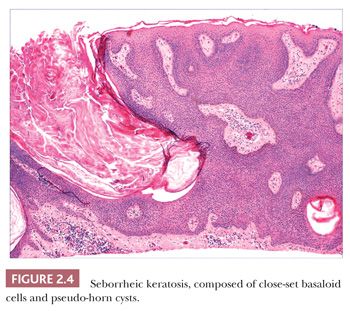
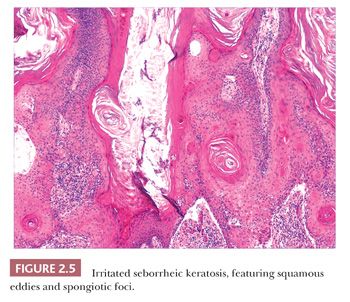
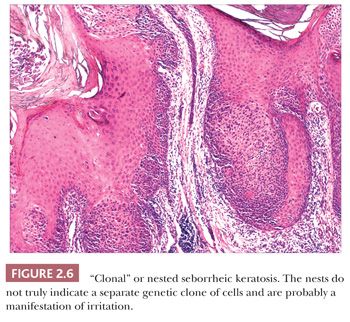
Seborrheic keratosis is one of the most common benign epidermal lesions of adult life. It typically presents from the fourth decade and beyond as a rough-surfaced papule, nodule, or plaque on virtually any cutaneous surface other than palms and soles. Sometimes, these lesions form large annular plaques or show changes of irritation or inflammation. Pigmented, papillomatous variants arising particularly in the malar regions of African American individuals are termed dermatosis papulosa nigra. Seborrheic keratoses are mostly exophytic, showing varying degrees of papillomatosis and acanthosis. They are composed of close-set basaloid cells and often feature “pseudo-horn cysts,” which are invaginations of the involved epidermis, filled with keratin, that in cross section appear to be intraepidermal “cysts” (Fig. 2.4). However, there can be considerable microscopic variability among seborrheic keratoses. They can be smooth surfaced and acanthotic, show reticulated downgrowths of interconnecting epidermal cords, or when irritated, feature spongiosis and whorled arrangements of keratinocytes (squamous eddies) (Fig. 2.5). So-called “clonal” seborrheic keratoses contain discrete clusters of keratinocytes within the epidermis; this may be another manifestation of irritation (Fig. 2.6). Papillomatous variants can closely resemble epidermal nevi. Therefore, when signing out such cases, the age of the patient should be taken into account; lesions resembling seborrheic keratoses in children or young adults are most likely either verrucae or epidermal nevi. It is also likely that lesions resembling seborrheic keratosis that arise in the anogenital region of young adults are actually verrucae (8). The sign of Leser-Trélat is the rapid appearance of pruritic seborrheic keratoses in association with internal malignancy, particularly adenocarcinomas of the gastrointestinal tract (9). A lymphocytic infiltrate is typically seen on microscopic examination of these lesions.
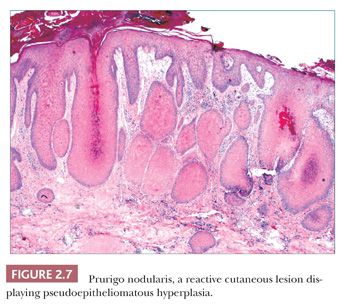
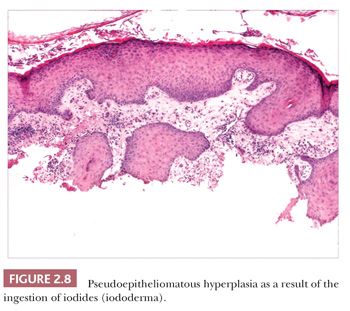
Pseudoepitheliomatous hyperplasia is characterized by extreme acanthosis of the epidermis with extension deep into the dermis. The lesions are often exophytic and verruciform. The microscopic image can raise concerns about squamous cell carcinoma, but there is a lack of cytologic atypia. A variety of conditions can be accompanied by pseudoepitheliomatous hyperplasia, including prurigo nodularis (an exaggerated host response to chronic rubbing and scratching of the skin) (Fig. 2.7), hypertrophic lichen planus, certain deep fungal infections (North American blastomycosis is the prototype), halogenodermas (e.g., bromoderma, iododerma) (Fig. 2.8), and granular cell tumors. The task of the pathologist in these cases is twofold: first, to recognize that the changes are reactive and not neoplastic, and second, to determine the underlying cause if possible. Thus, vertical streaking of thick papillary dermal collagen and chronic inflammation tend to accompany prurigo nodules; a lichenoid tissue reaction is seen in hypertrophic lichen planus, particularly at the tips of proliferative rete ridges; granulomas and intraepidermal neutrophilic microabscesses are seen in deep fungal infections; neutrophils often accompany halogenodermas; and dermal granular cell elements denote an accompanying granular cell tumor.
Other Benign Epidermal Tumors
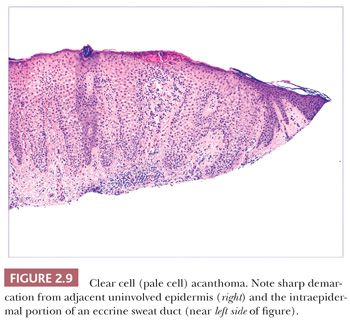
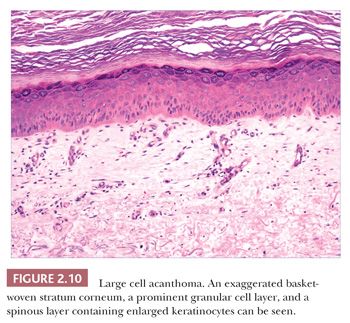
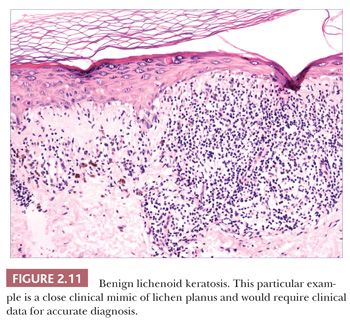
Some benign epidermal tumors are smooth surfaced. The clear cell acanthoma (pale cell acanthoma) is a circumscribed red nodule with peripheral scale that often arises on the leg of middle-aged or older adults (10). Microscopically, these lesions are acanthotic and composed of cells with pale-staining cytoplasm. The surface is smooth and often devoid of stratum corneum or stratum granulosum. A noteworthy finding is that the involved epithelium is sharply demarcated from the adjacent uninvolved epidermis and from adnexal epithelia, a feature of considerable diagnostic importance (Fig. 2.9) (11,12). Similar to hyperproliferative inflammatory processes such as psoriasis, clear cell acanthomas demonstrate increased levels of keratinocyte growth factor and downregulation of the high-affinity tyrosine kinase receptor for keratinocyte growth factor (13). Large cell acanthomas are flat to barely elevated, hyperpigmented patches with sharply demarcated borders that also arise in adult life (14). The epidermis may display an exaggeratedly basket-woven stratum corneum, whereas the constituent keratinocytes show cytoplasmic and nuclear enlargement (Fig. 2.10). Basilar hypermelanosis is often identified. These lesions are most often regarded as variants of solar lentigines (15). Aneuploidy was found in one study, but significant proliferation was not found using immunohistochemical methods, with a mean DNA index between those of actinic keratosis and Bowen disease (16). Benign lichenoid keratoses are characterized by vacuolar alteration of the basilar layer and, at times, a bandlike superficial dermal infiltrate, thereby mimicking the inflammatory dermatosis, lichen planus (Fig. 2.11). These are believed to arise in preexisting solar lentigines and seborrheic keratoses (17,18). The differential diagnosis includes lichenoid actinic keratosis, in which there is a greater degree of basilar keratinocyte atypia that extends beyond the zone of dermal inflammation. Occasionally, a small biopsy of lentigo maligna (or lentiginous melanoma in situ) can show a lichenoid tissue reaction that almost completely obscures the single-cell atypical junctional melanocytic proliferation. If the latter is suspected, additional levels or staining for MART-1 may be useful (19).
EPITHELIAL CYSTS
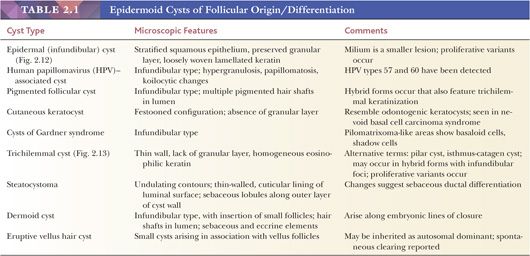
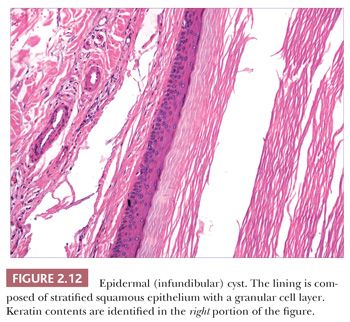
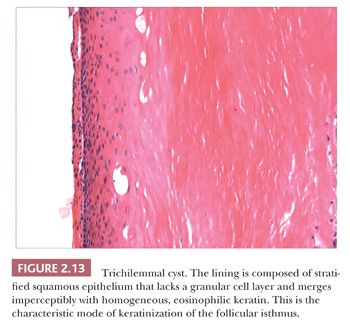
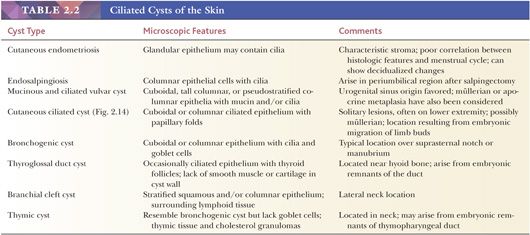
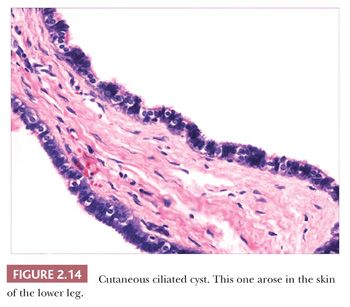
Classification schemes vary for the cystic epithelial lesions arising in skin. Several of these cystic lesions are conventionally included among the adnexal tumors, but often, cysts composed of stratified squamous epithelium (epidermoid cysts, or cysts of follicular origin or differentiation) (20–25) or those containing cilia (an uncommon feature of cutaneous lesions) (26–29) are classified separately. Tables 2.1 and 2.2 provide a simplified scheme for diagnosing these lesions.
