Nongenetic Influences on Drug Metabolism
LEARNING OBJECTIVES
BACKGROUND
The main goal of successful drug therapy is to ensure that a patient receives safe and effective drug treatment. Therapeutic drug concentrations need to be achieved to accomplish this goal. A solid understanding of the factors that can affect drug disposition is critical in achieving appropriate drug concentrations (Figure 8–1). Drug dose or dosing frequency may need to be modified according to changes in drug biotransformation or excretion caused by intrinsic or extrinsic factors. In this chapter, intrinsic and extrinsic factors known to affect metabolism, with a particular focus on cytochromes P450 (a principal component of drug-metabolizing enzymes), will be discussed.
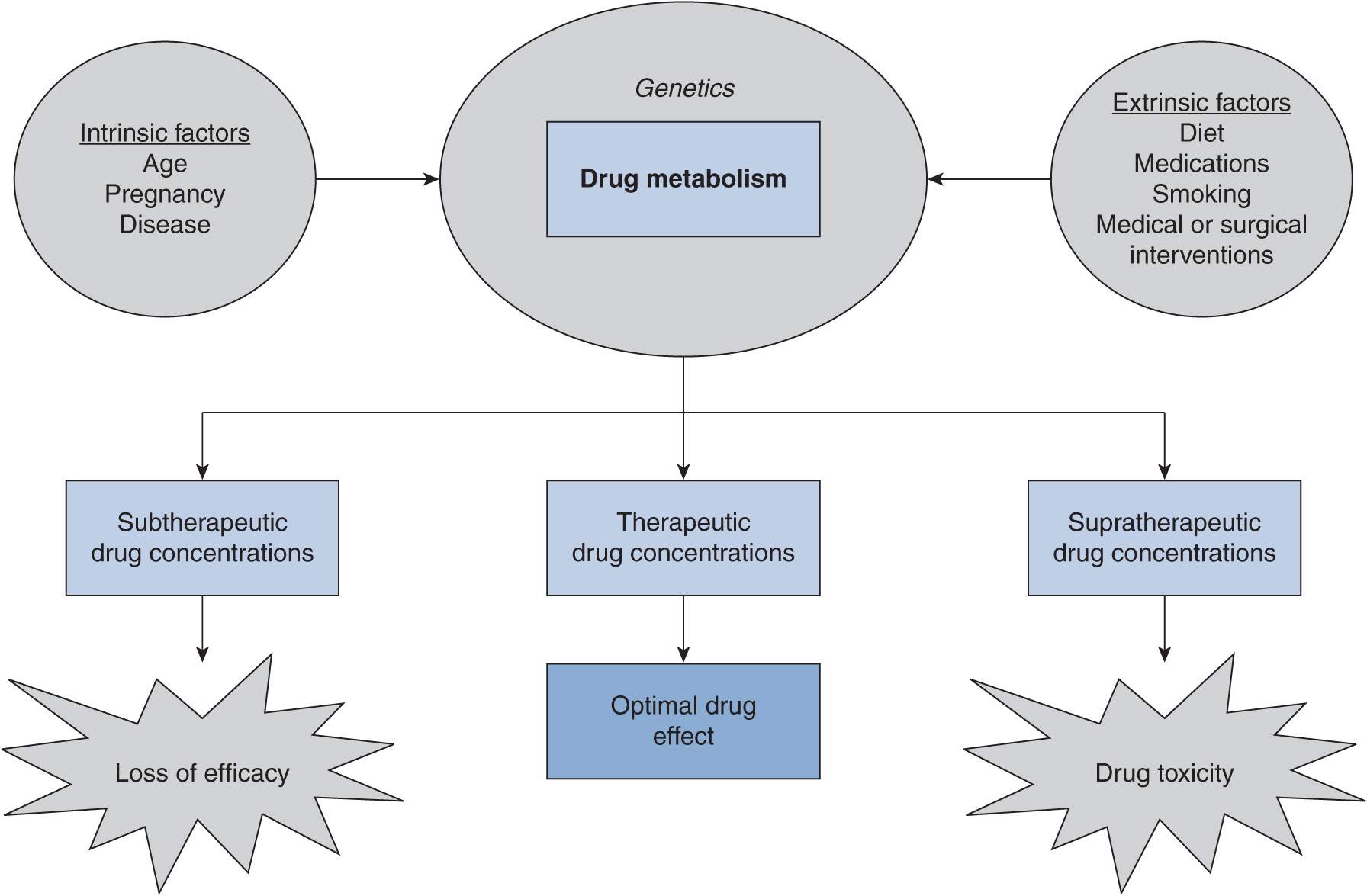
FIGURE 8–1 Intrinsic and extrinsic factors can influence the pharmacokinetics of a drug. The changes in drug clearance that occur can result in subtherapeutic, therapeutic, or supratherapeutic drug concentrations that in turn can lead to optimal drug effect or undesired drug effects.
