only about 10% of patients have symptoms of malabsorption (52). Although severe lesions similar to those of CS are reported, many patients show a distinctly patchy lesion with a variable villous abnormality that corresponds to the Marsh type 1 and type 2 lesions described previously (52). Approximately 75% of patients with dermatitis herpetiformis show clinical improvement of their skin lesions when they are on a gluten-free diet (48,49).
TROPICAL SPRUE
Tropical sprue is a chronic diarrheal process associated with steatorrhea that is seen in patients from South and Southeast Asia, West Africa, parts of Mexico, Puerto Rico and the Caribbean islands, and some areas of Central and South America (4,14,53,54). Affected patients have usually lived in these regions for years, but symptoms may be seen in people who have been there for as little as a few months. Patients usually respond dramatically to the administration of folate, vitamin B12, and tetracycline or other broad-spectrum antibiotics, implying that an infectious agent may be important in the etiology.
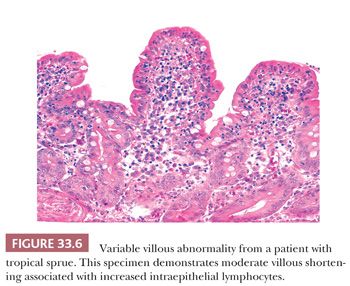
Small-bowel biopsy specimens show a variable villous abnormality. The usual histologic appearance includes mild to moderate villous shortening, increased numbers of chronic inflammatory cells in the lamina propria and epithelium, and crypt hyperplasia (Fig. 33.6). On occasion, a severe histologic lesion indistinguishable from CS may be seen.
INFECTIOUS GASTROENTERITIS
Jejunal mucosal lesions in infectious gastroenteritis are typically patchy with a variable villous abnormality; rarely, the villous abnormality can be severe, closely mimicking CS (4,13,55,56). In addition to increased chronic inflammatory cells, acute inflammatory cells are often seen both within epithelial cells and in the lamina propria (55). The acute onset and temporary nature of the symptoms, coupled with the acute inflammatory changes in biopsy specimens, usually help in the distinction from CS. However, the inflammatory changes can persist for months.
STASIS SYNDROMES
Stasis of small-intestinal contents may occur in surgical blind loops, bowel obstruction, small-intestinal diverticulosis, and intestinal pseudo-obstruction. With stasis, bacteria multiply, and this may be associated with malabsorption of fat and, on occasion, vitamin B12, resulting primarily from the bacterial degradation of bile salts and intestinal mucosal injury (4,15).
The morphologic changes are typically patchy, and they are composed of a variable villous abnormality associated with increased chronic inflammatory cells in the lamina propria and epithelium (8,57). Occasional neutrophils may be seen within the lamina propria and epithelium.
SMALL-BOWEL HISTOLOGY IN ZOLLINGER-ELLISON SYNDROME
Three patterns of focal structural injury are described in small-bowel biopsy specimens from patients with Zollinger-Ellison syndrome. First, in the inflammatory response, a patchy, variable villous abnormality associated with increased mononuclear cells and neutrophils in the lamina propria is observed; erosions can occur. In the second pattern, gastric foveolar metaplasia is seen. In the third pattern, the histology is normal by light microscopy but abnormal by electron microscopy (58).
MASTOCYTOSIS
Systemic mastocytosis is a rare disorder characterized by the infiltration of mast cells in the skin (urticaria pigmentosa), bones, lymph nodes, and other parenchymal organs (59). Over 90% of cases demonstrate a D816V KIT mutation (60,61).
A major criterion for diagnosis is detection of a dense infiltrate of mast cells, at least 15 in aggregate, in the bone marrow or at extracutaneous sites (61). Minor criteria for diagnosis include atypical or spindle-shaped morphology affecting more than 25% of the mast cells, detection of the D816V KIT mutation, cellular expression of CD2 and/or CD25, and a serum tryptase greater than 20 ng/mL. Diagnosis requires the major and one minor or three minor criteria (61).
The gut is involved in almost half of the affected patients, and malabsorption may occur (62,63). The histologic features in small-bowel biopsy specimens are inconsistent. Some specimens show a variable villous abnormality associated with increased numbers of mast cells, whereas others show no increase in mast cells but, instead, have increased numbers of eosinophils. Rare patients have had the histologic features and clinical syndrome of CS (63–65). Diagnostic gastrointestinal lesions are described in the small bowel or colon and have usually been associated with an endoscopic abnormality such as a mucosal nodule or nodularity, erosion and friability, or thickening or loss of folds (60). The neoplastic mast cells occur in aggregates and sheets and tend to be pericryptal or just below the luminal surface. The majority of cells are round with centrally placed nuclei and pale eosinophilic or clear cytoplasm. Some can be spindled, elongated, or large. Over 80% of gastrointestinal lesions in systemic mast cell disease also contained a marked infiltrate of eosinophils that can obscure the diagnostic abnormal mast cells and can easily be confused with eosinophilic gastroenteritis (60). Immunohistochemistry for CD117 and mast cell tryptase can be used to decorate the mast cells, and in systemic mastocytosis, these cells coexpress CD2 and/or CD25. The D816V KIT mutation predicts non-response to imatinib; therefore, patients are usually treated with antihistamines, prednisolone, interferon alpha, cladribine, and other investigational drugs (66).
Some authors think that a gastrointestinal biopsy specimen showing more than 20 mast cells per high-magnification field as determined by CD117 or mast cell tryptase immunostaining indicate mastocytic enterocolitis, which should be treated with antihistamines and/or mast cell stabilizers (67). This entity has been described in patients with intractable diarrhea, similar to diarrhea-predominant irritable bowel syndrome and also typically have normal endoscopic findings. Mastocytic enterocolitis has not been well characterized, does not have a consistent clinical or histologic appearance, and some experts do not make this diagnosis in practice (68). It is questionable if simple mast cell counting is sufficient to assess their role in the pathophysiology of diarrhea (69).
DUODENITIS AND PEPTIC ULCER DISEASE
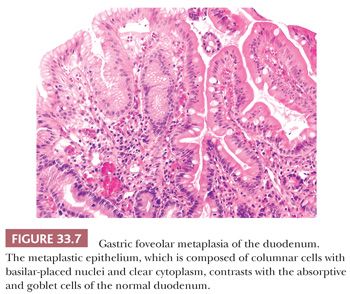
The normal duodenal mucosa often shows a mild decrease in villous height and increased mitotic activity when compared to normal jejunum. This is thought to be the normal physiologic response of the mucosa to the presence of acid in the gut lumen (3,70). Duodenitis may represent an exaggeration of this response, and it probably has the same pathogenesis as peptic ulcer disease (71), although it does represent a milder pre-ulcer form. Active duodenitis is generally limited to the first part of the duodenum (72), and it can demonstrate erosion, a variable villous abnormality associated with increased acute and chronic inflammatory cells and gastric foveolar metaplasia of villous epithelium (Fig. 33.7) (58,71). Proximal small-bowel lesions associated with nonsteroidal anti-inflammatory drugs (NSAIDs) can also show this histology and can be associated with increased intraepithelial lymphocytes (46,72,73). Nodular or diffuse hyperplasia of Brunner glands within the lamina propria may also be present in peptic duodenitis. A close association exists between peptic ulcer disease and gastric infection with Helicobacter pylori. Some authors think gastric heterotopia or metaplasia is an important predisposing factor for the effect of H. pylori because the organism is found in these ectopic gastric tissues by H&E, silver, or modified Giemsa stain techniques (74,75). The organism is not usually found on normal small-intestinal epithelium or on gastric mucosa in which intestinal metaplasia has occurred.
The histologic differential diagnosis of gastric metaplasia in duodenal villi includes gastric heterotopia (76). This usually appears as a mural nodule or mucosal polyp (76,77) on the anterior wall of the proximal duodenum. The histologic distinction between gastric metaplasia and heterotopia is made by the recognition of the specialized gastric gland cells in the latter. Once gastric mucosa occurs in the small bowel or elsewhere (either as a heterotopia or metaplasia), it can, under inflammatory influences, develop into a hyperplastic polyp of gastric type that is characterized by inflammatory expansion of the lamina propria, foveolar hyperplasia, and mucosal cyst formation (78). Gastric heterotopia, which is usually of no clinical significance, has been described throughout the gastrointestinal tract, including the rectum (79).
AUTOIMMUNE ENTEROPATHY
The term autoimmune enteropathy has been applied to an intractable watery diarrhea syndrome occurring in infants that has been associated with circulating autoantibodies against intestinal epithelial cells (41,80,81). A similar condition may also present as a refractory sprue-like syndrome in adults (82). The patients often have variable immunodeficiency and autoimmune phenomena, such as juvenile-onset diabetes mellitus, rheumatoid arthritis, and hemolytic anemia (83–85). The IPEX syndrome refers to an X-linked condition characterized by immune dysregulation, polyendocrinopathy, and enteropathy that has been associated with a mutation of the FOXP3 gene (41). Autoimmune enteropathy can also be seen with the APECED (autoimmune phenomenon, polyendocrinopathy, candidiasis, and ectodermal dystrophy) syndrome, which is transmitted as an autosomal recessive trait linked to mutations in the AIRE gene (86). The small-bowel mucosa shows a variable villous abnormality that is often severe and that resembles CS. Surface and crypt epithelial degenerative and regenerative changes occur, but many illustrated cases show few intraepithelial lymphocytes—a feature that may distinguish autoimmune enteropathy from CS. Adult cases have similar histology to the pediatric cases and often have an absence of goblet cells and/or Paneth cells (82). Some patients with autoimmune enteropathy have also had colitis. In some cases, the associated colitis has resembled lymphocytic colitis, whereas in others, the endoscopic and histologic picture is similar to ulcerative colitis (UC) (82,87,88). Autoimmune enteropathy can also be seen in patients with autoimmune atrophic pangastritis (89). Autoimmune enteropathy is usually severe and intractable, often requiring total parenteral nutrition. Scattered reports have indicated favorable responses to cyclosporine (90), tacrolimus (88), and infliximab (91). Some adult patients have responded to steroid and other immunosuppressive therapy (82,86).
Entities Associated with Variable Villous Abnormalities Illustrating Specific Diagnostic Changes
Collagenous Sprue. The term collagenous sprue describes the excessive subepithelial deposition of collagen associated with a severe villous abnormality noted in small-bowel biopsy specimens from some patients with malabsorption unresponsive to a gluten-free diet (92). Diagnostic criteria have varied, but most patients have had a maximum subepithelial collagen deposit exceeding 10 µm (93,94). Although some patients have followed a fulminant course succumbing to malnutrition and a few with this condition have ultimately responded to a gluten-free diet alone, the majority have required the addition of steroids and other immunosuppressive therapies to a gluten-free diet for clinical control (92–95). The collagen distribution is typically patchy and it may not be present early in the disease; multiple biopsies may be necessary to establish the diagnosis. Collagenous sprue can sometimes coexist with collagenous colitis/ileitis (96) and/or collagenous gastritis. Collagenous sprue should be considered in patients with refractory or unclassified sprue that are unresponsive to a gluten-free diet (93–95). Some patients with collagenous sprue have T-cell receptor gene rearrangements, suggesting that at least a proportion of these cases are actually lymphoproliferative disorders (refractory sprue type 2—see the following texts) but the occurrence of overt high-grade lymphoma has been rare (94). Recently, there have been reports of the antihypertensive drug olmesartan causing collagenous sprue-like changes as well as lymphocytic and collagenous gastritis and colitis (97). Some cases have looked more like autoimmune enteropathy, whereas others looked more like celiac disease. Discontinuation of the drug led to histologic and symptomatic improvement.