BENIGN ADNEXAL (APPENDAGEAL) TUMORS
There is a startling array of tumors that demonstrate differentiation toward the cutaneous appendages: hair follicle, sebaceous gland, apocrine gland, or eccrine gland. These vary in their degree of differentiation—the degree to which they mimic the normal appendage—and have been classified in descending order of differentiation as hyperplasias or hamartomas, adenomas, and epitheliomas. The theory is that pluripotential stem cells are capable of differentiating toward these epithelial structures.
Several systems have been suggested for classifying the cutaneous adnexal tumors (30–32). Tables of adnexal tumors have been constructed that appear to neatly group these lesions by degree of differentiation and by the appendage to which they appear to be differentiating (33). These schemes have been satisfying in many ways because they at least provide scaffolding upon which to build one’s understanding of these tumors. However, problems have arisen with this method, which can be summarized in the following manner:
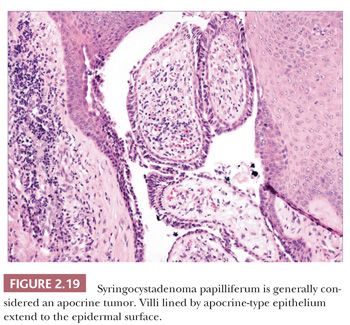
1. The degrees of differentiation are subject to debate. For example, should syringocystadenoma papilliferum be grouped with the adenomas rather than the epitheliomas? Should poroma be considered an epithelioma rather than an adenoma?
2. The original grouping of sweat gland tumors into either the apocrine or eccrine category was based on a technique called enzyme histochemistry as well as conventional microscopic morphology. In recent years, reevaluation of these tumors both morphologically and by immunohistochemical methods has resulted in reclassification of many of the sweat gland tumors. Thus, many chondroid syringomas (cutaneous mixed tumors) are now regarded as apocrine rather than eccrine in differentiation (34), and although acrospiromas can be either apocrine or eccrine, apocrine differentiation is favored for most of them (35,36).
3. Although there are certainly many “classic” examples of adnexal tumors, others defy categorization because a given lesion may not only show different lines of differentiation but also different degrees of differentiation. This seems to be a common problem in consultation practice.
4. Adnexal carcinomas do not always fit comfortably into existing classification schemes.
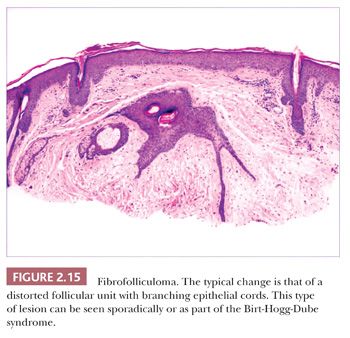
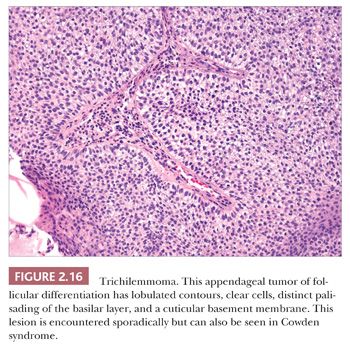
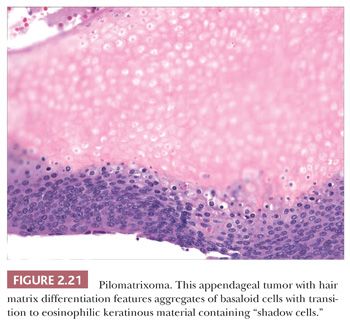
One could argue that the differences among the benign adnexal tumors are largely academic because the precise categorization of tumor rarely seems to make a difference in terms of clinical management. However, some of these tumors have a close relationship to genetically determined syndromes that may have significance in terms of proneness to internal malignancy or other health issues. Examples include multiple trichodiscomas and fibrofolliculomas (Fig. 2.15) (Birt-Hogg-Dube syndrome) (37), trichilemmomas (Fig. 2.16) (Cowden syndrome) (38), or sebaceous tumors (Muir-Torre syndrome) (39). Progression of apparently benign adnexal tumors to malignancy has been reported in several instances (examples include pilomatrixoma, acrospiroma, and poroma). Finally, although knowledge of the precise differentiation pathway may not make a difference in the immediate clinical situation, an understanding of these tumors may lead to discoveries that could have significant impact in other areas of medicine.
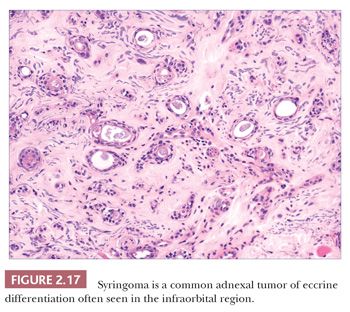
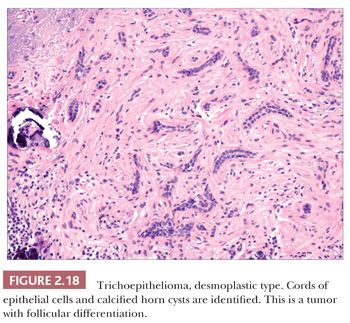
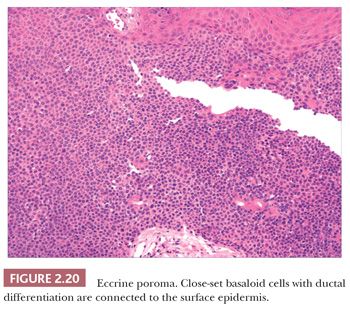
Although space considerations limit the ability to consider these lesions in depth, Table 2.3 provides a listing of the most important benign adnexal tumors of skin. Figures 2.17 to 2.21 display the microscopic features of five representative tumors: syringoma (Fig. 2.17), trichoepithelioma (Fig. 2.18), syringocystadenoma papilliferum (Fig. 2.19), eccrine poroma (Fig. 2.20), and pilomatrixoma (Fig. 2.21).
ADNEXAL CARCINOMAS
There is a group of malignant epithelial tumors that show some degree of differentiation toward recognizable adnexal structures, yet display infiltration of connective tissues as well as pleomorphism, cell necrosis, increased mitotic rate, and sometimes atypical mitotic figures. Some of these tumors resemble their counterparts among the benign adnexal tumors, whereas others are microscopically unique. Table 2.4 provides a listing of the important adnexal carcinomas grouped according to their lines of differentiation.

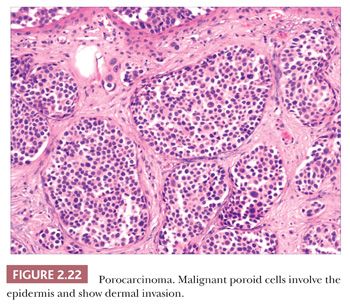
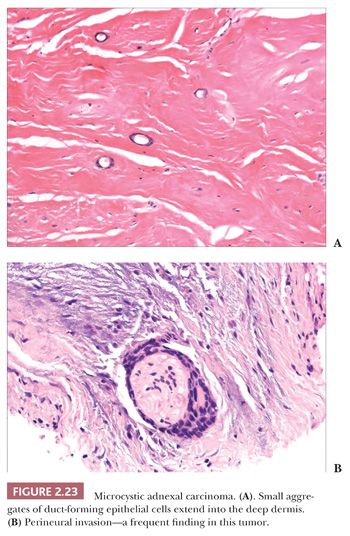
Although some of the adnexal carcinomas are readily identified as such, through their epidermal relationships or their derivation from/resemblance to benign adnexal tumors, others can closely resemble metastatic tumors from other sites, such as the breast. In these instances, clinical and radiographic data should be considered as important components of the diagnostic process. In addition, immunohistochemical methods have recently been brought to bear on the distinction between primary cutaneous adnexal carcinomas and metastatic carcinomas originating from internal sites; for example, cytokeratin 5/6 and p63 have been recommended because they appear to be preferentially expressed in neoplasms of cutaneous origin (40,41). Podoplanin (D2-40) has also been found to be useful in this situation in that there is expression of this antigen in most primary cutaneous neoplasms but not in metastatic carcinomas to the skin. This marker may be particularly useful when used in combination with p63 (42). Two other considerations should be mentioned with regard to the adnexal carcinomas. First, one must be cautious in judging the significance of mitotic activity and nuclear pleomorphism because both of these can be observed in benign adnexal tumors. For example, frequent mitoses can be observed in certain sweat gland tumors, such as acrospiroma, and some proliferating trichilemmal tumors can show both mitoses and foci of nuclear pleomorphism (43). Therefore, it is important to interpret these features in the overall context of the lesion, keeping in mind both the overall level of differentiation and the presence or absence of infiltration of the surrounding stroma. Second, intermediate levels of malignancy are increasingly being recognized among cutaneous adnexal tumors. An “intermediate-grade” lesion shows infiltration of connective tissues at the periphery of the tumor, whereas other histopathologic characteristics of malignancy (e.g., pleomorphism, cell necrosis, mitotic activity) may be less apparent. There is evidence that such tumors are prone to recurrences, sometimes accompanied by greater degrees of cytologic atypia. Examples of such tumors have been seen in association with pilomatrixomas, proliferating trichilemmal tumors, clear cell hidradenomas (acrospiromas), and poromas.
