Cardiovascular Pharmacogenomics
LEARNING OBJECTIVES
INTRODUCTION
Over the past decade, significant progress has been made in the field of cardiovascular pharmacogenomics. Pharmacogenomic literature is now available for most major cardiovascular disease states and includes medications that are at various stages in the pharmacogenomic research process. In some cases, the use of a patient’s genetic makeup to individualize drug therapy is far from being used in the clinic. However, in other cases, genetic information is included in prescribing information, and published guidelines exist to facilitate the use of genotype-guided drug therapy in clinical practice. This chapter will cover major cardiovascular disease states for which there exists a moderate to high amount of pharmacogenomic research. Within this framework, each disease state will highlight one or two genes of interest in which there are well-documented associations between genetic polymorphisms and variability in drug disposition, response, toxicity, or clinical outcomes. In addition, other pharmacokinetic and pharmacodynamic genes of interest will be presented in each section.
ACUTE CORONARY SYNDROMES
Acute coronary syndromes (ACS) describe a group of cardiovascular disorders including unstable angina (UA), non-ST-segment elevation myocardial infarction (NSTEMI), and ST-segment elevation myocardial infarction (STEMI).1 The common theme underlying these disorders is the rupture of a vulnerable atherosclerotic plaque followed by partial or complete thrombotic occlusion of an artery. Platelets play a key role in the pathophysiology of ACS and thrombus formation. As such, most ACS pharmacotherapy is geared toward inhibition of platelet adhesion, activation, and aggregation. Pharmacologic antiplatelet agents that are routinely used in the treatment of ACS include aspirin, thienopyridines (e.g., clopidogrel and prasugrel), and glycoprotein IIb/IIIa inhibitors (e.g., abciximab, eptifibatide, tirofiban).2 Recently, the Food and Drug Administration approved ticagrelor, a cyclopentyltriazolopyrimidine, to reduce the rate of thrombotic cardiovascular events in patients with ACS. The goal of antiplatelet therapy in ACS is to decrease ischemia, limit the extent of infarction, and decrease the risk of death and recurrent cardiovascular events. To date, clopidogrel is the agent that has been studied most extensively in antiplatelet pharmacogenomics and is the focus of this discussion.
Clopidogrel
Clopidogrel, typically used in combination with aspirin, is indicated for the treatment of UA and NSTEMI, including patients who undergo percutaneous coronary interventions (PCI) and those who are managed medically.3 Clopidogrel is also indicated for patients with STEMI, and patients with recent myocardial infarction, recent stroke, or established peripheral arterial disease.3 Despite being an effective anti-platelet agent, marked interindividual differences exist in the ability of clopidogrel to inhibit platelet aggregation, which is partly due to variability in clopidogrel pharmacokinetics.4 Of an absorbed clopidogrel dose, 85% is hydrolyzed by esterases to inactive metabolites. The remaining 15% of the dose undergoes a sequential two-step oxidative process in the liver to form an intermediate metabolite (2-oxo-clopidogrel), and then an active metabolite (R-130964). It is this active metabolite that irreversibly inhibits the P2Y12 receptor on platelets, thereby inhibiting adenosine diphosphate–mediated platelet aggregation. As shown in Figure 11–1, several cytochrome P450 (CYP) enzymes mediate clopidogrel bioactivation, and the CYP2C19 isoenzyme plays a major role in both oxidative steps of this process.5 Importantly, CYP2C19 genetic polymorphisms contribute to variability in clopidogrel pharmacokinetics that has translated into differences in clopidogrel pharmacodynamics and clinical outcomes.