Immunodeficiency Syndromes Excluding Acquired Immunodeficiency Syndrome. The primary immunodeficiency syndromes include selective IgA deficiency, X-linked agammaglobulinemia, X-linked hyper-IgM syndrome, CVID, severe combined immunodeficiency, IPEX (see earlier text), interleukin-10-receptor deficiency, and Wiskott-Aldrich syndrome (98). These syndromes are rare and usually present in childhood with recurrent and severe infections, oftentimes with gastrointestinal manifestations. The gastrointestinal pathology of the two most common forms, selective IgA deficiency and CVID, will be considered in more detail. Normal small-bowel villous morphology can be seen on routine light microscopy in selective IgA deficiency, although nodular lymphoid hyperplasia may be present (98,99). Reduced plasma cell numbers and decreased numbers of IgA-containing plasma cells can be demonstrated by immunocytochemical techniques. Giardiasis occurs with increased frequency, and care should be taken not to overlook them. There is also an association between selective IgA deficiency and CS. The absent or reduced plasma cells can help in recognition. CS- and IgA-deficient patients usually respond to gluten withdrawal, and failure to respond should increase the clinical suspicion for CVID (98,99).
Patients with CVID may have chronic diarrhea, malabsorption, and recurrent gastrointestinal giardiasis (92,98,100,101). The morphology of small-intestinal biopsy specimens may vary from normal to a severe abnormality mimicking CS (100–102). In contrast to CS, plasma cells in CVID are usually decreased and IgA-containing plasma cells are typically absent. Occasionally in CVID, the mucosa demonstrates nodular lymphoid hyperplasia that is associated with absent or markedly reduced numbers of plasma cells. Giardiasis commonly is found with either histology. Nodular lymphoid hyperplasia without plasma cell changes may also be seen in asymptomatic patients without an immunodeficiency syndrome, especially in children, in whom it may be considered a normal finding. Lymphocytic colitis with gastritis has been described in CVID (101). An injury pattern resembling acute graft-versus-host disease (GVHD), with numerous apoptotic bodies deep in the crypts, can also be seen in CVID and in selective IgA deficiency (101,102).
With routine light microscopy, nodular lymphoid hyperplasia of the gastrointestinal tract can be difficult to distinguish from non-Hodgkin lymphoma. Furthermore, non-Hodgkin lymphoma associated with nodular lymphoid hyperplasia has been described in patients with CVID (102,103). Immunocytochemistry and molecular analysis can aid in proper diagnosis and classification of gut lymphoid proliferations.
Whipple Disease. Whipple disease, a chronic systemic illness with numerous gastrointestinal features such as diarrhea and malabsorption, is caused by Tropheryma whippeli, a rod-shaped microorganism (104,105). The diagnosis of Whipple disease is usually based on the identification of PAS-positive, diastase-resistant inclusions in small-intestinal biopsy specimens. The diagnosis can be confirmed by a polymerase chain reaction (PCR) assay for the bacterial 16S ribosomal RNA, by electron microscopy, or by immunohistochemistry (IHC) (106–108). The organism has been cultured in human fibroblast cells (107). Whipple disease responds dramatically to antibiotic therapy, although there can be relapses; without treatment, it is fatal (107). The characteristic histopathology of Whipple disease is the infiltration of various organs with macrophages containing the T. whippeli bacillus. This infiltration has a predilection for the lamina propria of the small intestine, the mesenteric lymph nodes, the cardiac valves, and the central nervous system (107,109).
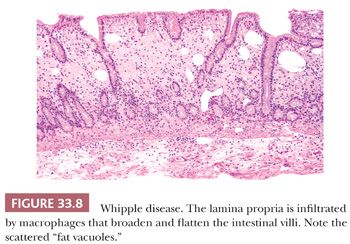
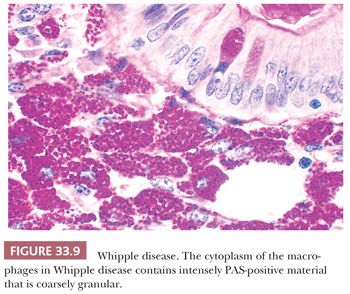
Positive gut mucosal biopsy specimens demonstrate infiltration of the lamina propria; muscularis mucosae; and, in some cases, submucosa by macrophages with a foamy gray-blue cytoplasm (Fig. 33.8). The number of intraepithelial lymphocytes is not increased. In addition, fat vacuoles are seen in the lamina propria, although many that are illustrated as such are clearly dilated lymphatic vessels. The diagnostic changes of Whipple disease are easily seen on H&E; the PAS stain, although dramatic, is probably not necessary to make the diagnosis. The macrophage cytoplasm contains intensely PAS-positive material that is coarsely granular and not bacillary in shape (Fig. 33.9). With antibiotic treatment, the pattern of mucosal infiltration can become patchy (110). The macrophages recede to the muscularis mucosae, submucosa, and basilar portions of the lamina propria, and the cytoplasmic inclusions become “tissue paper–like” with PAS stain, reminiscent of the inclusions of Gaucher cells. PAS-positive and Whipple IHC-positive macrophages can persist for years after treatment; however, PCR assays on intestinal tissues usually convert to negative in less than 12 months (106,107,111).
The histologic differential diagnosis of Whipple disease includes disseminated histoplasmosis (112) and MAIC infection (113). The macrophages in disseminated histoplasmosis contain faintly blue, dotlike inclusions that are usually surrounded by a clear halo. PAS and silver stains demonstrate budding yeast forms. The similarity of Whipple disease to MAIC infection has perhaps been overemphasized in the literature. The H&E appearance may be similar, but MAIC involvement of the small bowel tends to be patchy, whereas Whipple disease is diffuse, and the lipid vacuoles of Whipple disease are not seen in MAIC. PAS-positive inclusions are present within macrophages in both Whipple disease and MAIC; however, the shapes are different. In Whipple disease, the inclusion is bright-staining and coarsely granular; in MAIC, the PAS stain reveals a faintly positive bacillary form. Distinction between the two entities is made by use of the acid-fast stain that is positive in MAIC but negative in Whipple disease.
Eosinophilic Gastroenteritis. The term eosinophilic gastroenteritis has been used to describe a collection of clinical syndromes that are usually seen in children or young adults and that have in common infiltration of the gastrointestinal tract by large numbers of eosinophilic leukocytes and the absence of parasitic or extraintestinal diseases that may cause eosinophilia (114–117). Infiltration primarily in the mucosa of the esophagus is associated with dysphagia with mucosal furrows or rings endoscopically (118). Mucosal involvement of the stomach and small intestine is associated with diarrhea and malabsorption, whereas eosinophils predominantly in the submucosa and muscularis propria are associated with intestinal obstruction. Ascites is a major manifestation when the eosinophils chiefly infiltrate the subserosa (114–117,119,120). With the exception of eosinophilic esophagitis, eosinophilic gastroenteritis remains quite rare (117).
The histologic diagnosis of eosinophilic gastroenteritis may be difficult. Infiltration of the submucosa, muscularis propria, and subserosal connective tissue by eosinophils is always abnormal and, when it is corroborated clinically, is diagnostic of eosinophilic gastroenteritis; however, this type of evaluation does, in general, require a resection or full-thickness biopsy specimen. The diagnosis of the mucosal pattern of eosinophilic gastroenteritis in biopsy specimens can be particularly challenging to the pathologist. Scattered intramucosal eosinophils are normal in the gastrointestinal tract, and their mere presence should not prompt a diagnosis of eosinophilic gastroenteritis. However, collections of eosinophils not associated with other inflammatory cells, groups of eosinophils associated with focal mucosal architectural distortion or injury (e.g., cryptitis, crypt abscesses), and infiltration of the muscularis mucosae by eosinophils are all abnormal, and, in a corroborative clinical setting, these are diagnostic of eosinophilic gastroenteritis. The mucosal involvement in eosinophilic gastroenteritis is notoriously patchy; therefore, if the clinical suspicion is great, multiple or additional biopsy specimens should be obtained.
Numerous intramucosal eosinophils can be seen in other conditions. A search for parasites should always be conducted when eosinophils are prominent, but this is particularly true with obstructive eosinophilic enteritis (ileitis), which is sometimes caused by Sarcocystis species (121) or Ancylostoma caninum (122). Eosinophils may occasionally be a major component of primary IBD and non-Hodgkin lymphoma involving the gut. Additional histopathologic features and clinical findings distinguish these entities from eosinophilic gastroenteritis; furthermore, eosinophilic gastroenteritis essentially is never associated with the architectural or metaplastic changes of chronicity.
Enteropathy-Associated T-Cell Lymphoma, or So-Called Ulcerative Jejunoileitis. Ulcerative jejunoileitis is a rare and poorly understood condition that is characterized by mucosal ulcers associated with a severe small-intestinal villous abnormality. Patients have abdominal pain, fever, intestinal obstruction, perforation, or hemorrhage that often occurs in association with malabsorption (123). Examples of ulcerative jejunoileitis have been reported under a number of synonyms, including diffuse ulceration of the jejunum and ileum, chronic ulcerative nongranulomatous jejunoileitis, idiopathic chronic ulcerative enteritis, malignant histiocytosis (124), enteropathy-associated T-cell lymphoma (125), and epitheliotropic lymphoma of the small bowel (126). Although some researchers exclude patients who have had clinical CS (i.e., malabsorption initially responsive to dietary gluten withdrawal) (123), in my opinion, patients with ulcers and a severe villous abnormality share a common clinical course and histologic appearance regardless of an initial response to gluten withdrawal. Both patient groups demonstrate clinical malabsorption, a similar histologic appearance (ulcer associated with a flat small-bowel mucosa and atypical lymphoid cells), poor prognosis (75% are dead within 2 years), and close association with concomitant or subsequent malignant lymphoma (124–127).
The striking association with lymphoma has prompted some investigators to conclude that all cases of ulcerative jejunoileitis probably represent intestinal non-Hodgkin lymphoma (the synonyms include malignant histiocytosis and enteropathy-associated T-cell lymphoma) (124–127). Isaacson and Wright (124) chose the term malignant histiocytosis to describe this lymphoma because the morphology, erythrophagocytosis, ultrastructural appearance, and immunocytochemical reaction for α1-antitrypsin in the “atypical cells” favored origin from tissue macrophages (128). The same investigators later demonstrated T-cell immunophenotype and T-cell receptor β-chain gene rearrangements (127,129). T-cell lineage has been confirmed (130,131), thus accounting for the popularity of the far more accurate term enteropathy-associated T-cell lymphoma. “Benign” ulcer complications of malabsorption may well be seen, but I have been impressed with the focality of lymphoma and with the difficulty in making that diagnosis with certainty. Any ulcer in patients with CS or an unclassified malabsorption syndrome associated with a flat mucosa must be viewed as a potential harbinger of intestinal lymphoma. Therefore, lesions should be extensively sampled and worked up for lymphoma, including PCR testing for T-cell and B-cell gene rearrangements, and patients should receive careful clinical follow-up.