PREMALIGNANT AND MALIGNANT EPIDERMAL TUMORS
Actinic Keratosis
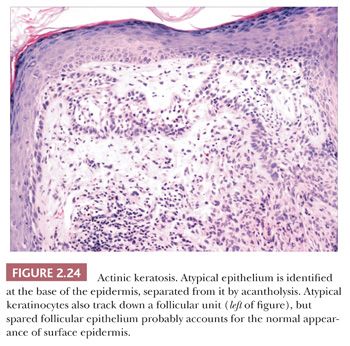
This is a common, sun-induced lesion that most often presents as a rough-surfaced to crusted patch, particularly over the head and neck and extremities. Lesions are variably elevated and can be erythematous, flesh colored, or hyperpigmented. They typically arise in middle-aged to older individuals but can be encountered in young adults as well and can even be seen in childhood in the rare, recessively inherited disorder xeroderma pigmentosum, which is a condition characterized by abnormal repair of ultraviolet-damaged DNA. Similar keratoses have arisen in individuals exposed to inorganic pentavalent arsenic. Actinic keratoses can progress to invasive squamous cell carcinoma at rates ranging from 0.1% to 10% (44). Metastases from actinic keratosis–related squamous cell carcinomas are uncommon, ranging in incidence from 0.5% to 3.0% (45,46). Microscopically, actinic keratoses often show atypia of the basilar or immediately suprabasilar keratinocytes, with nuclear crowding, pleomorphism, and irregular epithelial budding. Surface maturation of the involved epidermis is often preserved, at least initially, and adnexal epithelium is spared; in fact, it is believed that cells derived from these adnexal structures (e.g., hair follicles, eccrine sweat ducts) are responsible for the preservation of normal-appearing surface epidermis (Fig. 2.24) (47). However, with progression, greater involvement of the adnexa and surface epidermis occurs, so that eventually the entire thickness of epidermis may become atypical. Clinical and microscopic variants include bowenoid actinic keratosis, an actinic keratosis that has achieved full-thickness epithelial atypia (i.e., squamous cell carcinoma in situ) and resembles Bowen disease (see next section); atrophic or hypertrophic actinic keratosis; the spreading, pigmented actinic keratosis (showing overlapping features with solar lentigo) (48,49); and the acantholytic actinic keratosis, in which loss of cohesion among atypical basilar keratinocytes creates an image resembling various acantholytic dermatoses (50). The latter lesion can evolve into an invasive, acantholytic (“pseudoglandular”) squamous cell carcinoma. Lichenoid actinic keratoses combine the elements of typical actinic keratosis with a bandlike infiltrate that may partly obscure the dermal-epidermal interface (51). Diagnostic challenges may arise when attempting to distinguish actinic keratosis variants from nonmalignant acantholytic dermatoses (e.g., Grover disease, Darier disease), benign lichenoid keratosis, large cell acanthoma, solar lentigo, or early lentigo maligna. Other malignant or premalignant dermatoses that can microscopically resemble actinic keratosis include superficial basal cell carcinoma and forms of porokeratosis. It is not clear whether there is a difference in prognosis between the bowenoid actinic keratosis and true Bowen disease. One study has shown a similar histochemical profile for Bowen disease and bowenoid actinic keratosis using markers for p53 and p16 (52).
Bowen Disease (Squamous Cell Carcinoma in Situ)
Squamous cell carcinoma in situ of the skin, defined as full-thickness epidermal atypia, can appear in several forms. The best-established type is Bowen disease. These lesions appear as erythematous, sometimes pigmented plaques that can develop anywhere on the cutaneous surface, including sun-exposed and non–sun-exposed sites. Plaques can be irregular in shape but tend to be sharply demarcated, and foci of sparing are often identified. Bowen disease can apparently be triggered by several factors. Ultraviolet exposure may be one of these and is at least responsible for the bowenoid actinic keratosis referred to earlier. However, the formation of these lesions in sun-protected sites indicates that other carcinogens may be at work; the best established of these are inorganic arsenic exposure and certain types of HPV, particularly HPV-16. Lesions of Bowen disease uncommonly become invasive, but there is evidence that once invasive, these lesions have a high metastatic rate—higher than would be the case for squamous cell carcinomas arising in actinic keratoses. Bowenoid papulosis is a condition in which single or multiple papules or plaques develop in the anogenital region (53,54). These are histologically atypical, although they often lack the florid atypia seen in fully developed Bowen disease (53). In this respect, they more closely resemble lesser grades of vulvar intraepithelial neoplasia and currently are most often incorporated within the latter classification system. Lesions of Bowen disease rarely become invasive, and in fact, they may spontaneously regress or respond to conservative therapies. However, once invasive, there is a risk of metastasis that varies from 13% to 37%. Squamous cell carcinoma in situ arising on mucocutaneous sites is termed erythroplasia (erythroplasia of Queyrat, in the case of those arising on the glans penis) (55).
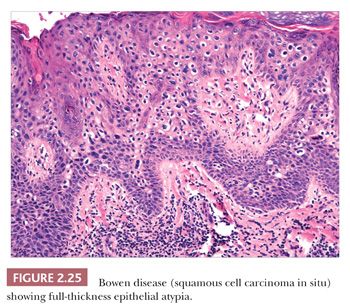
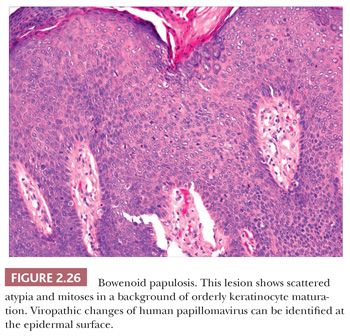
Microscopically, lesions of Bowen disease display full-thickness loss of keratinocyte maturation, with cells showing large, hyperchromatic nuclei; multinucleated keratinocytes; and mitotic figures (some atypical) at all levels of the epidermis (Fig. 2.25). Scattered vacuolated cells can mimic the image of Paget or extramammary Paget disease (this change is especially common at the edges of a lesion), or clusters of keratinocytes may produce a “nested” configuration. There may be “skip areas” of uninvolved epidermis. Typically, there is involvement of follicular epithelium extending to the level of the sebaceous duct. Similar changes are seen in forms of erythroplasia. In bowenoid actinic keratoses, there may be follicular sparing or changes of partial thickness, predominantly basilar atypia at the periphery of the lesions. It is not entirely clear whether the biology of untreated bowenoid actinic keratoses is similar to that of Bowen disease arising in sun-protected sites. In bowenoid papulosis, there are scattered atypical keratinocytes and frequent mitotic figures that are often in the same stage of development, but these tend to occur on a background of more orderly keratinocyte maturation (Fig. 2.26). The differential diagnosis of Bowen disease, particularly the pagetoid variant, includes Paget disease or extramammary Paget disease, but the latter can usually be recognized by its location, lack of pleomorphism among non-Paget epithelial elements, compression of the basilar layer by Paget cells, and the staining characteristics of Paget cells (e.g., positive staining for mucin, carcinoembryonic antigen, low–molecular-weight cytokeratin). Malignant melanoma with pagetoid features also lacks keratinocyte atypia; shows frequent nesting, particularly along the junctional zone, without compression of the basilar layer; and produces a quite different immunoprofile because the neoplastic cells express S-100 protein and other melanocytic antigens. Occasionally, irritated seborrheic keratoses can show some nuclear pleomorphism and mitoses at upper levels of the epidermis, resembling carcinoma in situ and thereby creating considerable diagnostic difficulty. In our experience, this situation is most common among lesions from elderly individuals and is a particular problem when only partial biopsies are provided (56,57).
Keratoacanthoma
In the mid-twentieth century, several authors, notably Rook and Whimster (58) and Musso (59), delineated a proliferative epithelial lesion with resemblances to squamous cell carcinoma that demonstrated spontaneous involution and self-healing. This lesion was termed keratoacanthoma. Although widely accepted for years as a pseudomalignancy, many experts currently regard this lesion as a variant form of squamous cell carcinoma (60). This has occurred, in part, because some lesions initially considered keratoacanthomas have metastasized (61) and, in part, because many immunohistochemical, ultrastructural, and genetic investigations have failed to reveal reliable differences from squamous cell carcinoma (62–64). Follicular derivation and a relationship to HPV infection has been supported by some studies (65).
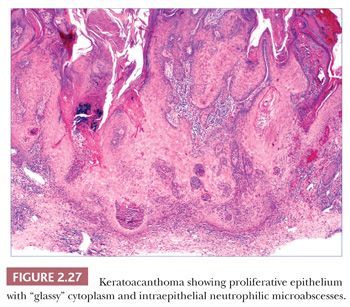
Keratoacanthomas develop most often in middle-aged to elderly adults, frequently (but not exclusively) on sun-exposed skin. They are rapidly evolving lesions, often developing over a period of 6 to 8 weeks, and present as dome-shaped nodules with central keratin craters. Barring therapeutic intervention, their natural history is to remain stationary for a time and then involute over a 6-month to 1-year period, leaving behind a ragged, crateriform scar. Special varieties include the giant keratoacanthoma known as keratoacanthoma centrifugum marginatum, an expanding, annular lesion; multiple self-healing epitheliomas of Ferguson-Smith; and the rare eruptive keratoacanthomas of Grzybowski. In cross sections of a fully formed lesion, there is a central keratin crater with a buttresslike arrangement of adjacent, uninvolved epidermis at either site of the crater. Underlying proliferative epithelium extends into the dermis, composed of cells with glassy-appearing cytoplasm and bland nuclei with low nuclear-to-cytoplasmic ratios. Nuclear pleomorphism and mitotic activity may be observed at the base of the lesion, and perineural infiltration is sometimes identified (66). Intraepidermal neutrophilic microabscesses are commonly found (Fig. 2.27). Although the accompanying dermal infiltrate may contain eosinophils, these can occasionally be found in squamous cell carcinomas and cannot be considered an absolutely reliable diagnostic feature. Subungual tumors often display extensive apoptosis and only scarce neutrophils or eosinophils in the accompanying infiltrate (67). Findings in involuting keratoacanthomas include apoptotic keratinocytes, a shallow crater with atrophic underlying epithelium, and dermal scar.
Not all pathologists will make a diagnosis of keratoacanthoma, instead preferring a designation such as “squamous cell carcinoma, keratoacanthoma type.” I will still render a diagnosis of keratoacanthoma, but the criteria for this diagnosis are fairly strict: (a) the entire lesion must be sampled and displayed in cross section; (b) the classic microscopic features must be observed, as listed earlier; (c) there should be a clear clinical description of the lesion, that is, a rapidly evolving crateriform lesion, with keratoacanthoma as the primary clinical diagnosis, provided by an experienced clinician, preferably a dermatologist; and (d) there should be no history of immunosuppression.