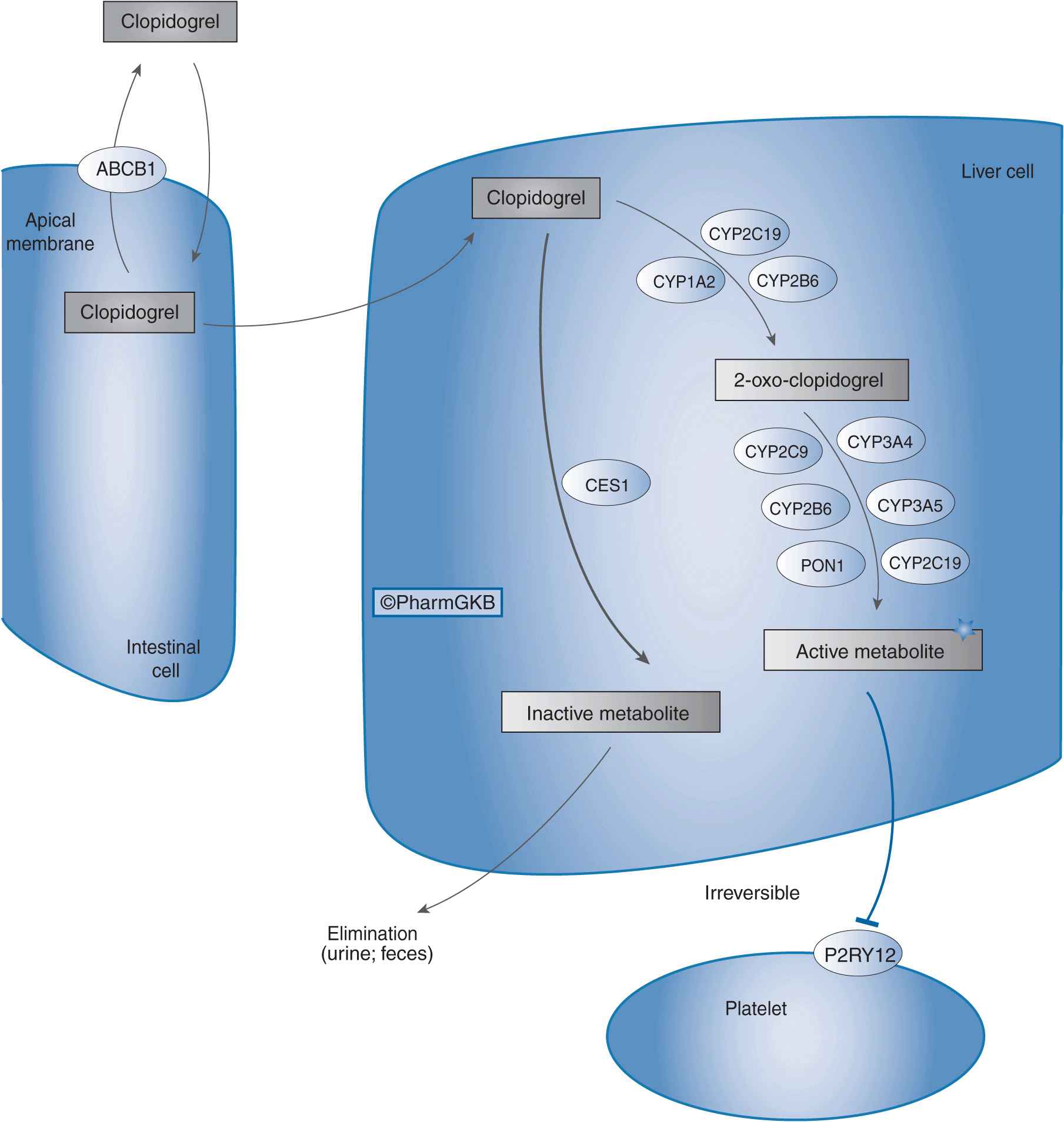
FIGURE 11–1 Clopidogrel pharmacokinetic and pharmacodynamic pathway. ABCB1: efflux transporter; CYP2C19, CYP1A2, CYP2B6, CYP2C9, CYP3A4, CYP3A5, CYP2B6, and PON1: metabolizing enzymes; CES1: esterase; P2RY12: drug receptor. Figure copyright PharmGKB; reprinted with permission from PharmGKB and Stanford University.24
Pharmacogenetic Focus: CYP2C19
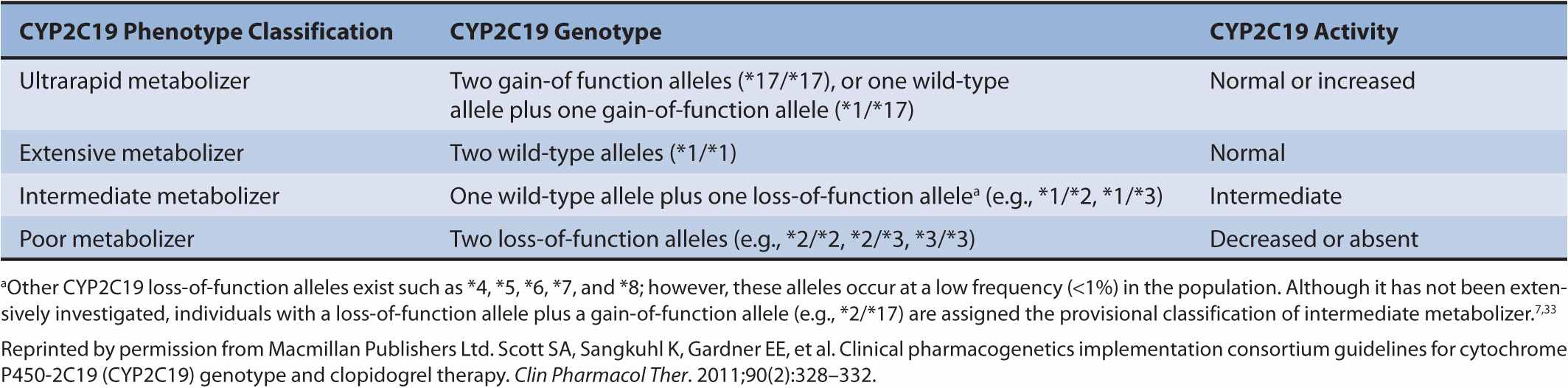
The CYP2C19 alleles most often studied in clopidogrel pharmacogenetics are CYP2C19*1, CYP2C19*2, CYP2C19*3, and CYP2C19 *17. CYP2C19*1 is the normal function (wild-type) allele, CYP2C19*2 (c.681G>A) and CYP2C19*3 (c.636G>A) are loss-of-function alleles, and CYP2C19*17 is a gain-of-function allele.6,7 These alleles result in different CYP2C19 metabolic phenotypes including ultrarapid, extensive, intermediate, and poor metabolizers (Table 11–1). Polymorphic CYP2C19 allele frequencies vary considerably by race. For example, the CYP2C19*2 allele frequency is 27% in Asians, 18% in African Americans, and 14% in Caucasians.8 The CYP2C19*3 allele frequency is 9% in Asians, but virtually nonexistent in African Americans and Caucasians.8 In contrast, the CYP2C19*17 allele is common in Caucasians (18%) and African Americans (18%), but less common in Asians (4%).9
TABLE 11–1 Relationship between CYP2C19 genotypes and phenotypes.