Enteropathy-associated T-cell lymphoma can be divided into two subtypes: a pleomorphic lymphoma (80% of cases) and a lymphoma made up of mononuclear small to medium cells (20% of cases) (18). The pleomorphic subtype is more often seen in patients with CS; is CD3 positive; is CD56-, CD4-, and CD5-negative; and expresses CD8 in 20% of cases. The monomorphic lymphoma demonstrates a much lower rate of coexisting CS and shows a high prevalence of CD56 (90% of cases) and CD8 (80% of cases) immunoreactivity.
Another controversial point concerns whether the villous abnormality in ulcerative jejunoileitis is, in fact, CS; a low-grade intramucosal T-cell lymphoma; mucosal injury resulting from cytokines released by abnormal T cells; or some other as yet unrecognized small-intestinal disorder (132–134). It is now standard to separate refractory sprue into two types (18). Refractory sprue type 1 shows no atypical lymphocytes, a normal distribution of CD3- and CD8-positive intraepithelial lymphocytes, and polyclonal T-cell receptor assays; usually responds to azathioprine, mesalamine, budesonide, or prednisone; and shows a low rate of progression to lymphoma (18,135,136). Refractory sprue type 2 cases may have atypical lymphoid cells, show loss of surface CD3 staining, can lose staining for CD8 (>50% CD3+/CD8− intraepithelial lymphocytes) and CD5, have a monoclonal T-cell receptor gene rearrangement, do not respond to drug therapy, and have a high case fatality rate due to enteropathy-associated T-cell lymphoma (18). Demonstration of T-cell receptor gene rearrangement in the mucosa not involved by the lymphoma provides a compelling argument that the “enteropathy” may not be CS but that it could instead represent de novo low-grade epitheliotropic (“cryptic”) and/or mucosatropic T-cell lymphoma (18,21,126,133–138). This concept may revolutionize the current perception and treatment of adult-onset unclassified sprue and CS that has become nonresponsive to dietary gluten withdrawal. Refractory sprue type 2 patients with abnormal immunostains (loss of CD8 and CD5) and T-cell receptor gene rearrangements could potentially benefit from lymphoma-directed treatment regimens rather than the standard malabsorption treatments (18,139), and chemotherapy and stem cell transplantation have been used to treat it (21,140).
Parasitic Infestations. Although a large number of parasites may infect the gastrointestinal tract, this discussion concentrates on those that are typically associated with intestinal malabsorption. Infection with Cryptosporidium species, Microsporidia species, and Isospora species are discussed later (see “Interpretation of Endoscopic Biopsy Specimens from Immunosuppressed Patients, Including Patients with Human Immunodeficiency Virus and Acquired Immunodeficiency Syndrome”).
GIARDIASIS OR GIARDIA LAMBLIA (ALSO REFERRED TO AS G. INTESTINALIS AND G. DUODENALIS) INFECTION. Infection by G. lamblia is common, especially in underdeveloped countries or in areas where the water supply has become contaminated by feces (141–143). Symptomatic patients demonstrate a spectrum of complaints, ranging from mild abdominal discomfort with intermittent diarrhea to frank intestinal malabsorption.
Giardiasis is usually diagnosed by duodenal aspirate or stool examination, enzyme-linked immunosorbent assays, direct immunofluorescent antibody microscopy on stool samples, or PCR (142–147). Although pathologists usually encounter Giardia in duodenal biopsy specimens, the organism has been found in the gastric antrum, ileal specimens, and even colonic biopsy specimens (146). The small-bowel mucosal histology in biopsy specimens is usually normal; however, a patchy villous abnormality of variable severity can be observed (Fig. 33.10) (141,146,147). The histologic diagnosis rests on the demonstration of trophozoites either along the surface of epithelial cells in biopsy specimens or in touch preparations (Fig. 33.11). The organism is approximately the same size as the enterocyte nucleus. In profile, the trophozoite of G. lamblia is sickle-shaped; seen en face, it has a characteristic pear shape with a tapered posterior region. It has prominent paired nuclei, paired median rods, a curved median body, and four pairs of flagellae. These morphologic features are most easily recognized on Giemsa-stained or trichrome-stained touch preparations. In tissue sections, fragments of mucus may closely mimic the appearance of G. lamblia trophozoites.

Although trophozoites are readily seen on H&E staining, verification with the trichrome technique can be helpful in some cases. The counterstain in most trichrome stains is iron hematoxylin, a stain that makes the organism more conspicuous. Furthermore, the red staining readily distinguishes it from blue-staining mucus. Because giardiasis is also associated with CVID, biopsy specimens may show absent or decreased numbers of plasma cells, with or without nodular lymphoid hyperplasia.
STRONGYLOIDIASIS OR STRONGYLOIDES STERCORALIS INFECTION. Several species of Strongyloides infect humans (141,148,149). Intestinal infection, which is usually the result of infestation with the nematode S. stercoralis, develops when the worms bury themselves within the mucosa of the duodenum and jejunum. Within the mucosa, the females then lay their eggs, which develop into rhabditiform larvae that pass in the stool. In most cases, these larvae mature into the infective form in the soil, and they then can penetrate intact skin, thus completing the life cycle. In some patients, the rhabditiform larvae may develop into infective filariform larvae before they are passed. These infective larvae may invade the intestinal mucosa or the perianal skin and set up a cycle referred to as autoinfection or hyperinfection syndrome with dissemination (149).
Although many infected patients have no gastrointestinal symptoms, some may have diarrhea or malabsorption (148,150), and, in the immunocompromised patient, autoinfection occurs. Such infections are severe and often fatal (141,148,149,151). The diagnosis is usually made by demonstrating larvae in the stools; the pathologist is unlikely to see the organism in biopsy material. In autopsy or surgical specimens, the adult female and the eggs can be seen in the small-bowel mucosa. With autoinfection, the filariform larvae may also be identified in fresh stool, colonic mucosal biopsy specimens, or in the bowel wall, where they are sometimes accompanied by an eosinophilic and/or granulomatous inflammatory reaction (151,152). Patients with long-standing infection may develop a chronic colitis resembling UC. The presence of eosinophilic and/or granulomatous inflammation and an absence of crypt abscesses should alert one to the possibility of strongyloidiasis (153).
CAPILLARIASIS OR AONCHOTHECA (CAPILLARIA) PHILIPPINENSIS INFECTION. Nematodes of the species Aonchotheca philippinensis can infect humans and can cause a protein-losing enteropathy that can be fatal (154). Infections have been described in patients from the Philippines, Thailand, Iran, Korea, and Egypt (141,154,155). The worms infest the jejunum and upper ileum, and they have only rarely been described in biopsy specimens. The adult worm and eggs bear a morphologic resemblance to those of trichuriasis. The diagnosis is usually made by identifying eggs, larvae, or adult worms in the stool.
Waldenström Macroglobulinemia. Intestinal involvement by Waldenström macroglobulinemia is rare but distinctive (156,157). Grossly, the small-bowel serosa shows small, white nodules and, occasionally, distended serosal lymphatics. The mucosal surface of the bowel may be white, nodular, granular, or mottled. Microscopically, biopsy specimens have shown marked mucosal and submucosal lymphangiectasia associated with short, broad villi. A coarse and sometimes fragmented eosinophilic material is present in the dilated lymphatics, the lamina propria, and macrophages. Immunocytochemistry can be used to confirm that the eosinophilic material is, in fact, the macroglobulin. Although the material may resemble amyloid, a Congo red stain is negative. The material will stain with PAS. The B-cell lymphoproliferative disorder associated with Waldenström macroglobulinemia shows a high frequency of MYD88 L265P somatic mutation (158).
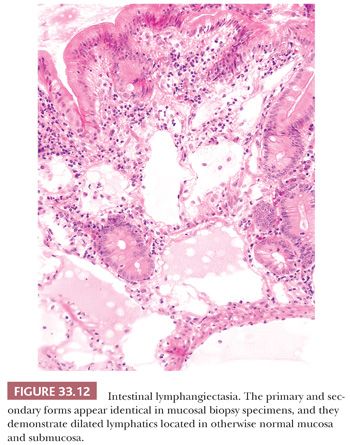
Intestinal Lymphangiectasia. Intestinal lymphangiectasia is characterized by focal or diffuse dilation of the mucosal, submucosal, and subserosal lymphatics. These are not uncommonly encountered during upper endoscopy in asymptomatic individuals where they can appear as pinpoint white dots, whitish discoloration at the tips of folds, or whitish macules or papules and are of no clinical consequence (159). Rarely, lymphangiectasia may be associated with protein-losing enteropathy, hypoalbuminemia, hypo-proteinemic edema, and lymphocytopenia (160,161). It can occur in a primary or secondary form. The primary form has a predilection for children and is caused by a congenital obstructive defect of the lymphatics (161). Secondary lymphangiectasia is associated with many diseases, including retroperitoneal fibrosis, pancreatitis, constrictive pericarditis, primary myocardial disease, intestinal Behçet disease, intestinal malignancy, Waldenström macroglobulinemia, and sarcoidosis (4,156,161,162). In both forms, the histology in mucosal biopsy specimens is identical—dilated lymphatics located in otherwise normal tissue (Fig. 33.12). Therapy includes the treatment of underlying conditions; dietary manipulation; and, in some localized forms of lymphangiectasia, resection (160,162).
Abetalipoproteinemia. Abetalipoproteinemia along with hypobetalipoproteinemia and chylomicron retention disease make up the familial hypocholesterolemias, rare genetic diseases that cause malnutrition, failure to thrive, growth failure, and vitamin E deficiency in pediatric patients (163). Hypobetalipoproteinemia caused by mutation in the APOB gene, abetalipoproteinemia due to mutation of the microsomal triglyceride transfer protein gene, and chylomicron retention disease associated with SAR1B gene mutation all have in common the inability to normally form chylomicrons for fat transport (163). As a result, fat accumulates in the absorptive cells (164). Biopsy specimens have a normal, villous architecture. Enterocytes, however, have cytoplasm packed with droplets of lipid that appear optically clear or foamy with H&E staining. The changes are most prominent at the tips of the villi (163,164). This enterocyte vacuolization, although it is characteristic, is not pathognomonic because similar vacuolar change has been described in megaloblastic anemia, CS, and tropical sprue (165). It is occasionally observed in patients without any apparent disease process.
Acrodermatitis Enteropathica. Acrodermatitis enteropathica is inherited as an autosomal-recessive trait linked to a gene identified as SLC39A4 (166). It manifests in children and has been linked to zinc deficiency. The patients are typically afflicted by cutaneous lesions (perioral and extremity skin lesions, alopecia, nail dystrophy), diarrhea, and malabsorption; they usually respond favorably to the administration of zinc sulfate (167–169). Small-bowel morphology varies, with some investigators reporting a severe villous abnormality similar to that in CS, which improved with zinc therapy, and others reporting normal or only minimally abnormal small-intestinal mucosa by routine light microscopy. Ultrastructural changes that consist of rodlike, fibrillar inclusions in Paneth cells are considered diagnostic for acrodermatitis enteropathica.
Tufting Enteropathy. The term tufting enteropathy has been applied to a sometimes familial intractable diarrhea syndrome in children (41,170–172) which is caused by mutations of EpCAM (173). The symptoms usually begin in the neonatal period with the patient requiring total parenteral nutrition. Small-bowel biopsy specimens have demonstrated a variable villous abnormality that is usually not associated with epithelial lymphocytosis as well as a distinctive surface epithelial change consisting of epithelial crowding, disorganization, and focal tufting.