Squamous Cell Carcinoma
This is the second most common cutaneous malignancy (basal cell carcinoma being most common). It usually presents as an indurated papule or nodule with crusting or ulceration. Squamous cell carcinomas most often arise in actinically damaged skin, particularly in actinic keratoses, but they also develop in mucocutaneous sites, chronic ulcers, burn scars, sinuses, or skin lesions subjected to chronic inflammation or scarring (e.g., discoid lupus erythematosus). They are prone to occur in immunocompromised patients, particularly in renal transplantation patients. The metastatic rate of actinically induced squamous cell carcinomas is low, ranging from 0.5% to 3.0% (46), although from a pathology perspective, the finding of actinic keratosis contiguous to a squamous cell carcinoma is not a useful predictor of metastatic behavior (tumor thickness and depth of invasion are most important in this regard) (68). Metastatic rates are higher among neoplasms arising in other settings, such as mucous membranes or burn scars.
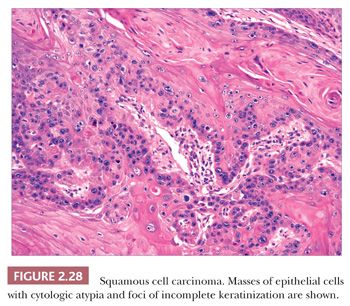
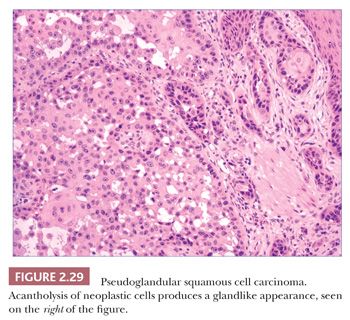
In typical examples, there are masses of epithelial cells within the dermis that display nuclear pleomorphism, atypical mitotic figures, and foci of incomplete keratinization termed horn pearls (Fig. 2.28). These epithelial islands extend from the surface epidermis, which usually also displays varying degrees of atypia ranging from actinic keratosis to squamous cell carcinoma in situ. Loss of cohesion of keratinocytes within tumor islands produces an acantholytic, or pseudoglandular, appearance (Fig. 2.29). One variant termed pseudovascular adenoid squamous cell carcinoma can closely mimic angiosarcoma; it requires careful inspection and sometimes immunohistochemical staining (cytokeratin positive, CD31−) for confirmation, although it, too, may demonstrate biologic aggressiveness (69). Spindle cell squamous cell carcinoma raises a differential diagnosis that includes spindle cell malignant melanoma and atypical fibroxanthoma. Epidermal connections are not always demonstrable, and it may be necessary to employ an immunohistochemical panel that includes S-100 protein, CD10, or other markers in addition to cytokeratins. Infiltrative, desmoplastic squamous cell carcinomas may have a higher rate of local recurrence and metastasis compared with neoplasms lacking these features (70). Other variant forms that may be encountered include signet ring cell squamous cell carcinoma (71,72) and carcinosarcoma (sarcomatoid carcinoma), the latter combining the features of carcinoma and assorted types of sarcomas (73). Invasive squamous cell carcinomas arising from Bowen disease often display basaloid tumor islands with nuclear pleomorphism, resembling the in situ carcinoma of origin or displaying features that more closely resemble an adnexal carcinoma (74,75).
Although the histopathologic diagnosis of cutaneous squamous cell carcinoma is often straightforward, differential diagnostic problems may arise in several circumstances. Issues regarding spindle cell squamous cell carcinoma have already been mentioned. Signet ring cell squamous cell carcinoma can resemble the rare signet ring cell carcinoma of the eyelids, a tumor with a high rate of regional or distant metastases (76). Unlike squamous cell carcinoma, the cells of the latter neoplasm may be positive for milk fat globule protein as well as estrogen and progesterone receptors. Furthermore, squamous cell carcinoma can be confused with basal cell carcinoma, or vice versa, in several circumstances (e.g., in instances of bowenoid squamous cell carcinoma or metatypical basal cell carcinoma) (77).
Verrucous Carcinoma
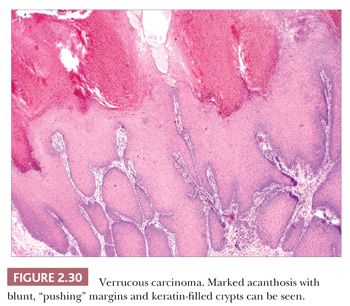
This particularly well-differentiated variant of squamous cell carcinoma develops on the sole of the foot, where it is sometimes called epithelioma cuniculatum (a reference to the deep lesional crypts that bear a resemblance to rabbit burrows), but it is also seen in the anogenital region, oral cavity, and occasionally in other locations or in association with chronic sinuses and ulcers. Lesions on the soles are often clinically thought to represent verrucae, but they are particularly resistant to the usual wart remedies. Although metastases are rare, these lesions can invade and destroy deep structures; therefore, complete removal is indicated (78,79). Anaplastic transformation can occur spontaneously and, despite earlier teachings, does not always accompany x-ray therapy (80,81).
Microscopic features include hyperkeratosis, parakeratosis, and marked acanthosis that is both exophytic and endophytic. These lesions tend to have blunt, “pushing” margins at the base, although the epithelium often includes keratin-filled crypts containing neutrophils. Cytologic atypia tends to be minimal (Fig. 2.30) but, when found, particularly in the context of dermal invasion, suggests transformation to a biologically more aggressive neoplasm. A relationship with HPV infection is supported by the finding of HPV types 16 and 18, sometimes types 6 and 11, and other HPV types in these lesions (82,83). Resemblances to verrucae, keratoacanthoma, or forms of pseudoepitheliomatous hyperplasia can create diagnostic difficulties, especially when partial or shallow biopsies are submitted for evaluation.
Basal Cell Carcinoma
Basal cell carcinoma is the most common cutaneous malignancy, accounting for approximately 70% of malignant skin tumors (84). With the exception of genetic disorders such as nevoid basal cell carcinoma syndrome (85) or some examples of xeroderma pigmentosum, it typically occurs among adults. Although most often seen in sun-exposed sites, basal cell carcinomas also develop in sun-protected areas. The classical lesion is an ulcerated papule or nodule with a pearly, telangiectatic border, but clinical appearances can vary, and other forms include a sclerotic plaque (morpheaform basal cell carcinoma), an erythematous scaly patch (superficial basal cell carcinoma), and a pigmented lesion capable of mimicking malignant melanoma (pigmented basal cell carcinoma). Basal cell carcinomas are best known for local recurrence or tissue destruction rather than metastasis. However, metastasis has been reported in approximately 0.1% of cases, with involvement of regional lymph nodes, lung, or bone (77). This event is more likely among neglected tumors.
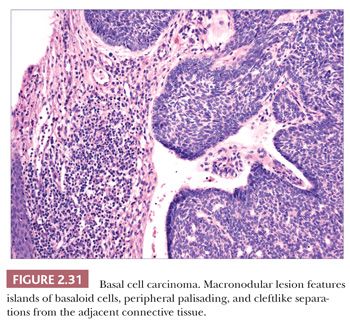
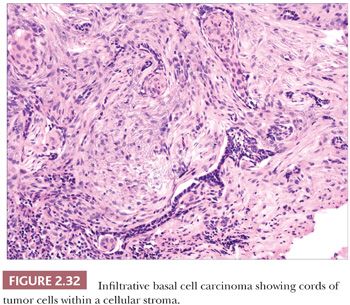
The most common histopathologic type of basal cell carcinoma is nodulocystic, composed of rounded lobules of small hyperchromatic cells. There is peripheral palisading of cells, and cleftlike spaces often separate portions of the tumor from the adjacent fibromyxoid stroma (Fig. 2.31). There are several important variant forms. The infiltrative basal cell carcinoma features narrow cords of tumor cells within a cellular stroma (Fig. 2.32) (86). When these tumor cells are embedded in a dense, collagenized, hypocellular matrix, the term morpheaform basal cell carcinoma is often used (87). The micronodular basal cell carcinoma consists of widely scattered small tumor islands, some of which may consist of only a few clustered cells. These may extend deep into the dermis/subcutis and are quite difficult to eradicate (88). Basosquamous carcinomas are somewhat controversial; many lesions that receive this designation probably represent keratinizing basal cell carcinomas, but there is another group composed of neoplastic cells having overlapping features of basal and squamous cell carcinoma that otherwise defy precise categorization (89). Studies indicate that tumors with infiltrative, micronodular, or basosquamous features tend to pursue an aggressive clinical course, and basosquamous carcinomas appear to have a propensity to metastasize.
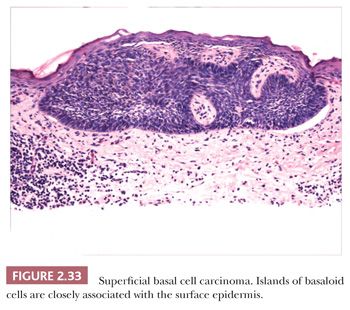
Other microscopic variants include the superficial basal cell carcinoma, in which buds of basaloid cells are identified at the base of the epidermis (Fig. 2.33); the pigmented basal cell carcinoma, in which abundant melanocytes and pigment production occur, usually within nodulocystic or superficial tumor types; and reticulated variants, such as the adenoid basal cell carcinoma and fibroepithelioma of Pinkus. Basal cell carcinomas with sebaceous differentiation (basosebaceous basal cell carcinoma) can mimic primary sebaceous neoplasms, and examples of these undoubtedly occur in Muir-Torre syndrome. Unusual forms of basal cell carcinoma feature signet ring, granular, or clear cell changes. Basal cell carcinomas must be distinguished from adnexal tumors such as trichoepithelioma and the basaloid variant of sebaceous carcinoma, basaloid follicular hamartoma, and “cloacogenic” carcinoma. Clear cell variants of basal cell carcinoma can mimic balloon cell melanoma or trichilemmal carcinoma; those with granular cell changes can be distinguished from true granular cell tumors by their distinct fibromyxoid stroma and cytokeratin positivity.