In relation to clopidogrel pharmacokinetics, CYP2C19 loss-of-function alleles, primarily CYP2C19*2, have been associated with decreased active metabolite plasma exposure in healthy volunteers and in patients with ACS.10–12 For example, in healthy volunteers who received a 300 mg loading dose of clopidogrel, active metabolite plasma exposure was 55% lower in poor metabolizers compared with that in extensive metabolizers, and 26% lower in intermediate metabolizers compared with that in extensive metabolizers.12 This pharmacokinetic difference resulted in smaller absolute reductions in platelet aggregation in poor and intermediate metabolizers as compared with those in extensive metabolizers.12 Importantly, the link between CYP2C19 genetics and clopidogrel pharmacokinetics and pharmacodynamics has been replicated in over 20 studies in healthy volunteers and patients with ACS.7,13,14 For example, a genome-wide association study in an Amish population found 13 polymorphisms within the CYP2C18–CYP2C19–CYP2C9–CYP2C8 cluster on chromosome 10q24 to be associated with poor clopidogrel pharmacodynamic response.15 The most significant polymorphism in this cluster was in strong linkage disequilibrium with the CYP2C19*2 allele. On further analysis, the CYP2C19*2 allele was significantly associated with clopidogrel-mediated inhibition of platelet aggregation in a gene dose-dependent manner (i.e., *1*/1 > *1/*2 > *2/*2).15 Furthermore, platelet response to clopidogrel was highly heritable, and about 12% of the variation in clopidogrel response was explained by the CYP2C19*2 allele.15 Taken together, it is clear that CYP2C19 genotype-mediated differences in clopidogrel metabolism result in differential effects on platelet aggregation.
Beyond pharmacokinetics and pharmacodynamics, the CYP2C19*2 loss-of-function allele has been consistently associated with an increased risk of adverse cardiovascular outcomes (e.g., cardiovascular death, myocardial infarction, and stent thrombosis) in ACS/PCI patients treated with clopidogrel.12,15–23 Similar findings have been observed for CYP2C19*3, although this allele is rare in non-Asian populations.12,17,20,21,23 A meta-analysis of 9,685 patients treated with clopidogrel, predominantly for PCI, showed an increased risk of the composite end point of cardiovascular death, myocardial infarction, or stroke in carriers of one CYP2C19 loss-of-function allele (HR, 1.55; 95% CI, 1.11–2.17) or two CYP2C19 loss-of-function alleles (HR, 1.76; 95% CI, 1.24–2.50) as compared with that in noncarriers.25 In addition, the risk of stent thrombosis was substantially increased in carriers of one loss-of-function allele (HR, 2.67; 95% CI, 1.69–4.22) or two loss-of-function alleles (HR, 3.97; 95% CI, 1.75–9.02) as compared with that in noncarriers. In contrast, CYP2C19 loss-of-function alleles have not been associated with an increased risk of cardiovascular events in studies with a low incidence of PCIs or in patients taking clopidogrel for other reasons such as atrial fibrillation.26,27 The wealth of clinical outcome data shows that CYP2C19 loss-of-function alleles, particularly CYP2C19*2, are important determinants of major adverse cardiovascular events in clopidogrel-treated patients with ACS/PCI.25,28–30 Perhaps even more concerning, the risk of stent thrombosis is substantially increased in clopidogrel-treated individuals with genetically mediated CYP2C19 poor metabolism.