NONNEOPLASTIC POLYPS AND NODULES OF THE SMALL BOWEL
PANCREATIC HETEROTOPIA, ADENOMYOMA, AND MYOEPITHELIAL HAMARTOMA
The most common nonneoplastic polyps of the small bowel are hyperplastic polyps of gastric mucosa arising in gastric foveolar metaplasia or gastric heterotopia (see in the following texts) (174). Pancreatic heterotopia, adenomyoma, and myoepithelial hamartoma are variants of the same process. Pancreatic heterotopia is characterized by the presence of pancreatic acinar, islet, or ductular elements (usually in association with smooth muscle proliferation) outside of the topographic boundaries of the pancreas. Adenomyoma and myoepithelial hamartoma are synonymous terms differing from pancreatic heterotopia by the absence of both acinar and islet-like tissue. The distribution of acinar and islet elements in heterotopic pancreas varies, and some areas of a pancreatic heterotopia may be indistinguishable from adenomyoma.
The prevalence of heterotopic pancreas in different series has varied from 0.55% to 13.7% (175). The common sites are the stomach, duodenum, and jejunum, but ectopic pancreatic tissue may also be encountered in Meckel diverticula, the ampulla of Vater, the gallbladder, the umbilicus, the fallopian tube, the mesentery and the mediastinum (176). Most examples are encountered incidentally at surgery, and the pathologist is often asked to identify these lesions on a frozen section. On rare occasions, epigastric pain, weight loss, hemorrhage, gastric outlet obstruction, and intussusception have been attributable directly to the presence of the heterotopic pancreas (175). Neoplasia arising in association with pancreatic heterotopia has been reported (176,177).
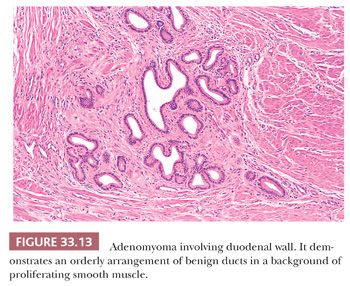
Pancreatic heterotopia presents grossly as a submucosal nodule, an intramural mass, or a nodular lesion involving the serosa. The color is typically yellow or yellow-white; on cut section, the surface is lobulated. The size ranges from 0.2 to 4 cm (176). Occasionally, it contains a central mucosal dimple, which is an important diagnostic clue radiologically, endoscopically, and grossly (176). Histologically, those cases of pancreatic heterotopia associated with acinar elements or islet cells pose little problem in differential diagnosis. Those composed purely of ducts and smooth muscle may be difficult to diagnose, and they are often confused with metastatic adenocarcinoma. However, the adenomyomatous pattern shows an orderly arrangement or lobular pattern of benign ducts set in a background of proliferating smooth muscle (Fig. 33.13). In those cases showing a gross central mucosal dimple, the pattern of ducts recapitulates the major duodenal papilla.
HYPERPLASIA OF BRUNNER GLANDS
Brunner glands are branched tubuloalveolar glands confined predominantly to the duodenum in humans. Most Brunner glands are submucosal, but about one-third of their volume can be found above the muscularis mucosae where the glands empty into the crypts of Lieberkuhn (3).
Hyperplasias of Brunner glands exist in the following three forms (178–180): (a) diffuse glandular proliferation, imparting a coarse nodularity to most of the duodenum; (b) limited discrete nodules in the proximal duodenum; and (c) solitary nodules, often referred to as “adenoma” of Brunner glands. Most are encountered as incidental findings during an investigation of the upper gastrointestinal tract. Histologically, these Brunner gland proliferations suggest a hamartoma or reactive hyperplasia in that they consist of increased numbers of normal-appearing Brunner glands, accompanied by variable proliferations of smooth muscle. Inflammatory cells are often present, and some larger lesions may ulcerate.
Differentiating normal Brunner glands from hyperplasia is difficult. If a gross nodule or polyp is present and is composed solely of Brunner-type glands, then referring to it as a hyperplasia is probably correct. The distinction between adenoma and hyperplasia is arbitrary, and no substantial evidence exists that suggests that any of these proliferations are truly neoplastic; thus, the term Brunner gland nodule is preferred. Dysplastic changes or carcinoma having arisen from either normal Brunner glands or Brunner gland nodules remains controversial (181).
INTERPRETATION OF COLONIC MUCOSAL BIOPSY SPECIMENS IN THE EVALUATION OF PATIENTS WITH SUSPECTED INFLAMMATORY BOWEL DISEASE
With the wide availability of total colonoscopy and flexible sigmoidoscopy, pathologists can expect an increasing number of colorectal biopsy specimens. The pathologist plays an important role in the diagnosis and management of patients with colitis. The numerous uses of mucosal biopsy to evaluate these conditions are summarized in Table 33.2.

COLONIC BIOPSY SPECIMEN PROCESSING AND NORMAL COLONIC HISTOLOGY
Colonic biopsy specimen orientation is not as critical as in small-bowel samples; however, the interpretation is still facilitated by good orientation. A procedure similar to that used in small-intestinal biopsy specimen procurement and processing can be used. Two to three step-section slides stained with H&E are examined.
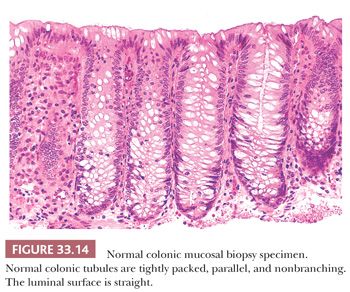
Normal colonic histology is illustrated in Fig. 33.14 (182). The luminal surface is straight. Colonic tubules are tightly packed, parallel, and nonbranching, and all closely approximate the muscularis mucosae. The appearance is similar to test tubes in a rack. Goblet cells are numerous. The lamina propria contains a modest amount of mixed inflammatory cell infiltrate, including plasma cells, lymphocytes, eosinophils, and macrophages. An occasional intraepithelial lymphocyte can be seen. The muscularis mucosae are thin and regular. The submucosa is generally devoid of inflammation. Scattered intramucosal lymphoid follicles are normally encountered, especially in younger individuals. In areas of lymphoid follicles, the architecture may be mildly distorted, the muscularis mucosae may be incomplete, and some of the lymphoid follicles may spill over into the submucosa. Overlying the lymphoid aggregates are flattened surface cells termed M cells. In this M-cell region, the epithelium normally contains more mononuclear inflammatory cells, and the amount of intraepithelial mucin is decreased (181,182). Paneth cells can be seen in the base of colonic crypts, but they are considered a normal finding only in the cecum and the proximal ascending colon (182,183).
Changes associated with bowel preparation or the trauma of biopsy itself can be expected. Bowel preparation decreases the amount of intracellular mucin, causes a mild increase in the number of mitotic figures, and triggers surface degeneration in the form of apoptosis; it can even be associated with rare neutrophils in the surface epithelium and crypts (182–186). Edema and recent hemorrhage into tissue that are not associated with other degenerative or inflammatory changes are best attributed to biopsy trauma.
Muciphages (i.e., macrophages containing PAS-positive material) are often present within the lamina propria of the large intestine, especially in the distal rectum. I do not consider their presence abnormal because they do not correlate with any inflammatory or infiltrative disorder (187,188); however, other authors think they represent a nonspecific response to mucosal injury. Muciphages have been confused with Whipple disease and MAIC infection. The macrophages in Whipple disease are mucicarmine-negative, and acid-fast stains are positive in mycobacterial infection.
PATTERNS OF COLONIC INFLAMMATION
Patterns of inflammation (e.g., chronic colitis, diffuse active colitis, focal active colitis, ischemic-type colitis, trauma change, apoptotic colopathy, and intraepithelial lymphocytosis) can be identified, and these can be helpful in assessing patients by creating differential diagnostic possibilities.
Chronic Colitis: Differential Diagnosis
The pattern of abnormality in chronic UC in remission (quiescent UC) is probably the easiest to identify in biopsy specimens. The predominant features are mucosal atrophy and mucosal architectural distortion (Fig. 33.15) (183,189,190). The luminal border is irregular. The number of crypts decreases; in addition, the remaining crypts appear short (i.e., they do not touch the muscularis mucosae), they lose their parallel arrangement, and they become branched and budded. The goblet cell population is usually preserved. Chronic inflammatory cells are only mildly increased in the lamina propria. Paneth cells may be present. The muscularis mucosae, if present in the specimen, is usually hypertrophied. These changes, although they are consistent with a diagnosis of chronic UC, must be interpreted in light of the clinical and endoscopic findings because identical changes can be seen in focal healed or healing areas of other chronic colitides such as CD, ischemia (including chronic irradiation injury), tuberculosis, and schistosomiasis.
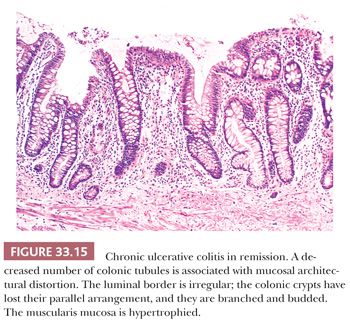
Care must be taken when interpreting biopsy specimens obtained from normal mucosa adjacent to lymphoid follicles, from normal mucosa containing the innominate groove, and from the lower portion of the rectum near the anorectal transition zone. These areas may normally show some crypt shortening and loss of crypt parallelism, and this should not be interpreted as chronic colitis (184). Likewise, histologically normal biopsy specimens must not be reported as showing chronic nonspecific inflammation consistent with chronic UC. Unless one or more of the features discussed in the preceding paragraph is also present, not making a diagnosis of chronic UC based only on an evaluation of inflammatory cells in the lamina propria is a good rule.
Active Colitis: Differential Diagnosis
The term active colitis is used to describe an inflammatory condition in which neutrophils are present in the lamina propria, within the epithelial cells (cryptitis), or within the crypt lumens (crypt abscesses). Included under this heading are (a) UC in an active phase, (b) most examples of Crohn colitis, and (c) infectious colitis and/or acute self-limited colitis (191,192). Recognition of an inflammatory pattern, coupled with clinical and endoscopic correlation, allows a fairly specific diagnosis to be made in many biopsy specimens.
Diffuse Active Colitis
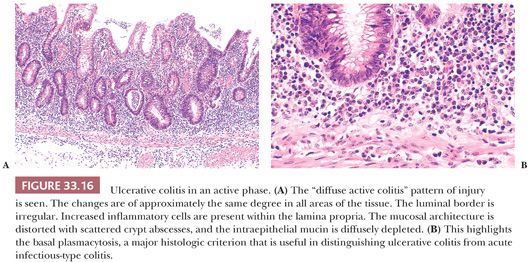
Untreated UC in an active phase represents the prototypic diffuse active colitis. Biopsy specimens usually demonstrate a diffuse abnormality, meaning that the changes are of approximately the same intensity in all areas of the tissue (Fig. 33.16). The luminal border of the mucosa is irregular (190,193,194). Increased numbers of chronic inflammatory cells are present in the lamina propria, and these may spill over into the superficial portion of the submucosa. The intracellular mucin in the goblet cells is depleted (195). Cryptitis and crypt abscess formation are often prominent (194). Surprisingly, even in UC of extremely short overt clinical duration, some atrophy, branching, and budding of crypts are already apparent in many specimens (184,191,192,194). This crypt distortion, coupled with basal plasmacytosis (i.e., increased numbers of plasma cells in the lower fifth of the mucosa), has been proposed to be the most useful criterion for differentiating UC from infectious colitis and/or acute self-limited colitis (190–194). The most a pathologist can conclude from a biopsy specimen showing this pattern is that the changes are consistent with UC in an active phase because the diffuse active colitis pattern can also be seen in some examples of Crohn colitis (193) and in some cases of documented infectious colitis (196), although the latter could represent an infectious exacerbation of underlying latent primary IBD. The diffuse active colitis pattern can also be seen in a form of colitis associated with diverticular disease (197); this entity is distinguished from classic UC by its rectal sparing and its presence exclusively in areas of diverticula.