TUMORS OF CONNECTIVE TISSUE
Because several soft tissue tumors also occur in or extend to the skin and are discussed elsewhere, this section will concentrate on those neoplasms that primarily involve the dermis.
CONNECTIVE TISSUE NEVI
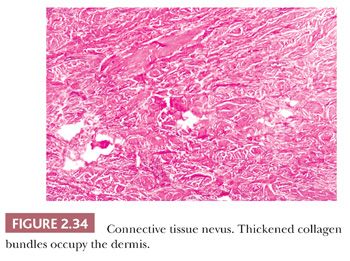
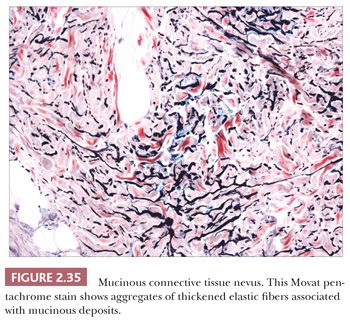
These lesions are hamartomas in which collagen is increased in quantity or thickness and may be accompanied by alterations in elastic tissue. They can occur sporadically or as a heritable, autosomal dominant trait. Connective tissue nevi present as indurated papules (90,91) or nodules that may be organized in plaques or appear in disseminated form. The epidermis tends to be uninvolved, although lesions with combined features of epidermal nevus or Becker nevus have also been reported (92). In one variant, the Buschke-Ollendorff syndrome, connective tissue nevi are associated with osteopoikilosis—sclerotic, stippled densities in long, round, and flat bones. Microscopically, thickened collagen bundles are observed, either in compact or complexly interwoven form (Fig. 2.34). Stains such as Verhoeff-van Gieson often show elastic fibers that are abnormally distributed; increased in number and density (nevus elasticus); or fragmented, diminished, or even absent (nevus anelasticus) (93,94). Increased quantities of mucin can sometimes be identified (nevus mucinosus) (95), and we have observed a few cases in which patchy increases of elastic fibers have been associated with increased quantities of mucin deposition in the same locations (Fig. 2.35). The changes in connective tissue nevi can be subtle, especially in hematoxylin and eosin–stained sections. One must also take into account the anatomic region from which a biopsy has been obtained, because dermal collagen may be particularly thick in portions of the trunk or overlying large joints. Therefore, the diagnosis depends heavily on a high index of suspicion and a good clinical history.
THE ANGIOFIBROMA FAMILY, INCLUDING FIBROUS PAPULE OF THE FACE AND ACQUIRED DIGITAL FIBROKERATOMA
The angiofibroma family is a group of generally acrally located lesions that share as common features dermal fibrosis and vascular proliferation or dilatation. Lesions commonly regarded as belonging to this group include fibrous papule of the face, pearly penile papules, the angiofibromas of tuberous sclerosis, sporadic angiofibromas, digital fibrokeratomas, and familial myxovascular fibromas. I will concentrate on two of these: fibrous papule of the face and acquired digital fibrokeratoma.
Fibrous Papule of the Face
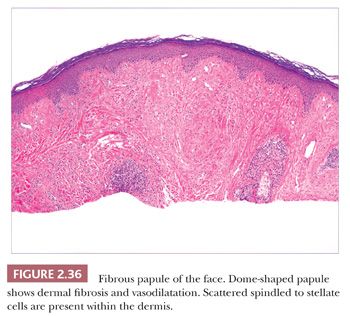
This lesion is typically a firm, dome-shaped lesion arising on the nose or central portions of the face. It is not unusual to encounter several such lesions in an individual patient (96). Fibrous papules are most often flesh colored but may be erythematous or show focal pigmentation. Despite their small size and the small shave biopsies that they usually elicit, these lesions are often recognizable on low-power microscopy. They tend to have gently dome-shaped contours and show both dermal fibrosis and vasodilatation (Fig. 2.36). On occasion, these lesions can show considerable edema. Scattered enlarged, angulated, stellate or multinucleated cells can often be observed in the papillary dermis. These were once suspected to be effete nevus cells, an impression that seemed to be reinforced by the occasional finding of unequivocal nevomelanocytes or S-100 positivity in these lesions (97). However, ultrastructural and immunohistochemical studies indicate that these stellate cells are most often factor XIIIa–positive dermal dendrocytes and that nevus cells, when present, probably represent a coincidental finding (98,99). In recent years, several histologic variants have been reported, including hypercellular, clear cell, pigmented, pleomorphic, inflammatory, granular cell (100), CD34+ (101), and epithelioid (102) types. The cells in clear cell variants have been positive for vimentin, CD68, and NKI/C3 (103), whereas those in epithelioid fibrous papule are positive for procollagen and negative for NKI/C3 (102). These lesions can be mistaken for hemangiomas, telangiectasias, and atypical melanocytic lesions; the latter occurs especially when there is overlying junctional melanocytic hyperplasia, which is a finding that is not unusual in fibrous papules.
Acquired Digital Fibrokeratoma
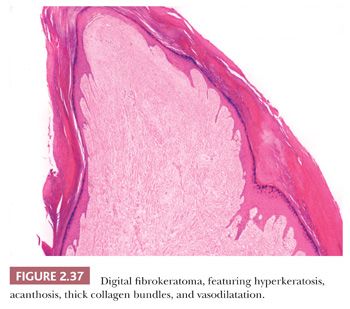
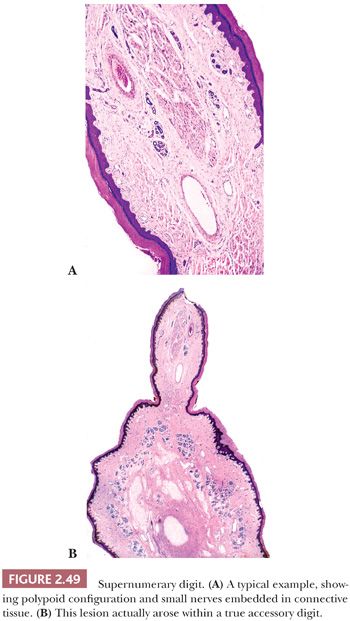
This lesion presents as a dome-shaped or pedunculated nodule found most commonly in adults on the hands, feet, fingers, and toes (104). The so-called garlic clove fibromas (Koenen tumors) that arise in periungual or subungual tissues of patients with tuberous sclerosis (and sometimes in normal individuals) are also regarded as types of digital fibrokeratomas (105,106). Microscopically, these lesions often appear as elongated or dome-shaped papules, with hyperkeratosis and acanthosis overlying thick collagen bundles that tend to be oriented perpendicularly to the overlying epidermis. Small blood vessels are a prominent feature but may at first glance be inconspicuous because of the density of surrounding collagen. Variant forms may show increased cellularity (fibroblasts) or edema (Fig. 2.37). The differential diagnosis includes particularly supernumerary digits (rudimentary polydactyly), which can closely resemble digital fibrokeratoma clinically and microscopically. However, that lesion is typically present at birth, may be bilateral, and shows prominent nerve bundles in the mid to deep dermis with features of neuroma. In fact, this lesion is believed to occur as the result of autoamputation of a true accessory digit.
DERMATOFIBROMA
Also known by the more generic term fibrous histiocytoma, the dermatofibroma is one of the most readily recognizable of the connective tissue tumors, although the variability of its features can create diagnostic difficulties. These lesions present as firm, round papules or small nodules, commonly on the lower legs but also on the arms and trunk. Multiple lesions are not uncommon in otherwise normal individuals, but multiple dermatofibromas have been reported with lupus erythematosus (107), human immunodeficiency virus (HIV) infection (108), and myasthenia gravis (109). Lesions involute even with partial therapy, and recurrences are uncommon, although they are more frequent with certain histologic subtypes, particularly cellular dermatofibromas. Recent genetic and cytogenetic studies seem to indicate that dermatofibromas are neoplastic (110,111), although there is a persistent viewpoint that they may represent a kind of reparative process (112).
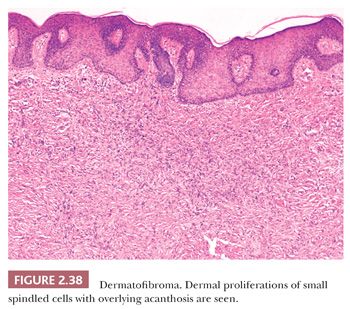
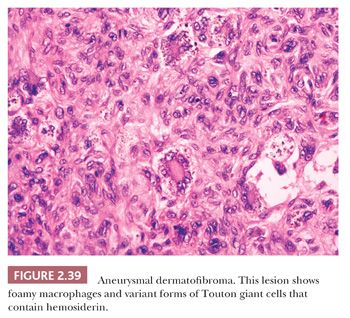
Microscopically, there is a proliferation of spindled cells between thickened collagen bundles within the dermis, forming a “lens-shaped” zone whose long axis is parallel to the overlying epidermis. The constituent cells apparently consist of dermal dendrocytes, fibroblasts, and “histiocytes” (macrophages) in varying proportions (Fig. 2.38). At the periphery of the lesion, these spindled cells surround cross-sectional profiles of collagen, a phenomenon sometimes described as “collagen entrapment.” With deeper involvement, the proliferations of cells either extend into the subcutis with bulbous, pushing margins or infiltrate within connective tissue septa. The epidermis overlying dermatofibromas is typically acanthotic, often with flattening of the bases of rete ridges, although there may also be rete ridge effacement in superficially located tumors. The phenomenon of stromal induction is sometimes seen, in which the involved connective tissue “induces” the formation of basal cell hyperplasia—resembling basal cell carcinoma, or even the formation of follicular units or sebaceous glands. Deeper variants such as the deep penetrating dermatofibroma may lack significant epidermal changes (113). Variant forms include the lipidized dermatofibroma, with numerous foamy macrophages, hemorrhage, and Touton-like giant cells; epithelioid cell histiocytoma (114–116); fibrotic types that can closely resemble sclerotic fibroma (117); aneurysmal dermatofibroma, combining features of lipidized dermatofibroma with large blood-filled spaces (Fig. 2.39) (118); and a rare granular cell dermatofibroma (119). Occasional dermatofibromas contain cells with large, bizarre, and/or hyperchromatic nuclei (dermatofibroma with monster cells) (120), but these lesions nevertheless typically behave in a benign fashion. Occasional mitotic figures can be found in dermatofibromas, but again, they do not appear to denote lesions with more aggressive biologic behavior (121). Factor XIIIa is commonly expressed in these lesions, although it sometimes appears that expression is confined to dendritic cells accompanying the tumor rather than the major constituent cells (122). This marker also may not be expressed in the aneurysmal variant (122). On the other hand, the cells of epithelioid cell histiocytoma are often strongly factor XIIIa positive. CD68 and smooth muscle actin may be expressed as well, the latter often in tumors with a myofibroblastic component. The differential diagnosis in a given case can include neurofibroma or spindle cell melanocytic lesions (S-100 positive), leiomyoma (muscle-specific actin and desmin positive), or dermatofibrosarcoma protuberans (CD34+).