While much of the focus of clopidogrel pharmacogenetics has been on CYP2C19 loss-of-function alleles, recent data suggest that the CYP2C19*17 gain-of-function allele is a determinant of clopidogrel pharmacodynamics.12,31–33 Specifically, this allele has been associated with enhanced clopidogrel-mediated inhibition of platelet aggregation, presumably due to increased formation of the active metabolite.12,31–1133 Clinical outcome studies have reported lower cardiovascular event rates and an increased risk of bleeding in carriers of the CYP2C19*17 allele.32,34 However, these associations have not been consistently replicated across clinical studies.15,20,35 Therefore, the exact role of CYP2C19*17 in clopidogrel response and clinical outcomes remains to be determined.
Other Pharmacokinetic and Pharmacodynamic Genes of Interest
Figure 11–1 shows the CYP enzymes, drug transporters, and drug targets that are involved in clopidogrel clinical pharmacology. In terms of drug metabolism, studies have evaluated the impact of CYP3A4/5, CYP2B6, CYP1A2, and CYP2C9 polymorphisms on clopidogrel disposition and/or response.10,12,35–39 However, there are conflicting results regarding the impact of polymorphisms within each of these genes on clopidogrel clinical pharmacology. Recently, the esterase paraoxonase-1 (PONI) was identified as an enzyme involved in the second step of the clopidogrel bioactivation process, and the PON1 Q192R polymorphism was shown to affect clopidogrel metabolism.40 Specifically, the 192R allele was associated with increased enzyme efficiency, and was correlated with clopidogrel active metabolite concentrations and clopidogrel-mediated inhibition of platelet aggregation.40 Furthermore, in clopidogrel-treated ACS patients, the risk of stent thrombosis was substantially higher in patients with the Q/Q and Q/R genotypes as compared with that in patients with the R/R genotype.40 Although these findings are intriguing, subsequent studies have not replicated these associations.41–43 In terms of drug transporters, P-glycoprotein (ABCB1) mediates clopidogrel efflux in the gastrointestinal tract. The ABCB1 3435T variant allele was associated with decreased clopidogrel bioavailability, diminished pharmacodynamic platelet effects, and worse patient outcomes.20,44,45 However, other studies have found no association between ABCB1 3435 genotype and clopidogrel response.15,21,34 Some data suggest that the combination of CYP2C19 and ABCB1 polymorphisms may provide complementary information regarding clopidogrel pharmacodynamics and clinical outcomes.20,45 Lastly, polymorphisms in P2RY12, the gene which encodes the drug target of clopidogrel, have not been consistently associated with clopidogrel response in clinical studies.20,46–48
Clinical Implications
Owing to the accumulation of clopidogrel CYP2C19 pharmacogenetic and clinical outcome data, the Food and Drug Administration updated the clopidogrel prescribing information in March 2010 to include a boxed warning regarding the reduced effectiveness of clopidogrel in patients who are CYP2C19 poor metabolizers.49 The warning advised, but did not mandate, the use of other antiplatelet agents or alternative clopidogrel dosing strategies in these individuals. The warning also informed health care professionals that genetic tests are available to identify differences in CYP2C19 function; however, genetic testing prior to clopidogrel use was not mandated. Following this revision of clopidogrel prescribing information, the American College of Cardiology Foundation (ACCF) and American Heart Association (AHA) published a report to provide guidance on how to approach the boxed warning.50 The task force acknowledged that clinicians should be aware of the effects of CYP2C19 polymorphisms on clopidogrel metabolism and response; however, the task force found the evidence base to be insufficient to recommend routine genetic testing. In this regard, the task force highlighted the following issues and gaps in knowledge regarding clopidogrel pharmacogenetics: (1) available data suggest that the positive predictive value of CYP2C19 genotyping for adverse clinical events in ACS/PCI patients is low (12–20%);12,17 (2) CYP2C19 polymorphisms account for only a small portion of variability in clopidogrel response (~12%);15 and (3) there is a lack of clinical trial data showing that routine CYP2C19 genetic testing improves clinical outcomes. Nonetheless, the task force suggested that CYP2C19 genotyping may be warranted before initiating clopidogrel in certain ACS/PCI patients, such as those with a moderate or high risk of poor outcomes (e.g., patients undergoing complex or high-risk PCI procedures).