INTRINSIC FACTORS
Aging
A plethora of research focuses on age-based differences in drug metabolism. In pediatric patients, changes in the ontogeny of drug-metabolizing enzymes account for many of the differences in drug disposition between children and adults. Studies show differential expression patterns of CYP enzymes in neonates, infants, children, and adolescents.1 Developmental changes such as changes in gastric acid production, gastric emptying, renal function, pancreatic function, body composition, and total body water also play a role in the evolving absorption, distribution, metabolism, and excretion processes in infants and children. These developmental changes contribute to variability in drug disposition in the pediatric population.2
On the other side of the age spectrum are the elderly, who are generally described as those aged 65 years or older. In the elderly population, the aging process may involve a progressive decrease in organ and tissue function (in particular, the kidney) that can lead to altered drug disposition. Although liver size and hepatic blood flow generally decrease with age, routine clinical tests of liver function reveal that hepatic function does not change significantly.3 In addition, in vitro studies investigating CYP enzyme activity in human hepatocytes find no differences in the activities of the 10 most predominant CYP isoforms between the age groups of 20–60 and above 60 years.4 In clinical studies, the effect of age on CYP enzymes appears to be isoform-dependent. Plasma paroxetine concentrations were evaluated in depressed patients aged 69–95 years and no age-related changes in paroxetine disposition (metabolized by CYP2D6) were detected.5 In studies involving the clearance of antipyrine (metabolized by CYP3A4 and CYP3A5 [collectively referred to as “CYP3A4/5” due to their high degree of homology and function], CYP1A2, CYP2C8, and CYP2C9), an approximate 20–25% decline in antipyrine clearance is observed between ages 25 and 76 years.6 Therefore, although some loss of drug-metabolizing enzyme function may occur with extreme aging, the changes are not dramatic and are isoform-specific.
Pregnancy
Pregnancy results in major physiological changes in a woman’s body. These include increased blood volume, increased water composition, increased urinary output, reduced intestinal motility, increased gastric pH, and changes in sex hormone concentrations. Because any of these factors can contribute to significant changes in the absorption, distribution, metabolism, and excretion of drugs, specific changes to CYP-mediated metabolism are difficult to discern. In studies measuring the ratio of excreted parent drug to metabolite, the metabolic ratio increases for CYP1A2 and CYP2C19 substrates, indicating lower activities for these isozymes.47 Conversely, metabolic ratios decrease for CYP2C9, CYP2D6, and CYP3A4/5, indicating higher activities for these isozymes.47 Examples of drugs that may require dose modification during pregnancy because of changes in hepatic drug metabolism include methadone, nifedipine, and lamotrigine.48 Since potentially lifesaving drugs may be necessary during pregnancy, these changes in drug-metabolizing enzyme activities should be taken into consideration when determining appropriate drug doses. However, there are few, if any, well-controlled clinical studies that have resulted in specific product dosing recommendations for pregnancy. Additional peripartum and postpartum changes in drug metabolism can also occur,48 but too few data are available to make general recommendations about alteration of drug therapy in these settings.
EXTRINSIC FACTORS
Diseases
Numerous disease states can affect drug disposition. The mechanisms by which drug disposition is altered by disease can result from direct physiological changes or alterations in major drug-metabolizing enzymes. The liver and kidney are the two primary organs affecting drug metabolism and excretion, and the effects of diseases on the liver and kidney will be discussed in the next section. The reader should bear in mind that other organ systems may also be affected, although these will not be discussed in this chapter.