FIBROMATOSIS
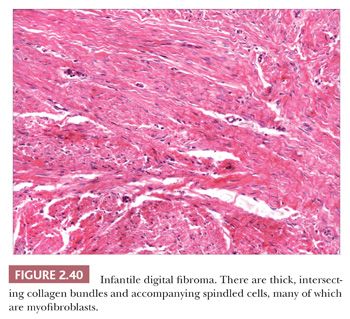
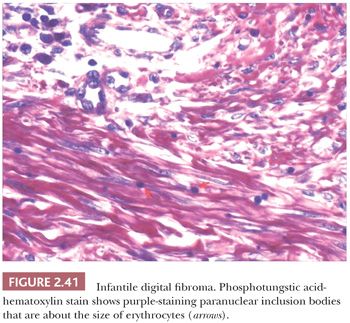
Forms of fibromatosis can extend into dermis and enter into the differential diagnosis of spindle cell cutaneous lesions, including scar. One unique form of the disease in skin is termed (recurrent) infantile digital fibroma. These fibromas manifest as solitary or multiple nodules of the fingers and toes, typically appearing in the first 3 years of life. Although recurrences are common, spontaneous regression may occur (123). On biopsy, there are thick, intersecting collagen bundles and accompanying spindled cells (Fig. 2.40) that contain paranuclear cytoplasmic inclusion bodies roughly the size of erythrocytes. These are eosinophilic with hematoxylin and eosin stain and purple with phosphotungstic acid-hematoxylin (Fig. 2.41). The inclusion bodies are actin positive (124), and ultrastructurally, they consist of bundles of microfilaments with dense bodies. This finding suggests that the constituent cells are actually myofibroblasts, and infantile digital myofibroblastoma has been proposed as a more accurate name for this tumor (125).
DERMATOFIBROSARCOMA PROTUBERANS
Dermatofibrosarcoma protuberans (DFSP) has been regarded as a fibrous tissue tumor of intermediate malignancy because it is best known for relentless growth and proneness to recurrence rather than metastasis, although the latter definitely occurs and is well documented (126). It is largely a tumor of adults, but there is a closely related pediatric tumor termed giant cell fibroblastoma. DFSP most commonly arises on the trunk or proximal extremities, presenting as one or more cutaneous nodules with surrounding induration, indicating subcutaneous growth that is often extensive. This growth pattern sometimes makes complete excision difficult, and recurrences are common. Metastases may develop in about 3% of cases, often with involvement of lymph nodes or the lungs (127).
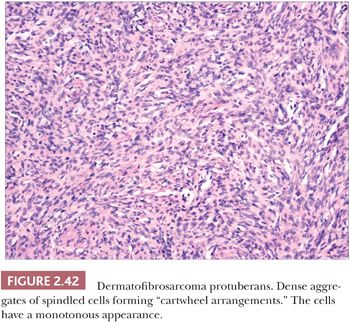
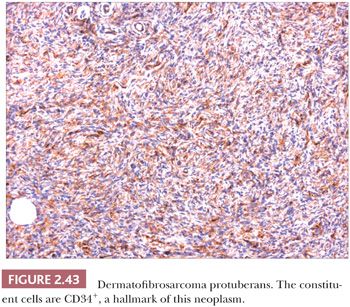
Microscopically, the tumor is composed of dense aggregates of spindled cells that form “cartwheel” arrangements (Fig. 2.42) (128). The cells have a monotonous appearance, featuring tapered nuclei and, most often, only sparse mitoses. Subcutaneous involvement is characterized by growth within septa and between adipocytes in horizontal fashion, producing a honeycomb appearance (129). The spindled cells may extend well into the dermis; changes in the overlying epidermis occur (130), but the type of acanthosis seen in dermatofibroma, with flattening of the tips of rete ridges, is rarely observed. Little or no polarizable collagen can be found among neoplastic cells (131). A pigmented variant, known as the Bednar tumor, also features scattered dendritic melanocytes (132). Occasionally, myxoid changes are seen, sometimes, but not invariably, in recurrent lesions (133,134). Fibrosarcoma-like areas occur, and their presence has been associated with local recurrence and metastasis (135,136). CD34 positivity of tumor cells is considered an immunohistochemical hallmark of DFSP (Fig. 2.43) (137). Fusion of the collagen gene COL1A1 to the platelet-derived growth factor beta-chain (PDGFB) gene has been repeatedly described and is considered a characteristic genetic marker of DFSP (138).
The differential diagnosis of these tumors includes myofibroma, malignant fibrous histiocytoma (pleomorphic sarcoma), spindle cell melanoma, neurofibroma, and (in the case of myxoid variants) myxoid liposarcoma, in addition to dermatofibroma. Attention to morphologic details and immunohistochemical analysis usually permit distinction from these other entities.
ATYPICAL FIBROXANTHOMA
Atypical fibroxanthoma (AFX) is a cutaneous mesenchymal tumor that microscopically resembles the soft tissue tumor malignant fibrous histiocytoma/pleomorphic sarcoma (MFH) but has a distinctive clinical presentation and usually a better clinical outcome (139). AFX usually arises in sun-exposed skin of the head and neck region of older adults. It uncommonly develops in younger patients or in other locations; some reports of occurrence in younger individuals may actually have been describing atypical variants of dermatofibroma (e.g., dermatofibroma with monster cells). The lesion often presents as an elevated, ulcerated nodule. It responds well to complete surgical excision. Recurrences are uncommon, and metastases are rare. However, metastases have been reported (e.g., to lymph nodes or parotid gland); this usually occurs in deep-seated, recurrent tumors or those that have arisen after radiation therapy (140).
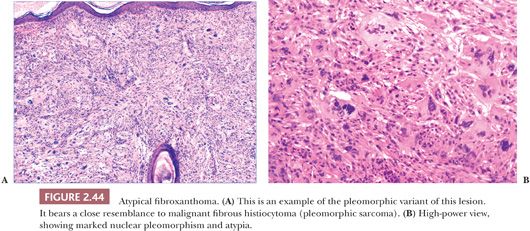
Microscopically, AFX shows a dermal proliferation of polygonal or spindled cells that occasionally contact the overlying or adjacent epidermis, although they lack evidence for origination from the epidermis. Some of the cells possess vacuolated, foamy cytoplasm. Pleomorphic variants may feature large, irregularly shaped nuclei with coarse chromatin, prominent nucleoli, and atypical mitotic figures (Fig. 2.44) (141,142). Other variants include angiomatoid and clear cell types (142) and AFX with osteoclast-like giant cells (143–145). A spindle cell variant with limited pleomorphism can be difficult to distinguish from a variety of other malignant spindle cell tumors (146). Immunohistochemical study often proves helpful in diagnosis but usually because negative results help to rule out such potential mimics as spindle cell melanoma (S-100 positive), spindle cell squamous cell carcinoma (cytokeratin positive), or leiomyosarcoma (more uniformly actin positive and desmin positive). A completely reliable positive marker for AFX is highly desirable but has been lacking. However, promising results have been obtained with antibodies to CD10 and CD99 (147). CD10 staining is not restricted to AFX and can be found, for example, in spindle cell squamous cell carcinoma. However, it may be of value when strong, diffuse membranocytoplasmic staining is the criterion for a positive result (148). Procollagen-1 is also frequently expressed in AFX (149).
Microscopic differentiation from MFH has been difficult if not impossible, and clinical features are most often decisive. It is widely believed that AFX is simply a superficial variant of that tumor, but this issue has not been fully resolved by studies of DNA content and proliferative activity (150,151). Although an earlier study suggested that expression of CD74 (LN-2) may help in distinguishing between the two, in that CD74 expression was found to be stronger in MFH (152), a more recent case series has not confirmed that finding (153).
NEURAL TUMORS
Although there is considerable variety among the neural tumors that can arise in skin, the types of lesions commonly found in a cutaneous location are limited, and it is unusual to find neoplasms of ependymal, meningeal, neuronal, or notochordal differentiation. The following represent the most important of the neural tumors from a dermatopathologic perspective.
NEUROFIBROMA
Neurofibromas frequently arise in skin and, in fact, are among the most familiar of cutaneous neoplasms, although variant forms can sometimes create diagnostic difficulties. They most commonly present as isolated soft papules or nodules arising in any cutaneous site (154,155), although in our experience, this presentation is most often encountered in middle-aged to older adults. Occasionally, several such lesions can be seen, in widely scattered or clustered form, in otherwise normal individuals. Multiple neurofibromas, particularly when developing in childhood and presenting together with café au lait lesions, strongly suggest a diagnosis of neurofibromatosis of the von Recklinghausen type (156,157). Plexiform neurofibromas can distort the superficial soft tissues, and can resemble a “bag of worms” both on palpation and as surgical specimens in cross section. Plexiform neurofibromas have traditionally been regarded as presumptive evidence of neurofibromatosis (158), but recent evidence suggests that this is not invariably the case (159–162).