More recently, the Clinical Pharmacogenetics Implementation Consortium of the National Institutes of Health’s Pharmacogenomics Research Network published guidelines for CYP2C19 genotype-directed antiplatelet therapy.7 These guidelines acknowledge the absence of randomized trials documenting that CYP2C19 genotype-guided antiplatelet therapy improves clinical outcomes. However, the guidelines put forth that the strength and scope of existing studies, along with the availability of potentially more effective antiplatelet agents, supports the use of CYP2C19 genotyping in clinical practice. The guidelines recommend two options for patient genotyping: (1) genotype all patients who undergo PCI or (2) genotype only those patients who are at moderate or high risk for poor outcomes (e.g., patients undergoing high-risk PCI procedures or who have prior stent thrombosis or diabetes). In ACS/PCI patients, standard clopidogrel doses are recommended in ultrarapid or extensive metabolizers. In ACS/PCI patients who are intermediate or poor metabolizers (i.e., defined as carriers of the CYP2C19*2 allele), either prasugrel (which is metabolized by multiple CYP isoenzymes) or alternative drug therapies that do not require metabolic activation (e.g., ticagrelor, ticlopidine, cilostazol) are recommended, assuming no contraindications to therapy. Another potential option is to increase the clopidogrel dose in intermediate or poor metabolizers, although additional clinical outcome data in ACS/PCI patients are needed in this regard.51,52 In terms of alternative ACS/PCI drug therapies, prasugrel is a newer thienopyridine that does not undergo extensive CYP2C19 metabolism, and whose disposition is not affected by CYP2C19 polymorphisms.53 In ACS patients, prasugrel was superior to clopidogrel on clinical outcomes, but with an increased risk of major bleeding.54 Ticagrelor is a non-thienopyridine reversible inhibitor of the P2Y12 receptor that was approved by the FDA in July 2011. It does not undergo CYP2C19 metabolism, and was superior to clopidogrel in decreasing the risk of cardiovascular death, myocardial infarction, or stroke in patients with ACS, irrespective of CYP2C19 or ABCB1 polymorphisms.26 In addition, ticagrelor was effective in patients who were nonresponsive to clopidogrel.55 Beyond drug therapy, clinical factors (e.g., diabetes, increased body mass index, use of proton pump inhibitors) and possibly other genetic variants (e.g., CYP2C19*3) should be considered in patient care decisions.
Clopidogrel pharmacogenetics has made significant strides in the last 5 years, and it is one of the more well-developed areas in cardiovascular pharmacogenomics. However, there remain a number of issues regarding the implementation of prospective CYP2C19 genotyping in clinical practice. The most pressing need is for prospective, randomized trials to assess whether genotype-guided antiplatelet drug selection will improve clinical outcomes compared with standard of care approaches. While the simple solution may be to prescribe non-CYP2C19-dependent medications such as prasugrel or ticagrelor to all ACS/PCI patients, these agents are not without limitations including cost (both are brand name products), specific contraindications, limited approved indications, and bleeding risks.8 Further complicating the issue, clopidogrel became generic in 2011, resulting in major cost savings and additional use. Therefore, more studies are needed to determine the cost effectiveness of generic clopidogrel plus CYP2C19 genotyping versus alternative brand-name antiplatelet agents. Other issues that will need to be addressed in the future include payer reimbursement for genetic tests, availability of point-of-care genotyping assays, clarification of the role of platelet responsiveness testing in drug therapy selection and management, and the development of algorithms that incorporate genetic and nongenetic factors into the decision-making process.8,50,51
Clinical Case: Clopidogrel
JS is a 68-year-old, 59-kg female who is being admitted to the emergency department after experiencing an episode of sustained, “crushing,” substernal chest pain that radiates down her left arm. She has a past medical history significant for uncontrolled type 2 diabetes, depression, hypertension, and hyperlipidemia. She has no known drug allergies, has excellent medication coverage through her health insurance, and takes lisinopril, aspirin, glipizide, simvastatin, sertraline, and ranitidine. Based on her presenting symptoms, increased cardiac bio-markers, and electrocardiographic changes, she is diagnosed with STEMI and sent immediately for coronary angiography and subsequent PCI. Prior to PCI, JS is given 325 mg of aspirin and 300 mg of clopidogrel along with abciximab. During PCI, the patient is found to have 95% occlusion of her left anterior descending artery in which a drug-eluting stent (e.g., paclitaxel-eluting stent) is deployed. After 1 week of medical therapy, the patient is stabilized and discharged on the following medications: clopidogrel 75 mg daily, aspirin 325 mg daily, lisinopril 20 mg daily, metoprolol succinate 25 mg daily, ranitidine 75 mg twice daily, Lantus insulin 35 U at bedtime, atorvastatin 80 mg daily, and sertraline 75 mg daily. Four weeks after discharge, JS is readmitted with sustained chest pain, elevated cardiac biomarkers, and electrocardiographic changes suggestive of STEMI. Again, she is immediately sent for coronary angiography in which she is given 325 mg of aspirin and 600 mg of clopidogrel along with abciximab. JS is found to have thrombosed her drug-eluting stent. Pharmacogenetic testing indicates that JS has the CYP2C19*2/*2 genotype. JS’s medication adherence is also assessed and she is found to have missed only one dose of clopidogrel since her admission.
Question: Based on the case and her pharmacogenetic results, what should be done with this patient’s long-term antiplatelet regimen?