Inflammation and Cytokines
Infectious diseases have long been recognized to alter hepatic CYP expression and activity. In 1978, Chang et al. first described higher theophylline half-lives in asthmatic children with confirmed upper respiratory tract infection caused by influenza A or adenovirus.7 This key report illustrated that infection can cause clinically significant alterations in drug disposition and suggested that changes in drug-metabolizing enzymes could be affected by the state of infection. Subsequent research in a mouse model showed that inflammation caused by infection causes production of a bacterial endotoxin (lipopolysaccharide [LPS]) that leads to down-regulation of CYP enzyme mRNA levels and activities that may vary during the course of infection.8
Inflammation is an essential component of the innate immune response to pathogens and tissue damage. The inflammatory process is mediated by cytokines, a group of secreted polypeptides that include interleukins (IL), interferons (IFN), tumor necrosis factors (TNF), and chemokines. The effect of inflammation on CYP enzymes in vitro and in vivo has been extensively reviewed.9–11 In particular, IL-1, IL-6, IFNs, and bacterial LPS cause decreased hepatic CYP expression.12 Coupled with the systemic release of cytokines, the liver responds to inflammation by increasing synthesis and secretion of acute-phase proteins such as fibrinogen, α1-acid glycoprotein, and C-reactive protein.13 Cytokine cascades are known to influence inflammatory and anti-inflammatory responses in multiple disease states, can be involved in both acute and chronic inflammation, and alter the pharmacokinetics or pharmacodynamics of numerous drugs.14 One example of a clinical impact on drug metabolism is when CYP enzyme activity is markedly decreased during allograft rejection.15 The impairment of CYP activity in biopsy samples is attributed to an increase in the intragraft production of proinflammatory cytokines. In another example, melanoma patients treated with high-dose IFN therapy have greater than 60% suppression of CYP1A2 activity.16
More recently, cancer has been recognized as a disease state that can alter CYP-mediated drug metabolism. Many cancers involve the continued release of cytokines, such as TNF-α, IFN-α, IL-4, IL-6, IL-8, IL-10, IL-12, and IL-17, that are involved in tumor cell proliferation, angiogenesis, metastasis, and adaptive immunity.17 Through the intermediate event of increased cytokine release, cancer can indirectly decrease hepatic CYP expression. Other physiological states, such as acute pain18 and depression,19 have also been shown to result in alterations in pharmacokinetics and pharmacodynamic responses. Since the acute-phase response involves cytokine release and inflammation, disease states with an acute-phase response may affect drug pharmacokinetics.
Chronic Liver Disease
The hallmark of chronic liver disease is gradual destruction of liver tissue that leads to fibrosis and hepatic cirrhosis. Fibrosis is the growth of scar tissue due to infection, inflammation, injury, or healing and frequently results in cirrhosis. Cirrhosis is characterized by lowered hepatic blood flow and eventual loss of liver function. Causes of liver disease include, but are not limited to, viral infections, xenobiotics, excessive alcohol consumption, metabolic disorders, and autoimmune disorders.
Although the exact mechanisms by which liver disease affects hepatic drug disposition are unknown, researchers have postulated four main mechanisms: reduced hepatocyte content and activity of metabolizing enzymes, decreased cell mass, reduced drug uptake, and diminished oxygen uptake.20 The mechanism that affects a particular drug’s disposition also depends on that drug’s clearance characteristics. As blood flow can be a major determinant of drug clearance, drugs with a high extraction ratio (such as lidocaine) are most likely to have clearance affected by both decrease in metabolic activity and a decline in functional hepatic perfusion. However, in drugs with a low hepatic extraction ratio, the most likely cause of lowered hepatic clearance is decreased metabolic activity or protein binding.