Focal Active Colitis
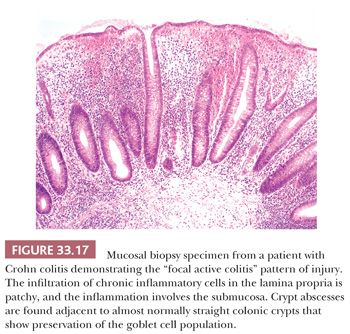
Focal active colitis refers to the patchy distribution of combined architectural change and inflammation in a mucosal biopsy specimen. Chronic colitis showing the diffuse chronic changes described earlier, coupled with patchy acute inflammation, is not considered focal active colitis. The focal active colitis pattern consists of limited areas of increased inflammatory cells associated with focal architectural distortion; characteristically, some areas of the biopsy specimen maintain an essentially normal appearance (Fig. 33.17). The focal active colitis pattern is usually not seen with UC and, when it is present, suggests Crohn colitis (184,198–200) or infectious colitis and/or acute self-limited colitis (184,190–193,195,200). However, the focal active colitis pattern can be seen in resolving UC under medical treatment (195,201), and areas of previously inflamed colon and rectum in UC can return, with therapy, to an almost normal histologic appearance. The focal active colitis pattern has been described in some patients with ischemia (198,199), and it has been linked to NSAIDs in which the focal active colitis is more basal in location (200) and to bowel preparation itself (185,186,198,201).
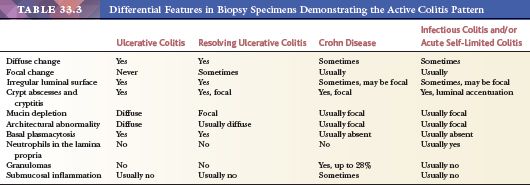
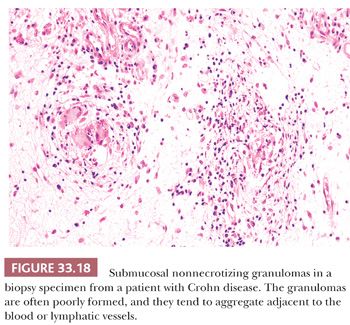
The major differential diagnostic features in biopsy specimens containing the diffuse and focal active colitis patterns are summarized in Table 33.3. Granulomas, which are typically found in CD, should be sought in all biopsy specimens, and some authors advocate serial sectioning for their detection (202) (Fig. 33.18). In my experience, granulomas are rarely missed; however, germinal centers, tangential cuts of blood vessels, tangential cuts of the pericryptal fibroblastic sheath, and inflammatory reactions to extravasated mucin are often misinterpreted as the granulomas of CD. In the absence of true granulomas, biopsy specimens from patients with CD often show the focal active colitis pattern without neutrophils in the lamina propria. However, some examples of CD may be indistinguishable from resolving UC in biopsy specimens (190,193).
A fibrinopurulent exudate in the specimen overlying, but separate from, the mucosa is always abnormal; the clinician must be informed that ulceration is likely in a more proximal location of the bowel. All inflammatory exudates should be examined under high magnification for trophozoites of Entamoeba histolytica because this is their preferred hiding place.
The definitive classification of IBD rests on clinicopathologic correlation. The pathologist should convey the histologic pattern of injury to the clinician, who then collates that information with the clinical history and data obtained from endoscopic and radiologic examination. Through consideration of all this information, an accurate diagnosis can be rendered.
The following histologic appearances have been described in culture-proven and/or toxin-proven infectious colitis: normal colon, nonspecific increases in chronic inflammatory cells, diffuse active colitis, ischemic-like changes, and the focal active colitis pattern of injury (203). Although the colonic mucosal biopsy appearance in infectious colitis can vary greatly, a large number do demonstrate the focal active colitis pattern. In general, invasive organisms cause greater changes in morphology than do those producing their effect by toxins.
Histologic evaluation, although helpful in suggesting an infectious etiology, can only rarely suggest a specific etiology. The definitive diagnosis of infectious colitis requires laboratory recovery of the offending organism or the demonstration of a fourfold rise in a specific antibody titer.
Even after extensive microbiologic workup, a subset of patients remains that is clinically presumed to have infectious colitis and that experiences spontaneous recovery in less than 6 months; in these patients, biopsy specimens demonstrate focal active colitis, although no infectious etiology can be identified. The term acute self-limited colitis has been used to describe such patients (191–193). I prefer the term infectious-type colitis to acute self-limited colitis because some examples of acute self-limited colitis may not be self-limited (184). Others prefer the term nonrelapsing colitis (193,194).
Acute Ischemic-Type Change
The characteristic pattern of acute ischemic-type injury consists of hemorrhage into the lamina propria that is associated with superficial epithelial coagulative necrosis with sparing of the deep portions of the crypts (183). These changes can occasionally be associated with more extensive necrosis of the superficial epithelium with inflammatory pseudomembrane formation. Surprisingly, acute and chronic inflammatory cells (e.g., plasma cells) are typically few in number in ischemic-type damage, and this feature can be helpful in differentiating ischemic-type damage from primary IBD.
The differential diagnosis of acute ischemic-type damage is very wide, including all causes of true ischemia, such as inadequate perfusion, narrowing of blood vessels for any reason, trauma/prolapse and obstructing lesions of the bowel, and bowel distention. Ischemic-type change is also associated with a wide variety of drugs, including vasopressors, oral contraceptives, NSAIDs, cocaine, and glutaraldehyde (used to clean endoscopes) (204–207). Some infectious agents, such as cytomegalovirus (CMV), Clostridium difficile, Clostridium septicum, and the enterohemorrhagic Escherichia coli, typically cause ischemic-type damage.
Traumatic-Type Change or Mucosal Prolapse
Traumatic-type histologic changes frequently coexist clinically with mucosal ulcers. The characteristic trauma-type histopathology is found in the mucosa adjacent to ulcers or in polypoid areas, and it consists of fibromuscular obliteration of the lamina propria that is associated with mucosal architectural distortion and capillary ectasia. The trauma-type histology can be seen in the solitary rectal ulcer syndrome, localized colitis cystica profunda, inflammatory cloacogenic polyp, and mucosa adjacent to the orifices of colonic diverticula; it is a frequent finding adjacent to neoplasia and in the vicinity of the ileocecal valve (208).
Apoptotic Colopathies
Surface colonic epithelial apoptosis and karyorrhectic debris in the superficial lamina propria are commonly seen in mucosal biopsy specimens, and these are widely attributed to bowel preparation (185,186). Apoptotic bodies in the deep crypts are rarely seen (<1 per 20 crypts) outside of pathologic conditions. Increased deep apoptotic bodies are characteristic in ischemic-type damage, CMV infection, chemotherapy/radiation, and certain drugs (e.g., mycophenolate mofetil) (209). Although apoptosis is seen in association with a variety of injurious agents, it is the characteristic form of cell death in cell-mediated immune cytotoxicity and can be seen in patients with grade 1 GVHD or other immunodeficiency syndromes and those with thymoma (102). A primary apoptotic colopathy has been proposed as a cause of chronic diarrhea; however, distinction from the bowel preparation effect is difficult, and it may be one only of degree (210).
Specific Infectious Colitides
Common Bacterial Agents. Colitis can be caused by a host of bacteria, including Campylobacter species, Salmonella species, Shigella species, Staphylococcus aureus, Neisseria gonorrhoeae, E. coli, Treponema pallidum, Yersinia species, and Mycobacterium species. Although the colonic mucosal biopsy appearance in these infections can vary greatly, ranging from essentially normal to lesions like those of idiopathic UC, a large number of specimens demonstrate the focal active pattern of injury outlined earlier that strongly suggests infectious colitis or acute self-limited colitis (191,192,203,211–216).
Histologic evaluation, although helpful in suggesting an infectious etiology, can only rarely suggest a specific agent. True granulomas can be seen in tuberculosis, syphilis, Chlamydia species infection, and Yersinia pseudotuberculosis infection. Microgranulomas are described in infection with Salmonella species, Campylobacter species, and Yersinia enterocolitica. Isolated mucosal giant cells, although they are nonspecific, have been described in Chlamydia trachomatis infection (191,217).
Hemorrhagic Colitis Syndrome. The clinical syndrome of hemorrhagic colitis is characterized by abdominal cramping, bloody diarrhea, and no or low-grade fever (218). Patients typically demonstrate right-sided colonic edema, erosions, and hemorrhage. Investigations of epidemic outbreaks have confirmed the association between hemorrhagic colitis and enterohemorrhagic E. coli (EHEC), the most important of which is E. coli O157:H7 (218–220).
Symptoms in patients with hemorrhagic colitis characteristically present several days after the ingestion of contaminated food, which is usually undercooked hamburger. In almost all patients, the disease resolves spontaneously, but severe cases can be complicated by hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (221,222). In some geographic areas, the majority of HUS cases can be linked to E. coli O157:H7 (223,224).
Most investigators have reported hemorrhage and edema within the lamina propria as the major histologic findings (218). Most patients also demonstrate focal necrosis of the superficial mucosa in association with hemorrhage and acute inflammation and preservation of the deep portion of the colonic crypts, an appearance that is similar to the pattern of injury described with acute ischemic colitis. Some specimens have shown the focal active colitis pattern of injury (see “Focal Active Colitis”). Specimens from patients with hemorrhagic colitis may also demonstrate inflammatory pseudomembranes similar to those seen in C. difficile–associated colitis (218). Because routine stool culture media do not distinguish E. coli O157:H7 from other strains of E. coli normally present in the stool, physicians suspecting hemorrhagic colitis caused by EHEC should specifically request that stools be screened for these organisms. IHC stains and molecular assays for EHEC have been described but are not widely available (203).
Because options for treating EHEC disease are limited, emphasis must be placed on public health and prevention. Consumer activism has resulted in safer techniques in processing for the meat supply. Improved knowledge of safe meat preparation and cooking have caused a reduction in incidence since 1999 (225). Rapid identification of outbreaks is important because it can reduce morbidity and mortality by enabling recall of tainted foods, closing of contaminated swimming pools, and the initiation of educational programs to reduce person-to-person spread (226).
Other E. coli Pathogens. E. coli are a predominant component of the gut microflora. Although the vast majority are harmless or even beneficial, at least five other major categories of E. coli intestinal pathogens, in addition to the EHEC described previously, are recognized as follows: enterotoxigenic (ETEC), enteroinvasive (EIEC), enteropathogenic (EPEC), enteroaggregative (EAEC), and diffusely adherent (DAEC) (227,228).