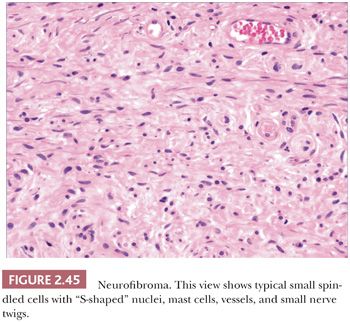
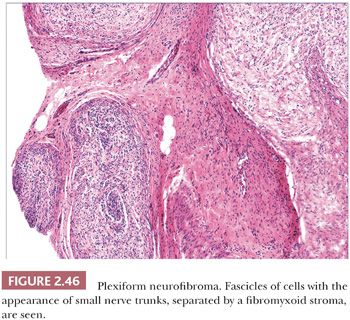
Microscopically, neurofibromas have a familiar appearance, consisting of scattered spindled cells with “buckled” or “S-shaped” nuclei, within a variably myxoid matrix (163,164). These are arranged haphazardly or in a vaguely storiform configuration. Scattered small vessels and mast cells are common (Fig. 2.45). Although at times the lesions appear to be well demarcated from adjacent dermal connective tissue, they are not encapsulated, and frequently, the lesions merge imperceptibly with dermal collagen. The constituent cells are largely composed of endoneurial fibroblasts and include Schwann cells and perineurial cells. Myxoid, pigmented, and diffuse types occur; at times, the small vessel component may be the predominating feature. There is a recently described variant termed dendritic cell neurofibroma with pseudorosettes (165). Plexiform neurofibromas show demarcated fascicles of proliferating Schwann cells, separated by a myxofibrous stroma, producing the appearance of miniature nerve trunks (Fig. 2.46) (166).
The most common differential diagnostic consideration is that of melanocytic nevus, sometimes termed neurotized nevus. Both are S-100 positive, although neurofibroma would lack other melanocytic markers and would be more likely to be immunoreactive for factor XIIIa (167). Because it makes little difference prognostically whether an isolated, banal lesion is termed neurofibroma or neurotized nevus, we typically do not launch into an expensive battery of immunohistochemical stains in an attempt to differentiate the two; lesions lacking identifiable nevomelanocytes (usually identifiable as nests or aggregates of cells) and accompanied by small vessels and mast cells are usually termed neurofibroma. A more significant problem is the occasional lesion with hypercellularity, mitotic activity, nuclear atypia, or cell necrosis; such lesions must be distinguished from malignant peripheral nerve sheath tumors. At times, this may be a difficult task, and a given lesion may have to be designated as a “peripheral nerve sheath tumor of indeterminate biologic potential” (166). However, staining for p53 and Ki-67 may help to distinguish malignant peripheral nerve sheath tumors from neurofibroma, which would be expected to be negative for these markers (168,169). Such tumors may also bear a resemblance to desmoplastic malignant melanoma; therefore, special attention should be paid to any changes at the dermal-epidermal junction, realizing, however, that a proportion (perhaps 20%) of desmoplastic melanomas lack both junctional changes and distinctive melanocytic immunohistochemical markers. Neurofibromas are commonly CD34+ (170), and this can create confusion with other CD34+ spindle cell tumors such as myxoid DFSP. Perineurioma can have similar features, although spindled cells are often organized in lamellated or concentric configurations. However, the cells of perineurioma are positive for epithelial membrane antigen and claudin-1 (171). The differential diagnosis for plexiform neurofibroma includes plexiform fibrohistiocytic tumor, plexiform dermatofibroma, and some Spitz nevi.
NEURILEMMOMA (SCHWANNOMA)
Neurilemmomas are less common in skin than are neurofibromas. They somewhat resemble solitary neurofibromas clinically, although they are often encapsulated and firmer to palpation, sometimes mimicking epidermal (infundibular) cysts. They are well known to generations of dermatologists as one of the “painful dermal tumors” (172,173). Multiple neurilemmomas can be associated with neurofibromatosis (174) or occur as a noninherited syndrome termed schwannomatosis (175).
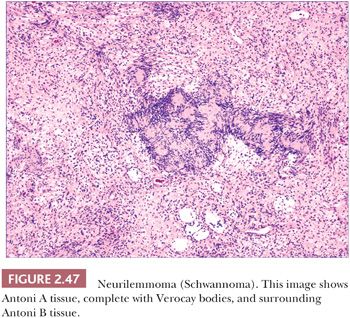
Microscopically, neurilemmomas are encapsulated and show two major histopathologic patterns: Antoni A tissue, consisting of densely aggregated spindle cells whose nuclei form palisaded arrangements, especially around syncytia of cytoplasm (Verocay bodies); and Antoni B tissue, consisting of sparsely cellular, edematous or myxoid foci (166,176). The latter foci have been considered to represent degenerative change in Antoni A tissue (Fig. 2.47). Mast cells are often numerous in these tumors. Microscopic variants include glandular neurilemmoma (probably the result of a metaplastic phenomenon) and melanotic, plexiform, and epithelioid neurilemmomas. Long-standing and/or traumatized lesions may feature enlarged, hyperchromatic nuclei with occasional mitoses, a change termed ancient Schwannoma (177). A variant termed neuroblastoma-like neurilemmoma can contain small ovoid cells that surround fibrillary material. However, in contrast to metastatic neuroblastoma, these lesions are strongly, diffusely S-100 and vimentin positive and fail to stain with NB84 or to label for synaptophysin (178,179). Malignant change in a cutaneous neurilemmoma is rare (180). The S-100 positivity of neurilemmoma allows distinction from other tumors that exhibit nuclear palisading, such as leiomyoma, and the uncommon neuroid basal cell carcinoma. The localized, circumscribed (“palisaded, encapsulated”) neuroma can have overlapping microscopic and immunohistochemical characteristics.
NEUROMAS
There are several types of neuroma that can arise in or involve the skin. Although traumatic neuroma has traditionally been considered the most common of these, in our own surgical material, localized, circumscribed neuroma (formerly “palisaded, encapsulated” neuroma) is seen most frequently. The latter lesion is generally painless and tends to occur on the central portion of the face (181). The so-called supernumerary digit is found at the base of the fifth finger near its ulnar border. The traditional view has been that this lesion represents a kind of “amputation neuroma” (182), the result of autoamputation of a true accessory digit during embryonic life. Although this view has been questioned (183), we recently received a specimen consisting of a true accessory digit, with a polypoid neuroma (supernumerary digit) extending from its surface. Morton neuroma, in reality a fibrosis of the plantar digital nerve, is occasionally encountered in dermatology practice (184,185).
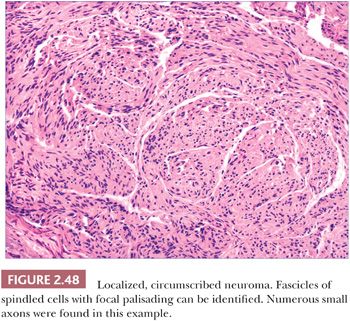
Localized, circumscribed neuromas are considered to be true neoplasms. They are well circumscribed and often possess an attenuated fibrous capsule. Fascicles of bland spindled cells are found, often with demonstrable palisading but without surrounding fibrous sheaths (Fig. 2.48) (186,187). The fascicles often, but not invariably, contain axons (188). S-100 protein staining is regularly demonstrated. Traumatic neuromas are in reality aberrant attempts of reinnervation following disruption of neural axons. Consequently, nerve bundles are surrounded and disrupted by fibrous bands. The supernumerary digit also features small nerves embedded in connective tissue (Fig. 2.49), but small oval structures resembling Meissner bodies are also identified.
NEUROTHEKEOMA
Gallager and Helwig (189) first introduced this term in 1980 to refer to a group of tumors of presumed nerve sheath origin. Since that time, it has become apparent that three somewhat distinctive lesions have become subsumed under this title: the nerve sheath myxoma, largely as described by Pulitzer and Reed (190); the classic neurothekeoma; and cellular neurothekeoma (191). These tumors predominantly affect younger individuals and are most frequently found on the head and neck and upper extremities. They typically present as firm, dome-shaped nodules.
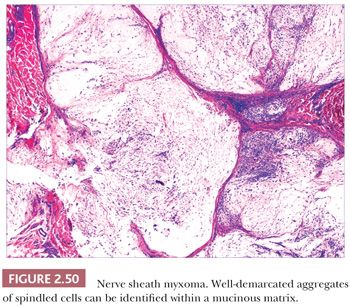
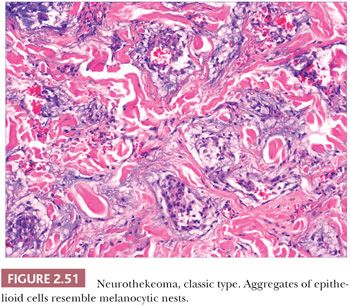
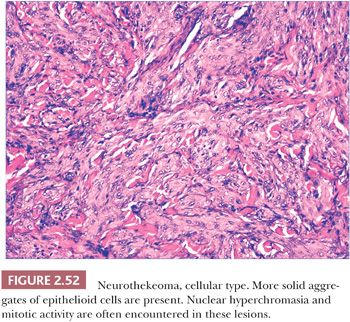
Microscopically, nerve sheath myxomas consist of well-demarcated aggregates of spindled cells that are separated from one another by a markedly mucinous matrix (Fig. 2.50). Classic neurothekeomas are composed of concentric arrangements of plump spindled to epithelioid cells resembling melanocytic nests. Cytologic atypia ranges from minimal to moderate (Fig. 2.51), and mitoses (but not atypical ones) are commonly encountered. In cellular neurothekeoma, there are more solid dermal aggregates of epithelioid cells without formation of discrete nests as seen in the classic form (Fig. 2.52). Nuclear hyperchromasia and mitotic activity are common in these lesions. Plexiform and desmoplastic variants have been described (192). On immunohistochemical staining, nerve sheath myxomas are variably positive for S-100 protein, CD57, and epithelial membrane antigen, indicating schwannian and/or perineural differentiation. However, classic and cellular neurothekeomas are negative for these antigens, and in fact, the cellular origin/differentiation of these tumors is problematic. Cellular neurothekeomas can be positive for alpha-isoform (smooth muscle) actin, NKI/C3, microphthalmia transcription factor, S-100 protein isoform A6, neuron-specific enolase, factor XIIIa, and podoplanin, thereby displaying “crossover” characteristics of melanocytic and fibrohistiocytic neoplasms (193). Local infiltration of deep tissues has been seen in large neurothekeomas, but to date, metastases have not been identified. A group of neurothekeomas has been described in which tumors were larger than usual, showing deep growth, infiltrative borders, vascular invasion, frequent mitoses, and significant cytologic atypia; nevertheless, adverse biologic behavior was not found among these lesions (194).