Answer: When assessing stent thrombosis that is possibly due to clopidogrel failure, many patient-related factors should be taken into consideration. First, medication adherence is one of the major causes of clopidogrel failure. Data suggest that one in six patients may delay filling his or her index clopidogrel after hospital discharge for drug-eluting stent implantation. Such delay has been associated with a significant increase in the risk of both death and myocardial infarction. Second, a patient’s medication regimen should be evaluated for potential drug–drug interactions that could impede metabolic activation of clopidogrel because of CYP2C19 inhibition. In the case of JS, she has excellent medication adherence, adequate drug coverage through her health insurance, and no major drug–drug interactions with clopidogrel. Based on the recommendations from the Clinical Pharmacogenetics Implementation Consortium of the National Institutes of Health’s Pharmacogenomics Research Network, JS would warrant CYP2C19 genotype testing as she possesses risk factors that increase the possibility of poor outcomes (e.g., diabetes and stent thrombosis). Based on her testing, JS has the CYP2C19*2/*2 genotype (i.e., poor metabolizer) meaning that she may lack the ability to metabolically convert clopidogrel to its active metabolite. What then would be the best long-term antiplatelet therapy for JS in addition to her aspirin? Options would consist of increasing her maintenance dose of clopidogrel to 150 mg daily or consider changing to ticlopidine, prasugrel, or ticagrelor. For higher dose clopidogrel, recent data from the GRAVITAS study suggest that patients with either one or two CYP2C19 loss-of-function alleles (*2) do not generally respond to double-dose clopidogrel. Ticlopidine is also not a therapeutic option for JS as it has limited data in patients with drug-eluting stents and is associated with significant adverse effects such as neutropenia, rash, and severe diarrhea. As JS has medication coverage through her health insurance, changing to either prasugrel or ticagrelor is an option. However, prasugrel is not recommended in patients with a body weight of 60 kg or less, age of 75 years or greater, or a propensity to bleed, as each of these risk factors increases the risk for major bleed. Since JS weighs less than 60 kg, prasugrel may not be an option. Compared with clopidogrel and prasugrel, ticagrelor is a twice-daily medication, so the potential exists for medication nonadherence. The aspirin dose will also need to be changed to 81 mg daily, as doses above 100 mg daily may reduce the effectiveness of ticagrelor. Nonetheless, based on the clinical evidence, ticagrelor may be the most optimal long-term antiplatelet therapy for this patient.
DYSLIPIDEMIA
Statins
Statins are the most commonly prescribed medications for the treatment of dyslipidemia.56 They lower LDL cholesterol by 18–55%, lower triglycerides by 7–30%, and raise HDL cholesterol by 5–15%.57 More importantly, statins reduce the risk of cardiovascular morbidity and mortality in patients with and without existing coronary heart disease.57,58 The statin drug class includes atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, and simvastatin. Statins competitively inhibit HMG CoA-reductase, the rate-limiting step in hepatic cholesterol biosynthesis. They also have pleiotropic effects including attenuation of endothelial dysfunction, inhibition of vascular inflammation, immunomodulation, and antithrombotic actions.59 Genetic polymorphisms have been shown to influence interindividual variability in statin pharmacokinetics, pharmacodynamics, and the risk of adverse effects.60 To date, a plethora of genes and polymorphisms has been investigated as potential predictors of statin response. However, in many cases, the pharmacogenetic associations are inconsistent and the clinical relevance is unclear. As such, the following section will focus on the more well-developed statin pharmacogenetic examples, namely, the role of solute carrier organic anion transporter family member 1B1 (SLCO1B1) drug transporter polymorphisms in statin clinical pharmacology, and the role of KIF6 genetics in the risk of cardiovascular disease and statin response. In addition, a brief overview of other genes involved in statin pharmacokinetics and pharmacodynamics will be presented.