A great deal is known about the microbiology, pathogenic mechanisms, virulence factors, molecular genetics, and the epidemiology of intestinal E. coli infections. However, surprisingly little is known about the histopathology of the human gut infection. Information remains scant because most infectious diarrheas are self-limited, so they do not require specific treatment even when an infectious organism is identified. Therefore, sophisticated diagnostic tests, such as organism identification, virulence factor determination, and endoscopy with biopsy, are reserved for outbreaks or for cases with unusual features (e.g., severe or protracted diarrhea, systemic symptoms, patients requiring hospitalization, or patients in whom the differential diagnosis includes other serious diseases such as IBD or ischemia).
Analysis of the information available yields a limited number of reaction patterns in E. coli–associated chronic infection. These include no histologic change (ETEC), observation of surface-adherent organisms (EPEC, EAEC, and possibly DAEC), mild nonspecific inflammation (possibly all subtypes), ischemic-like change (EHEC), and acute infectious (self-limited) colitis (EHEC, EIEC, and possibly ETEC).
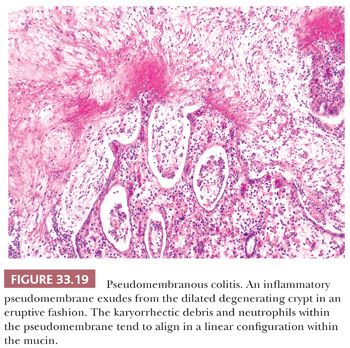
Antibiotic-Associated Colitis and Pseudomembranous Colitis. Toxin-producing C. difficile may cause some antibiotic-associated diarrheas, but it is more strongly associated with pseudomembranous colitis. Administration of any antibiotic that favors the growth of C. difficile can lead to pseudomembranous colitis (229). The symptoms usually develop during therapy, but they are occasionally delayed (229–231). The characteristic lesions occur only early in the disease. Grossly, the surface of the mucosa contains focal, plaque-like, cream to yellow pseudomembranes (232); some early lesions resemble aphthoid ulcers of CD (233). Histologically, patchy necrosis of the superficial portions of the colonic crypts is observed, not unlike that seen in ischemia, although true ischemia tends to show more extensive hyalinization of lamina propria (234). The affected crypts become dilated, and an inflammatory pseudomembrane exudes from the superficial aspects of the degenerating crypt in an eruptive or mushroomlike configuration (Fig. 33.19). This pseudomembrane extends laterally to overlie the adjacent, virtually normal colonic mucosa. The karyorrhectic debris and neutrophils within the pseudomembrane often align in a curious linear configuration within the mucin. Very early lesions, as well as the mucosa between diagnostic lesions, can occasionally show the focal active colitis pattern of inflammation associated with infectious colitis or acute self-limited colitis (232). With the progression of the disease, the plaques become confluent and the crypt necrosis becomes complete. At this point, pseudomembranous colitis becomes indistinguishable from ischemic colitis. Toxic megacolon and perforation can occur (230).
The diagnosis of C. difficile infection is made by stool analysis. Detection of glutamate dehydrogenase that is produced by C. difficile is sometimes used as a screening test, whereas nucleic acid amplification tests have largely replaced enzyme immunoassays for toxin A and B detection for definitive diagnosis (235,236).
Spontaneous recovery has been reported, but most patients are given vancomycin, metronidazole, or fidaxomicin (235,236). Recurrent C. difficile infection poses a significant therapeutic challenge. The first recurrence can be treated with the same regimen used for the initial infection. Pulsed vancomycin is recommended for the second recurrence, whereas fecal microbiota transplant should be considered for the third recurrence (235,236). Recent successes with fecal transplant suggest it may soon become a first-line treatment for C. difficile infection.
C. difficile has been linked to exacerbations of IBD (237). These patients have not shown the characteristic gross features of pseudomembranous colitis, and cases examined histologically have revealed only the features of the underlying primary IBD. A previously uncommon strain of C. difficile called the NAP-1 strain has been responsible for several hospital outbreaks and serious disease in otherwise healthy individuals. These reports of close-contact transmission, high recurrence rate, younger patient age, bloody diarrhea, and lack of antibiotic exposure suggest a changing epidemiology (229,238).
Viral Agents. Norwalk agent and rotavirus, common causes of viral gastroenteritis, are not known to cause morphologic changes in the colon (203). CMV and herpes simplex virus (HSV) may cause proctitis and colitis; they are described in more detail in the section “Interpretation of Endoscopic Biopsy Specimens from Immunosuppressed Patients, Including Patients with Human Immunodeficiency Virus and Acquired Immunodeficiency Syndrome.”
Other Infectious Agents. Fungi, parasites, and Mycobacterium species are discussed in their respective sections in this chapter.
SPECIFIC FORMS OF COLITIS
Collagenous Colitis and Lymphocytic Colitis
Collagenous colitis is a distinct clinicopathologic syndrome consisting of watery diarrhea that predominantly occurs in middle-aged or older (mean age of 59 years) women (male-to-female ratio of 1:7.5) (239). Colonoscopic and barium enema examinations in these patients are usually normal. Therefore, the diagnosis depends on the recognition of characteristic changes in biopsy specimens. The primary histologic change of collagenous colitis consists of a patchy increase in the thickness of the subepithelial collagenous plate (239,240). The normal colonic subepithelial collagen layer is approximately 5 µm in thickness, but, in collagenous colitis, it may increase to 15 µm or more (Fig. 33.20). Various collagen subtypes and tenascin have been identified in the subepithelial collagen plate (241,242), but their demonstration is not necessary in routine practice. The lamina propria is expanded by a mild to moderate chronic inflammatory infiltrate. Patchy injury to the surface epithelium, which is characterized by increased numbers of intraepithelial lymphocytes (>15 per 100 colonic epithelial cells), epithelial degeneration, and sloughing, may also occur. Atrophy, mucosal architectural distortion, and acute inflammation are usually not present or are minimal (239,240,243).
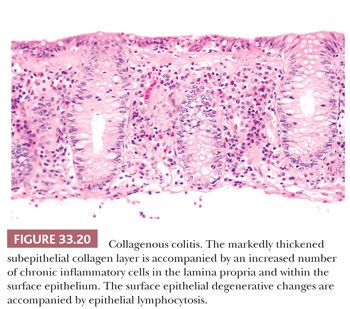
Differential diagnostic considerations include nonspecific changes, primary IBD, acute colitis, mucosal prolapse syndrome (solitary rectal ulcer syndrome), ischemic bowel disease, amyloidosis, and lymphocytic (“microscopic”) colitis. Features for comparing and contrasting these conditions are listed in Table 33.4.
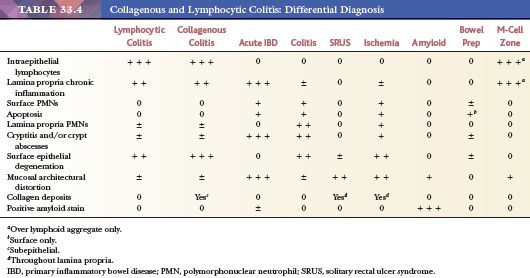
Read et al. (244) introduced the term microscopic colitis to describe chronic watery diarrhea of unknown origin occurring in middle-aged (mean age of 54 years) patients. As in collagenous colitis, the colonoscopic appearance and the barium enema are usually normal. The biopsy specimens demonstrated increased inflammatory cells in a pattern not specific for any established entity (244). Since this initial report, several additional investigators have refined the clinical, and especially the histologic, diagnostic criteria (243,245). The histologic changes include increased chronic inflammatory cells in the lamina propria and surface epithelium, degenerative changes of the surface epithelium, minimal architectural changes, and minimal acute inflammation (Fig. 33.21).
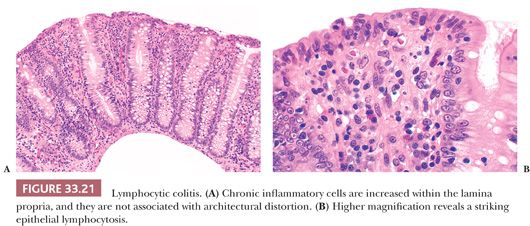
The changes of microscopic colitis resemble the surface epithelial and lamina propria changes of collagenous colitis; indeed, changes identical to microscopic colitis may be seen in biopsy specimens from patients with collagenous colitis if areas in which the collagen plate is not thickened are sampled. Because of the lymphocyte predominance in the inflammatory infiltrate, Lazenby et al. (243) proposed the term lymphocytic colitis to describe this entity. They noted that the term microscopic colitis could be confused with other diseases that may have a normal gross appearance but abnormalities on histologic analysis (e.g., CD and acute self-limited colitis). Lymphocytic colitis, therefore, has emerged as the preferred term for this entity.
The histologic differential diagnosis of lymphocytic colitis includes changes caused by enema (bowel preparation), the mucosal M-cell zone, acute colitis, primary IBD, and collagenous colitis. Features for comparing and contrasting these various conditions are included in Table 33.4.
The similarities between lymphocytic colitis and collagenous colitis are dramatic. They share a common symptomatology and endoscopic findings. The histologic similarities include increased intraepithelial lymphocytes, surface epithelial damage, and increased chronic inflammatory cells within the lamina propria (240,243,245). Both collagenous and lymphocytic colitis may be associated with autoimmune phenomena such as thyroid disease, rheumatoid arthritis, myasthenia gravis, and CS (246,247). Progression of lymphocytic colitis to collagenous colitis has been described (243). Although some controversy still exists about the subject, lymphocytic colitis and collagenous colitis appear likely to be either the same or very similar entities that perhaps represent different morphologic phases of one disease state (243,245,246).
Spontaneous resolutions of collagenous colitis and lymphocytic colitis have occurred, thus rendering the evaluation of therapeutic regimens difficult (248,249). A minority of patients with these colitides has been seen with relatively minor diarrhea, and medical control has been achieved with dietary restriction, bulking agents, and antimotility drugs (248–250). This type of so-called symptomatic therapy has failed, however, in the vast majority of patients. Probably all patients should have a trial of bismuth subsalicylate (251). Most patients require the addition of anti-inflammatory agents. Approximately 80% to 90% of patients eventually respond to therapy with sulfasalazine and corticosteroids such as budesonide (245,248–251). Treatment of the often-associated thyroid problems can also improve bowel function. Case reports of improvement after treatment with mepacrine hydrochloride, steroid enemas, metronidazole, and 5-aminosalicylate have been published (245,248–251).
NSAIDs and ticlopidine have been reportedly linked to some occurrences of collagenous colitis (252,253), and lymphocytic colitis has been reported in patients with immune deficiencies and those receiving the drugs Cyclo 3 Fort (254), ranitidine (255), ticlopidine, some proton pump inhibitors, and flutamide (253). However, most cases cannot be linked to drug ingestion, and the bulk of evidence suggests that lymphocytic colitis and collagenous colitis share a common immune-mediated etiology and pathogenesis. Both conditions have a striking histologic similarity to CS, a condition known to be autoimmune (243). In addition, both lymphocytic and collagenous colitis have been linked to other conditions thought to have an autoimmune pathogenesis, including CD, hypothyroidism, hyperthyroidism, inflammatory arthropathies, pernicious anemia, small-bowel villous atrophy (see the section “Lymphocytic Enterocolitis” earlier), iritis, and myasthenia gravis (256). Finally, both conditions respond dramatically to anti-inflammatory agents (251). A lymphocytic colitis-like histology was reported in an epidemic outbreak of Brainerd diarrhea linked to a water tank aboard a cruise ship, supporting the idea of an infectious trigger for some cases of lymphocytic colitis (see the following texts) (257).