Differences in drug disposition as a result of liver impairment have been demonstrated.21 Theoretically, any chronic disease that significantly affects liver function can affect the disposition of a drug that is highly dependent on hepatic drug metabolism for elimination. However, the occurrence of impaired hepatic drug metabolism is dependent on the type and severity of liver disease. In liver cirrhosis, hepatic drug clearance is generally reduced in proportion to the degree of liver dysfunction.20 Particular consideration should be given to drugs that target the liver, such as treatments for viral hepatitis. These drugs may be a substrate for one or more hepatic CYP enzymes as well as hepatic transporters whose function can also be affected by chronic liver disease. In patients with liver disease of varying degrees and from various etiologies, the metabolizing activities of CYP1A2, CYP2C19, CYP2D6, and CYP2E1 decrease with increasing severity of liver disease.22 However, the degree to which activity is lowered is specific to each CYP isozyme.22,23 Hepatic metabolic capacity usually remains intact because of multiple compensatory responses until advanced cirrhosis develops. Although there are some drugs that require dose reduction in the setting of moderate cirrhosis (e.g., morphine, verapamil, nifedipine, losartan, omeprazole), dosing adjustments are typically not required except in the setting of advanced cirrhosis.22
Chronic Kidney Disease
Chronic kidney disease is characterized by the gradual loss of renal function. One method of categorizing the severity of renal impairment is with creatinine clearance. Severity of renal impairment ranges from mild (creatinine clearance between 60 and 80 mL/min), moderate (creatinine clearance between 30 and 59 mL/min), and severe (creatinine clearance between 15 and 29 mL/min) to end-stage renal disease (creatinine clearance <15 mL/min or requiring dialysis).24
Chronic kidney disease that leads to impairment in renal function not only has consequences for drugs that are excreted by the kidneys but can also affect drugs cleared by nonrenal routes. Specifically, renal impairment causes physiological alterations such as changes in absorption, plasma protein binding, drug transport, or tissue distribution, and can affect hepatic and gut metabolism. In addition, renal dysfunction directly affects CYP-mediated metabolism and transmembrane transport of drugs in the liver. Although the exact mechanisms are unclear, it is hypothesized that systemic accumulation of uremic toxins (e.g., urea, parathyroid hormone, indoxyl sulfate, and cytokines) is responsible.25 These solutes could contribute to transcriptional, translational, or posttranslational downregulation of drug-metabolizing enzymes and transporters. Several studies using probe CYP substrates have been conducted in humans to determine the extent of CYP enzyme inhibition. A review of these studies is presented elsewhere.26 In brief, clinically relevant decreases in activities of CYP3A4/5, CYP2C9, CYP2D6, and CYP2C19 are demonstrated in patients with chronic kidney disease and end-stage renal disease.
Cardiac Failure
Cardiac failure is a condition in which the heart is unable to deliver sufficiently oxygenated blood throughout the body. Lower cardiac output leads to lower hepatic blood flow, and this can affect the clearance of drugs with a high extraction ratio (clearance highly dependent on blood flow).27 Logic would dictate that a monooxygenase system such as the CYP enzyme system would be suppressed if there are inadequate oxygen levels in the systemic circulation. In fact, the activity level of hepatic CYP2C19 is significantly decreased in patients with heart failure.27 A significant correlation between enzyme activity levels and plasma concentrations of TNF-α and IL-6 has also been demonstrated.27 As the concentrations of these cytokines increase, CYP enzyme activities decrease. Patients with heart failure have higher circulating concentrations of TNF-α and IL-6 that correlate with the severity of heart failure. Thus, the induction of proinflammatory cytokines with consequent decreases in CYP activity likely represents another mechanism by which cardiac failure leads to alterations in drug metabolism. Although the activity of hepatic CYPs appears to be suppressed, expression of certain cardiac CYP isoforms is upregulated in a rat model of cardiac hypertrophy.28 This indicates that the effects of cardiac failure on drug-metabolizing enzymes may be highly organ-specific.
Diet
Perhaps no extrinsic factor is more pervasive or influential on the disposition of oral medications than diet. When a solid oral drug is ingested, it is immediately vulnerable to disintegration, dissolution, and absorption in the gastrointestinal tract. Various components of diet can affect one or more of these steps as well as drug metabolism. Thus, it is essential to recognize the importance of the effect of diet on drug disposition. Meal volume, food composition (e.g., protein, fat, dairy products, presence of fortification), and food preparation techniques (e.g., charbroiling) can all play a role in the interaction between diet and drug disposition. The next section will focus on the effect of the composition of various foods on CYP enzymes, and thus factors that can influence drug metabolism.
Fruit Juices
Grapefruit juice is widely recognized to cause clinically relevant food–drug interactions, but it is only one of many juices that can do so. This recognition is often reflected by the prohibition of grapefruit juice consumption during the conduct of clinical trials. Consumption of grapefruit juice can significantly increase plasma concentrations of many drugs, including some HMG-CoA reductase inhibitors, HIV protease inhibitors, antihistamines, macrolides, cyclosporine, calcium channel antagonists, and benzodiazepines.29 Inhibition of CYP3A4/5 in the gut wall and small intestine, leading to lower presystemic drug metabolism and higher plasma concentrations, is at least partially responsible for this effect.30 In addition to inhibiting CYP3A4/5
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