Pharmacogenetic Focus: SLCO1B1
Organic anion-transporting polypeptide 1B1 (OATP1B1) is a sodium-independent uptake transporter expressed on the baso-lateral surface of hepatocytes. OAPT1B1 mediates the active uptake of drugs and endogenous compounds from portal blood into the liver.61,62 All of the statins are OATP1B1 substrates, and hepatic uptake of statins via OATP1B1 is the first step in their subsequent hepatic metabolism and biliary elimination (Figure 11–2).63 Owing to the key role of OATP1B1 in statin transport, alterations in OATP1B1 transporter function due to genetic polymorphisms may influence statin pharmacokinetics and pharmacodynamics.64
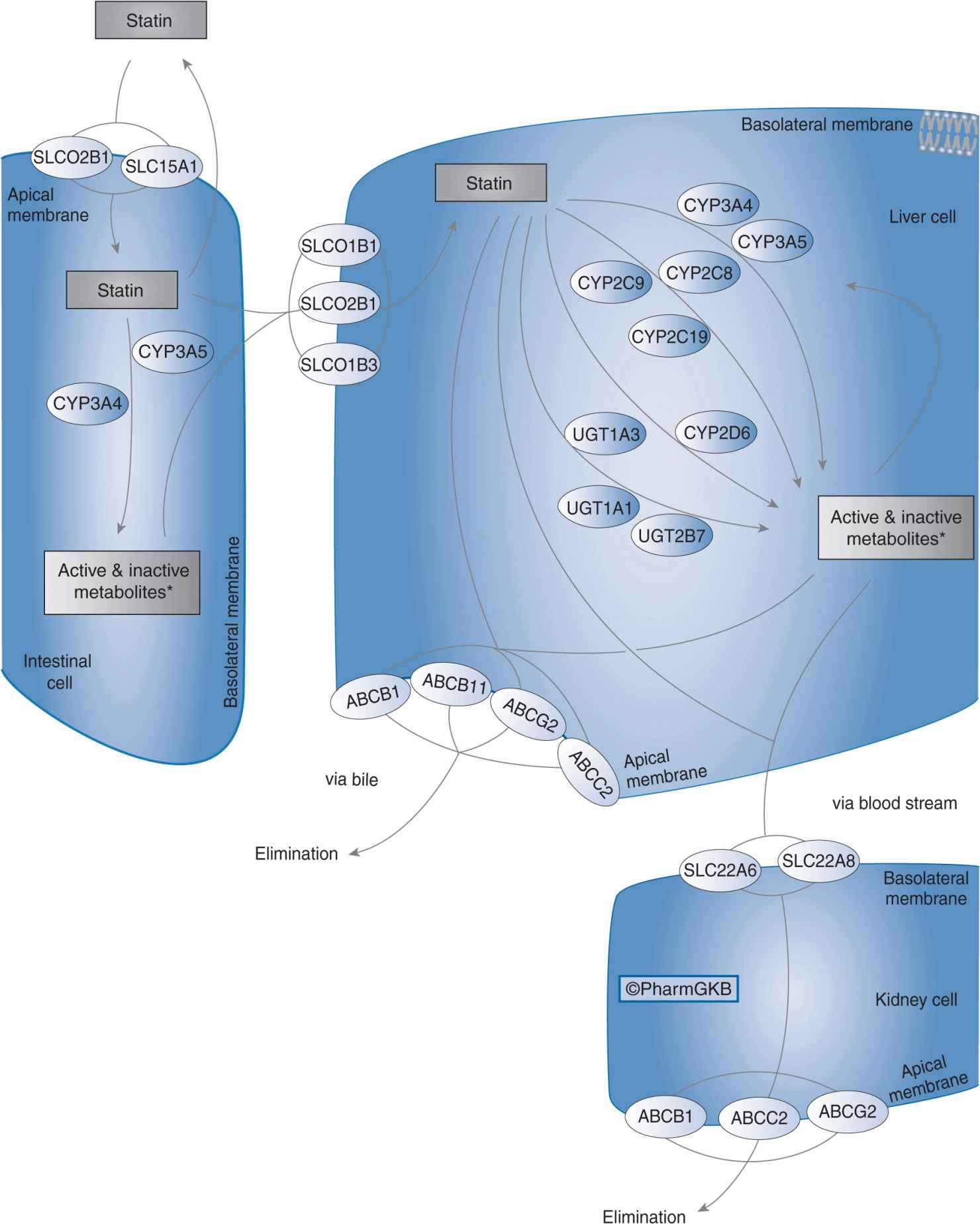
FIGURE 11–2 Statin pharmacokinetic pathway. SLCO1B1, SLCO2B1, SLCO1B3, SLC15A1, SLC22A6, and SLC22A8: uptake transporters; CYP3A4, CYP3A5, CYP2C9, CYP2C8, CYP2C19, CYP2D6, UGT1A3, UGT1A1, and UGT2B7: metabolizing enzymes; ABCB1, ABCB11, ABCG2, and ABCC2: efflux transporters. Figure copyright PharmGKB; reprinted with permission from PharmGKB and Stanford University.86
SLCO1B1 is the gene that encodes OATP1B1. The SLCO1B1 polymorphisms most often studied in relation to drug disposition are c.521 T>C (Val174Ala) and c.388 A>G (Asn130Asp). The frequency of the c.521C allele is 8–20% in Caucasians, 1–8% in those of African descent, and 8–16% in Asians.63 The frequency of the c.388G allele is 30–45% in Caucasians, 72–83% in those of African descent, and 59–86% in Asians.63 These polymorphisms are often studied in haplotype form (Table 11–2). The c.521C allele is associated with decreased OATP1B1 transporter activity, and the *5 and *15 haplotypes that contain this allele are referred to as low-activity haplotypes.65 The c.388G allele has been associated with increased transporter activity, and the *1B haplotype that contains this allele is referred to as a high-activity haplotype.66–68 In clinical studies, the c.521C allele (present in the *5 and *15 haplotypes) has been associated with decreased hepatic uptake and increased plasma exposure of atorvastatin, pitavastatin, pravastatin, rosuvastatin, and simvastatin.67,69–81 For example, the plasma exposure of simvastatin acid was 221% higher in healthy individuals with the variant c.521 C/C genotype compared with that in individuals with the wild-type T/T genotype.80 Of note, the c.521 T>C polymorphism does not affect the pharmacokinetics of fluvastatin, most likely due to the lipophilic nature of this statin.77 In some studies, the c.388G allele (present in the *1B haplotype) has been associated with increased hepatic uptake and decreased plasma concentrations of pravastatin and pitavastatin.63,67,82,83
TABLE 11–2 Description of SLCO1B1 haplotypes.