Aberrant Histology in Lymphocytic and Collagenous Colitis
As more cases are investigated, it is not unusual to see variations in histology commingled with the classic changes of lymphocytic or collagenous colitis. These variations include architectural change, cryptitis, Paneth cell metaplasia, ulcers, and inflammatory membranes (258–260). Approximately one-third of patients demonstrate cryptitis. Rarely, one encounters inflammatory membranes. Neither histology correlates with infection. Approximately 2% of patients have ulcers, with additional architectural abnormalities reported in up to 5%. Paneth cell metaplasia has been seen in 14% to 44% of patients and is more often seen in collagenous colitis patients in whom it may correlate with increased severity of diarrhea. Acute inflammation was seen with increased frequency in patients taking antibiotics. Ulcers are linked with concomitant NSAID usage or can be seen with fractured colon (ulcers caused by increased pressure during colonoscopy) in collagenous colitis (261). Another variant of lymphocytic and collagenous colitis with subepithelial giant cells has been described (262). Histology, aberrant or otherwise, does not seem to correlate with symptoms, results of medical treatment, or outcome (263). No patient with unusual histology, including those with architectural changes and Paneth cell metaplasia, has yet to develop primary IBD (e.g., CD or UC).
Brainerd Diarrhea
The term Brainerd diarrhea has been applied to outbreaks of diarrhea of unknown etiology that are characterized by acute onset and prolonged duration (257). The clinical and epidemiologic characteristics of these outbreaks were typical for a point-source epidemic infectious diarrhea. However, unlike typical infectious diarrhea, a chronic watery diarrhea syndrome developed with symptoms lasting longer than 6 months and often for years.
In general, the small-bowel biopsy specimens are histologically normal in Brainerd diarrhea. Colonic biopsy specimens reveal surface epithelial lymphocytosis without distortion of the mucosal architecture, surface degenerative changes, or a thickened subepithelial collagen plate. The degree of surface epithelial lymphocytosis is similar to that seen in collagenous colitis and lymphocytic colitis (257,264).
Patients with the clinical syndrome of chronic watery diarrhea of unknown etiology and patients reported as having lymphocytic colitis represent a heterogeneous group that may contain persons with unrecognized Brainerd diarrhea. With longer term follow-up, the Brainerd diarrhea cases appear to be self-limited, with patients recovering in 3 years or less. Surface epithelial lymphocyte counts should be performed on all colonic biopsy specimens from patients with chronic diarrhea, especially chronic watery diarrhea. Somewhat lower lymphocyte counts and a lesser degree of surface degeneration may distinguish Brainerd diarrhea from other types of lymphocytic colitis, but more study is required. Currently, Brainerd diarrhea cannot be recognized outside the setting of an epidemic.
Eosinophilic Colitis and/or Proctitis
Infiltration of the large intestine by large numbers of eosinophils correlates with a variety of clinical syndromes. One variant is probably an extension of the eosinophilic gastroenteritis discussed earlier. Peripheral eosinophilia is marked, and a history of atopy is common (265–267). A second type, termed allergic proctitis, is, in my opinion, a form of UC (268). Whenever large numbers of eosinophils are encountered in colonic biopsy specimens, this should prompt a thorough search for parasites, especially Strongyloides species, and for abnormal mast cells in systemic mastocytosis (see earlier discussion).
The most common type of primary colorectal eosinophilic infiltrate is confined to the mucosa, and it occurs in infants and young children as a result of dietary-related (protein) allergy (allergic proctitis and/or colitis) (269). These children typically have rectal bleeding with or without diarrhea and many show peripheral blood eosinophilia. Colonic biopsy specimens may show increased numbers of eosinophils within the lamina propria that are often accompanied by a mild focal active colitis. Precise biopsy classification may be difficult. In general, however, more than 60 eosinophils per 10 high-magnification fields and eosinophils in the muscularis mucosae or as the predominant cell in crypt abscesses are features suggestive of an allergic etiology (269). The clinical course is usually benign following withdrawal of the offending dietary trigger (270).
Graft-versus-Host Disease
Allogeneic bone marrow transplantation, allogenic and autologous hematopoietic stem cell transplantation, and umbilical cord blood transplantation, which have been used effectively for the treatment of hematologic malignancies, aplastic anemia, various immunodeficiency states, and genetic disorders, can be complicated by GVHD (271–275). Patients with autologous hematopoietic stem cell transplantation, especially those receiving this treatment for myeloma, can also develop GVHD (274). GVHD can occur in an acute or chronic form. The changes of acute GVHD (“apoptotic colopathy”) in mucosal biopsy specimens have been described (Fig. 33.22), and various grading systems are used (271,276,277); a commonly used system is summarized in Table 33.5. It should be noted that from a clinical standpoint, the most important point is the presence or absence of GVHD rather than what grade is present. Treatment is the same regardless of what grade of GVHD is assigned. Biopsy specimens should be carefully examined for the presence of CMV and adenovirus inclusions; an immunostain should be considered because viral infections may cause changes (apoptosis) that mimic acute GVHD (274). Moreover, infections and GVHD may coexist (277). CD123 expression in the lamina propria by IHC usually correlates with GVHD and can be useful in separating GVHD from infection and mycophenolate-related injury (278).
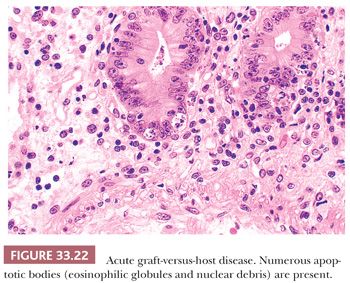

Apoptosis identical to that seen in grade 1 GVHD is encountered in chemotherapy and irradiation damage. Because patients receiving these transplants for malignant conditions are prepared with chemotherapy and irradiation, distinguishing the effects of therapy from GVHD may be impossible. However, changes of chemotherapy or irradiation resolve in 7 to 20 days (272,275,276); therefore, with knowledge of the clinical setting, an accurate diagnosis is possible. Apoptosis identical with grade 1 GVHD has been seen in patients with severe immunodeficiencies and T-cell defects (103,280–282); in patients with acquired immunodeficiency syndrome (AIDS), in whom it may represent a primary AIDS-related enterocolitis (282); and in patients receiving mycophenolate mofetil. One article suggests that increased eosinophils in the lamina propria (>15 per 10 high-magnification fields) coupled with a lack of endocrine cell aggregates and apoptotic microabscesses (both typical for GVHD) help to separate mycophenolate-related injury from GVHD (283). Grade 4 GVHD cannot be distinguished from typhlitis or ischemic bowel disease without knowledge of the clinical picture.
Chronic GVHD usually spares the small bowel and colon; however, rare cases of presumed chronic GVHD involving these organs have been described. The pathologic changes have included colonic submucosal fibrosis, mucosal calcification, and focal fibrosis of the lamina propria (271,272,277). Chronic colitis-type changes can be seen in a subset of patients with GVHD; however, the relationship between this histologic alteration and the clinical diagnosis of chronic GVHD requires more study (284).
Umbilical cord blood transplantation typically results in less severe GVHD. In addition, a peculiar form of culture-negative nonsteroid responsive diarrhea referred to as cord colitis syndrome has been described (275). Linked to Bradyrhizobium enterica infection (285), cord colitis syndrome responds to antibiotics and has a favorable prognosis. Mildly increased apoptosis has been described, but microgranulomatous inflammation in biopsy specimens from the upper and lower gastrointestinal tracts appears to be more typical (275).
Acute Ischemic Colitis
Ischemic bowel disease can closely mimic IBD, especially CD, because the bowel involvement is typically patchy, ulcers may be discrete or serpiginous, and healing ischemic damage may form strictures. This section deals with changes of early ischemic bowel disease in biopsy specimens. Ischemic bowel disease is discussed in more detail in the section “Ischemic Bowel Disease.”
Acute ischemic damage may be focal or diffuse (286). The characteristic pattern of injury consists of hemorrhage into the lamina propria that is associated with superficial epithelial necrosis, which usually spares the deep portion of the colonic crypt (287). This may be associated with an inflammatory pseudomembrane (Fig. 33.23); however, associated inflammatory changes are typically mild. Depending on the duration of ischemia, these early changes may progress to full-thickness mucosal ulceration, at which point the histology becomes nonspecific (183). Although infection with EHEC, such as E. coli O157:H7, can produce the injury pattern of acute self-limited colitis, in my experience, many cases look identical with early ischemic colitis (218). Clusters of “ischemic colitis” cases, especially if they occur in younger patients and are associated with a right-sided colonic distribution, should be investigated for the presence of these bacteria.
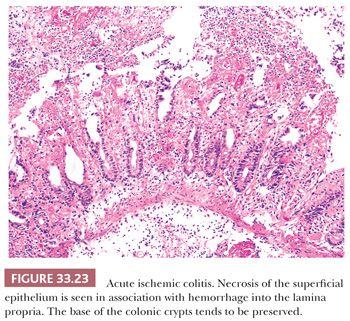
LESIONS THAT MIMIC INFLAMMATORY BOWEL DISEASE
Colorectal Mucosal Prolapse Syndromes
The solitary rectal ulcer syndrome, localized colitis/proctitis cystica profunda, inflammatory cap polyposis, prolapsing mucosal fold in diverticular disease, inflammatory myoglandular polyp, and inflammatory cloacogenic polyp are closely allied conditions that have been linked to mucosal prolapse (288–294). The affected patients often demonstrate abnormal function of the anal and pelvic floor musculature during defecation that leads to rectal mucosal prolapse or even intussusception (288,295,296). The resulting trauma is thought to cause the clinical symptoms and the pathologic changes. The term solitary rectal ulcer syndrome is a misnomer because the ulcers are often multiple, a preulcer polypoid phase is present, and similar lesions can also occur in the anal canal and sigmoid colon. Furthermore, colitis cystica profunda and inflammatory cloacogenic polyp are also misnomers. Because these conditions share a common histologic appearance, clinical presentation, clinical course, and pathogenesis, I prefer to consider them together under the heading mucosal prolapse syndromes.
The symptoms usually present in the third and fourth decades of life, with constipation and other difficulties in defecation (297). Rectal bleeding occurs commonly. The ulcer, when it is present, typically occurs on the anterior or anterolateral wall of the rectum; it is irregular in shape; and it often appears well demarcated. In patients without ulceration, the mucosa may be rough, red, and polypoid. Inflammatory cloacogenic polyp involves the lower rectum and the anal transition zone (289). The clinical impression in patients with mucosal prolapse syndromes is often incorrect, and it includes ulcer, CD, nonspecific proctitis, carcinoma, or villous adenoma (298).
The characteristic histopathologic changes (traumatic-type changes) found in the mucosa adjacent to ulcers or in the polypoid areas consist of fibromuscular obliteration of the lamina propria associated with mucosal architectural distortion (Fig. 33.24), often with a hyperplastic or villiform appearance (288,290,291,298). Inflammation is typically mild or absent, and the mucosal capillaries may be ectatic. Superficial erosions occur, and they can be associated with acute inflammation and the formation of inflammatory “pseudomembranes.” On occasion, colonic glands may be misplaced into the muscularis mucosae or submucosa, a histology often referred to as localized colitis/proctitis cystica profunda. Submucosal blood vessels may also be ectatic and hyalinized. The histology of specimens obtained from ulcerated areas is usually nonspecific, consisting of fibrinopurulent debris and granulation tissue.