MERKEL CELL CARCINOMA (NEUROENDOCRINE CARCINOMA OF THE SKIN)
Neuroendocrine carcinoma is one of the most biologically aggressive neoplasms of the skin. This primary cutaneous tumor is widely believed to be of Merkel cell origin. The Merkel cell is found in epidermis, oral mucosa, outer follicular sheath, and sweat ducts and is believed to be a specialized neuroepithelial cell that may have a touch receptor function. Merkel cell carcinoma (MCC) typically presents as a violaceous nodule arising in the head and neck region of elderly adults, although exceptions certainly occur (195). Development of MCC in lesions of Bowen disease (squamous cell carcinoma in situ) has been repeatedly described (196–198). Forty percent of these tumors recur locally, and 75% of patients with one or more recurrences develop regional nodal or distant metastases. In most instances, visceral involvement is accompanied by death within 6 months (199,200). In recent years, it has been learned that most MCCs, in this country and elsewhere, are associated with a particular polyomavirus, now known as the Merkel cell polyomavirus (MCPyV), which is integrated into the genome of the tumor. The virus can be detected in formalin-fixed tissues using PCR technology, but it has not been found in visceral high-grade neuroendocrine tumors from several different anatomic sites. The prevalence of this virus is high in immunosuppressed patients and may partly explain the increased incidence of MCC in these individuals (201,202). Treatments of Merkel cell carcinoma generally include surgical excision and/or radiation therapy.
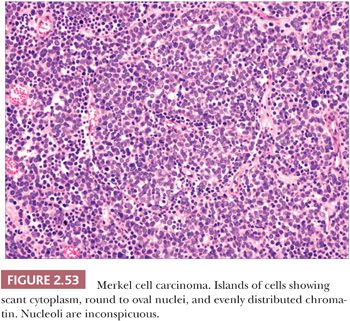
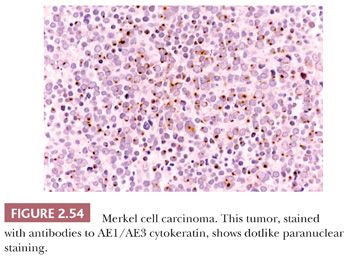
On biopsy, aggregates of small, round tumor cells are found within the dermis. The configurations produced by these cells vary from broad sheets to trabeculae to rounded islands showing organoid growth. Involvement of the overlying epidermis with nestlike arrangements of tumor cells is sometimes noted. Typically, the cytoplasm is scant, and the nuclei are round to oval with evenly distributed chromatin and inconspicuous nucleoli (Fig. 2.53); mitotic activity is often brisk (195,203). Larger, sometimes giant tumor cells with the same nuclear characteristics are occasionally seen. Divergent squamous and even sweat gland differentiation can be seen in some examples (204,205). Immunohistochemical study shows dotlike paranuclear cytokeratin staining (Fig. 2.54). Cytokeratin 20 is frequently positive, and cells also express neuron-specific enolase, chromogranin, synaptophysin, and a variety of other neural and neuropeptide markers (206).
MCC must be distinguished from neuroendocrine carcinomas metastatic to the skin. The most useful immunohistochemical markers for this purpose are cytokeratin 20 (usually positive in MCC and negative in metastatic neuroendocrine carcinoma) and thyroid transcription factor-1 (TTF-1), which produces the reverse result (207,208). The Azzopardi phenomenon—basophilic nucleic acid encrustation of tumoral blood vessels—has been observed in metastatic small cell pulmonary carcinoma but has only recently been reported in primary MCC of skin (209,210). Other neoplasms that can be confused with MCC (and the immunohistochemical markers that often permit distinction) include non-Hodgkin lymphoma (CD45+) and basal cell carcinoma (Ber-EP4 positive).
VASCULAR TUMORS
Many of the lesions labeled “hemangioma,” particularly those arising in childhood, probably represent malformations rather than true neoplasms. However, there is an array of vascular tumors with distinctive microscopic characteristics. Several of these have been linked to infectious agents. Angiosarcoma arises most often in elderly individuals, whereas Kaposi sarcoma can develop in several clinically distinct circumstances.
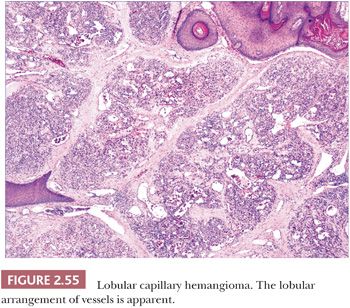
LOBULAR CAPILLARY HEMANGIOMA
For years, this hemangioma was termed pyogenic granuloma because of its resemblance to granulation tissue following infection. The latter name still persists among clinicians but is now a well-established misnomer. Lobular capillary hemangioma (LCH) is a true neoplasm with characteristic microscopic features (211). It typically presents at any age as an erythematous, often polypoid growth on the skin or mucous membranes. It is sometimes, but not always, precipitated by trauma. Development on mucosal surfaces during pregnancy is a recognized phenomenon. Subcutaneous and intravenous forms of LCH, as would be expected, have quite different clinical presentations (212,213). The entity known as acquired tufted hemangioma is likely another variant of LCH; this presents as enlarging erythematous macules and plaques on the neck and trunk of young individuals (214).
These lesions have in common the proliferation of small vessels that are divided into lobules by fibrous connective tissue (Fig. 2.55) (211). The vessels are lined by plump endothelial cells and surrounded by pericytes. The lobular arrangement of vessels can be obscured by edema and granulation tissue changes that have been superimposed by trauma; this is particularly likely to occur in protuberant lesions. An in-growing collarette of hyperkeratotic epidermis is often seen at the base of polypoid examples of LCH, corresponding to the collarette of scale noted clinically to surround these lesions. A central vessel with muscular wall resembling a vein may be observed, and fibrosis is observed in the involutional stage. Multiple recurrences (often related to partial removal), regional satellite formation, and disseminated lesions have all been reported, but these are nevertheless benign lesions that ultimately either respond to surgical methods or, in the case of disseminated lesions, may regress spontaneously.
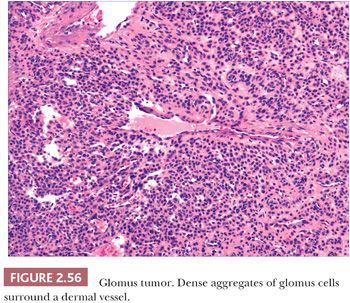
GLOMUS TUMOR
Glomus tumor is one of the most distinctive of the cutaneous vascular tumors. It is composed of elements recapitulating the Suquet-Hoyer canal, the arterial segment of the glomus, a specialized arteriovenous shunt found primarily in hands, feet, fingers, and toes. This structure is composed of a narrow lumen lined by a single layer of endothelium, surrounded in turn by four to six layers of glomus cells, which can be regarded as modified smooth muscle cells. Occasionally, these normal structures are found incidentally in biopsies of distal extremities obtained for other reasons and are mistaken for glomus tumors.
The glomus tumor usually presents as a purple nodule, often in a subungual location. These lesions are characteristically painful, a sensation that either arises spontaneously or is elicited by pressure. A variant of this tumor, glomangioma, is often solitary but may be multiple and can be inherited as an autosomal dominant trait (215). These lesions occur on the trunk or extremities and are most often painless.
The glomus tumor is well circumscribed or encapsulated and is composed of rather solid aggregates of glomus cells surrounding relatively small vessels (Fig. 2.56) (216), whereas glomangiomas are poorly circumscribed and feature prominent vessels surrounded by only a few layers of glomus cells (217). Immunohistochemistry shows that glomus cells are smooth muscle actin and muscle-specific actin positive and are negative for desmin and markers of endothelial cells (218,219). Multiple nerve fibers can be identified in solitary glomus tumors. Glomus tumor can bear a resemblance to acrospiroma, a benign adnexal tumor of sweat gland differentiation, but can easily be distinguished by the actin positivity of its constituent cells, in contrast to the expression of cytokeratin by the cells of acrospiroma.
Most glomus tumors are benign, but atypical examples exist. The study of atypical glomus tumors by Folpe et al. (220) resulted in a classification scheme that categorizes these lesions as follows: malignant glomus tumor, symplastic glomus tumor, glomus tumor of uncertain malignant potential, and glomangiomatosis. Malignant tumors were considered those with deep location and size greater than 2 cm, or atypical mitotic figures, or moderate to high nuclear grade and five or more mitotic figures per 50 high-power fields. Symplastic glomus tumors featured high nuclear grade but no other malignant features. Metastasis occurred only in the group of tumors fulfilling criteria for malignancy (220).
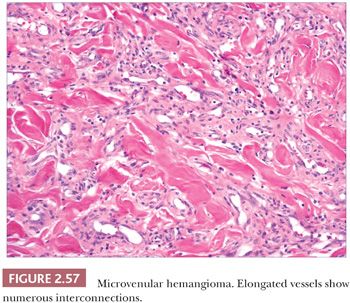
MICROVENULAR HEMANGIOMA
This lesion arises in sun-damaged skin, especially the arms, of young to middle-aged individuals. Clinically, it has features of a conventional hemangioma, although Kaposi sarcoma is occasionally considered in the differential diagnosis. Microscopic findings include elongated vessels with narrow lumina that display interconnections and branching configurations that may extend into subcutaneous septa (Fig. 2.57). These vessels have venous characteristics. Despite this somewhat unusual configuration, the lesion appears to be relatively confined on low-power inspection, shows minimal if any inflammation, and lacks nuclear atypia or mitotic activity. Significantly, changes expected in Kaposi sarcoma, such as the “promontory sign,” hyaline globules, plasma cells, and solid aggregates of spindled cells, are not observed (see “Kaposi Sarcoma,” pp. 65–66) (221–223).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