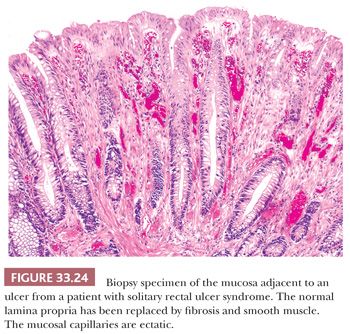
Differential diagnostic considerations include mucinous carcinoma, chronic UC, Cowden disease, Peutz-Jeghers polyp, and ulcers resulting from ergotamine suppositories. The misplaced glands of mucosal prolapse syndrome (colitis cystica profunda) may be associated with dissecting mucous pools, and they can be easily mistaken for invasive mucinous adenocarcinoma (294,298–301). Table 33.6 lists histologic features that aid in this distinction.

The mucosal abnormalities of mucosal prolapse syndromes also mimic chronic UC. Knowledge of the clinical picture and the recognition of the characteristic fibromuscular obliteration of the lamina propria aid in this distinction. The histologic appearance of colorectal polyps from patients with Cowden disease (multiple hamartoma syndrome/PTEN-associated polyposis) is identical to that of the mucosal prolapse syndrome (302). Mucosal prolapse syndrome and Cowden disease must be separated on clinical grounds. Rectal ulcers associated with the use of ergotamine suppositories may be grossly and microscopically identical to mucosal prolapse syndrome (303).
TERMINAL ILEUM BIOPSY SPECIMEN INTERPRETATION
Terminal ileal biopsy is increasingly a part of the endoscopic workup for patients with lower gastrointestinal symptoms (304–308). Lymphoid aggregates—a normal finding, especially in younger patients—can frequently be misinterpreted as inflammation and architectural distortion of chronic enteritis. The increased intraepithelial lymphocytes of the normal M-cell zone should not be misinterpreted as evidence of CS or lymphocytic enterocolitis (3). Fibromuscular proliferation in the lamina propria is quite common in the normal terminal ileum (208). Pigment deposition is found in a high percentage of the population. These pigments are environmentally derived, deposit in patchy collections of macrophages adjacent to lymphoid aggregates, and are of no clinical significance (3,6,7).
Minimal focal acute inflammation in surface epithelium is commonly encountered and difficult to interpret. This minimal active enteritis could be a response to bowel preparation or a trauma/prolapse effect. More intense collections of neutrophils with erosion, cryptitis, and crypt abscess formation usually coexist with architectural change and pyloric gland metaplasia. These changes are sometimes seen in patients with IBD demonstrating a pancolitis in which it could represent so-called backwash ileitis. In some classification schemes, this histology would suggest CD or primary IBD of an indeterminate type (see the following texts). In practice, this terminal ileal abnormality encountered as an isolated finding or in association with colitis/proctitis sparing the right colon can be considered consistent with CD, but similar lesions have been described in patients taking NSAIDs. Granulomas should be searched for in all terminal ileum biopsy specimens, but especially in patients with chronic active enteritis patterns with pyloric gland metaplasia (308).
Giardiasis can be seen in terminal ileal biopsy specimens (146). Intraepithelial lymphocytosis in the terminal ileum (>15 intraepithelial lymphocytes per 100 colonic enterocytes vs. normal of 6) (304) is abnormal and could be a manifestation of CS, or it may be seen in patients with lymphocytic enterocolitis (307).
INTERPRETATION OF ENDOSCOPIC BIOPSY SPECIMENS FROM IMMUNOSUPPRESSED PATIENTS, INCLUDING PATIENTS WITH HUMAN IMMUNODEFICIENCY VIRUS AND ACQUIRED IMMUNODEFICIENCY SYNDROME
Profound changes have occurred in the medical management of human immunodeficiency virus (HIV) and AIDS with highly active antiretroviral treatment. This therapy has markedly reduced the incidence of opportunistic infection, and it is invariably the first-line therapy not only for HIV and AIDS but also for the complicating infections. However, the interpretation of intestinal biopsy specimens from patients with untreated HIV and AIDS remains one of the premier challenges in surgical pathology. These patients are afflicted by a virtual smorgasbord of traumatic, infectious, and neoplastic conditions, and many of these lesions are subtle, so they can be easily missed unless specifically and systematically sought. Furthermore, multiple lesions characteristically coexist in the same biopsy specimen.
Traumatic lesions in HIV and AIDS are usually recognized by the clinician, and biopsy is not performed. The neoplastic diseases associated with HIV and AIDS, such as anal and perianal squamous carcinoma and malignant lymphoma, are covered in more detail elsewhere in this text. This section concentrates specifically on the infectious and inflammatory-like processes affecting the small bowel and colon in patients with HIV and AIDS.
COMMON INFECTIOUS AGENTS
Bacterial Infection
Infection with the usual bacterial intestinal pathogens is common in men who have sex with men (MSM) and in patients with HIV or AIDS (217,309,310). The diagnosis is made by identification of the organism in stool culture. Biopsy specimens often illustrate the spectrum of changes associated with infectious colitis or acute self-limited colitis as discussed earlier.
An increasing incidence of syphilis and lymphogranuloma venereum has been reported in HIV-positive MSM (311). These usually present as proctitis and can clinically mimic IBD or even neoplasia. Intense lymphohistiocytic infiltrates, prominent plasma cells, and lymphoid aggregates, without architectural changes and without marked active inflammation, are described in biopsy specimens. Silver stains and even IHC for T. pallidum are not sensitive, and diagnosis requires serum testing or special studies done on rectal swabs (311).
Parasitic Infestations
G. lamblia infects about one-third of MSM in the urban community, and it can be seen in patients with HIV or AIDS (309). E. histolytica shows an increased prevalence in MSM community and in patients with HIV or AIDS. It often represents a nonpathogenic commensal in MSM; in patients with HIV or AIDS, it can rarely cause fulminant colitis (309). The pathologic changes noted with each parasite are similar to those seen in the non-AIDS patient.
Viral Infections
CMV commonly infects patients with HIV or AIDS and is often present in biopsy specimens (309,310). CMV inclusions are usually encountered in endothelial cells, fibroblasts, and smooth muscle cells; only rarely are they seen in epithelial cells. Infected cells can be highlighted with immunostains. CMV inclusions in the gut usually reflect disseminated CMV infection; the precise role of CMV in the pathogenesis of intestinal disease is unknown and is difficult to assess (312). However, marked endothelial CMV infection with “vasculitis” and luminal thrombosis is the probable cause of severe necrotizing gut injury and perforation, typically in the ileocecal area.
HSV is associated with painful discrete ulcers, vesicles, or pustular lesions in the distal rectum or perianal skin (309,310,313). It is easily cultured and immunostained. The inflammatory pattern in the rectum shows (a) ulceration with neutrophils within the lamina propria, (b) cryptitis, and (c) crypt abscess formation. HSV inclusions are not seen in the colon or rectum, but they are found in the anal transition zone epithelium or perianal skin.
Adenovirus-associated colitis has also been described in patients with HIV or AIDS (309,310). The mucosa shows moderate architectural changes and chronic inflammation. Infected colonic epithelial cells can be seen at the surface, and they have the appearance of dystrophic goblet cells with a crescent-shaped or sickle-shaped amphophilic nucleus that occasionally contains inclusion bodies (203,314). Adenovirus can be verified by IHC or electron microscopy.
Fungal Infections
Although oral and esophageal candidiasis is common in HIV or AIDS (309,310), seeing this organism in biopsy specimens from the small intestine or colon is unusual unless disseminated candidiasis has occurred.
UNUSUAL INFECTIOUS AGENTS COMMONLY FOUND IN PATIENTS WITH ACQUIRED IMMUNODEFICIENCY SYNDROME
Bacterial Infections
Intestinal spirochetosis (infections with Brachyspira aalborgi or Brachyspira pilosicoli) can be seen in patients with HIV or AIDS (315). My experience is that it is encountered more frequently in immunocompetent patients in whom its significance as a cause of colitis and appendicitis is controversial (315–319). The microscopic appearance is subtle—a thickening or accentuation of the colonic brush border that deeply stains with hematoxylin is caused by the spirochetes that align in parallel, embedding themselves into the luminal border of the absorptive cells (Fig. 33.25). They do not attach to goblet cells. Identification of intestinal spirochetosis can be enhanced by the use of the Warthin-Starry or Dieterle stain or by electron microscopy. The organism will immunostain using antibodies to Treponema species.
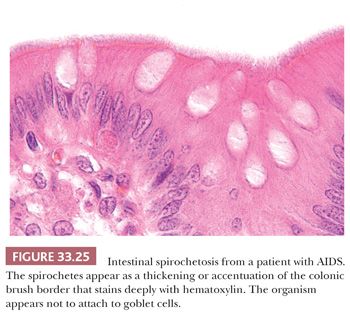
Gastrointestinal tract involvement by MAIC is usually part of disseminated infection (320,321). The organism is readily cultured from stool and even blood. MAIC may affect any part of the gastrointestinal tract. The characteristic histology is infiltration of the lamina propria by foamy macrophages, which can closely mimic Whipple disease (Fig. 33.26A) (113,320,321). In addition, MAIC is also PAS-positive, further adding to the possibility of a misdiagnosis. MAIC is easily distinguished from Whipple disease because MAIC is characteristically patchy, the fat vacuoles of Whipple disease are absent, the PAS stain shows faintly staining bacillary forms (Fig. 33.26B), and the acid-fast stain is positive. The infiltrative macrophages may be subtle, and the recommendation is that an acid-fast stain be done on all mucosal biopsy specimens from patients with HIV or AIDS.
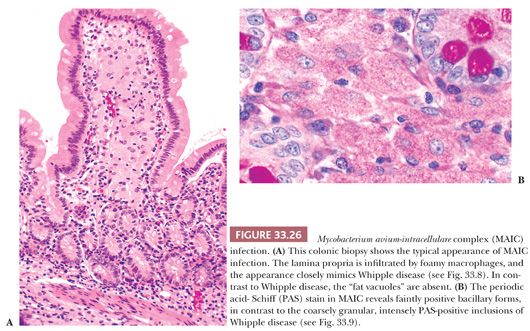
Mycobacterium tuberculosis infection in the GI tract has histologic features that can vary from well-formed focally necrotizing granulomas to neutrophil-rich histiocytic infiltrates with suppuration to mycobacterial spindle cell pseudotumors (311).
Diarrheogenic bacterial enterocolitis has been described in patients with HIV or AIDS (322). Colonic biopsy specimens demonstrate surface epithelial inflammation and degeneration associated with Gram-negative bacteria adherent to the surface colonocytes (Fig. 33.27). The histology resembles that seen in EPEC infection (323).
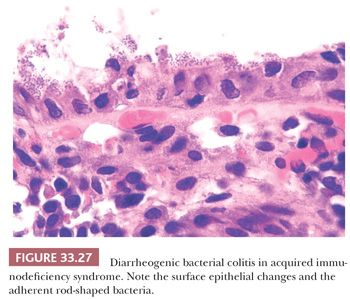
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