Statins inhibit cholesterol biosynthesis in the liver. Therefore, it might be expected that changes in the hepatic uptake of statins due to SLCO1B1 polymorphisms would result in variability in the lipid-lowering efficacy of these drugs. However, SLCO1B1 polymorphisms have not been consistently associated with the lipid-lowering effects of statins in clinical studies.74,84,85 In contrast, SLCO1B1 polymorphisms appear to play a greater role in mediating the risk of statin-associated adverse effects, namely, myopathy and rhabdomyolysis. The SLCO1B1 c.521 T>C polymorphism is associated with increased statin plasma concentrations, and statin-induced myopathy is a concentration-dependent toxicity.63 In this regard, a genome-wide association study showed that the SLCO1B1 c.521 T>C polymorphism was significantly associated with severe myopathy in a case-control study of patients treated with simvastatin 80 mg.84 Specifically, the odds ratio for myopathy was 16.9 (95% CI, 4.7–61.1) in patients with the c.521 C/C genotype as compared with the wild-type T/T genotype. Furthermore, over 60% of myopathy cases in this study were attributed to the c.521C allele.84 This study highlights the utility of genome-wide association studies in identifying genetic predictors of less common adverse events, such as statin myopathy. Another study showed that the SLCO1B1*5 haplotype was associated with milder forms of simvastatin- and atorvastatin-induced muscle side effects.85 Taken together, the SLCO1B1 c.521 T>C polymorphism has been proven to be a significant predictor of both mild and severe statin-induced muscle side effects.
Clinical Implications
Currently, SLCO1B1 genotyping to identify patients who may be at risk for statin-induced myopathy is not routinely used in clinical practice. However, in the future, prospective SLCO1B1 genotyping could potentially serve as a risk assessment tool. Genotype-based risk stratification may be most applicable during the first year of statin therapy, when the risk of myopathy is the highest. SLCO1B1 genotyping may be most relevant for patients with an increased risk for myopathy such as those with kidney or liver disease, muscle disease, advanced age, low body weight, on high-dose statin therapy, or on concomitant interacting medications (e.g., transplant and HIV populations).63,84 In patients with SLCO1B1 low-activity polymorphisms, potential alternative treatment options may include the use of low-dose statin therapy, use of other statins with a lower risk of myopathy (e.g., pravastatin), use of statins that do not appear to be affected by SLCO1B1 polymorphisms (e.g., fluvastatin), use of nonstatin lipid-lowering agents, or enhanced safety monitoring.63,87,88 SLCO1B1 genotyping may also serve as a useful tool to understand the etiology of severe myopathy or rhabdomyolysis when other clinical risk factors do not exist or are ruled out.
Pharmacogenetic Focus: KIF6
The KIF6 gene encodes the kinesin-like protein 6. This protein mediates the intracellular transport of organelles, protein complexes, and mRNA along microtubules.89 A nonsynonymous polymorphism, Trp719Arg, exists in KIF6. The 719Arg allele is common in the population, with frequencies of 37% in Caucasians, 84–90% in Africans/African Americans, and 48–57% in Asians.90 The KIF6 Trp719Arg polymorphism has been associated with an increased risk of coronary events in the placebo arms of two large clinical trials, the Cholesterol and Recurrent Events (CARE) trial and the West of Scotland Coronary Prevention Study (WOSCOPS).91 In the CARE trial, carriers of the KIF6 719Arg allele (i.e., Trp/Arg and Arg/Arg) had a 50% increased risk of recurrent myocardial infarction compared with noncarriers (i.e., Trp/Trp genotype). In WOSCOPS, carriers of the KIF6 719Arg allele had 55% increased odds of coronary heart disease compared with noncarriers.91 Further analysis of WOSCOPS revealed that carriers of the 719Arg allele derived a greater benefit from pravastatin therapy as compared with noncarriers. Specifically, pravastatin therapy resulted in a 5.49% absolute risk reduction in 719Arg carriers compared with a 0.09% absolute risk reduction in noncarriers. Along the same lines, in the Pravastatin or Atorvastatin Evaluation and Infection Therapy: Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) trial, 719Arg carriers had a greater benefit from high-dose statin therapy (atorvastatin 80 mg daily) compared with that from moderate-dose statin therapy (pravastatin 40 mg daily) than did 719Trp homozygotes.92 Taken together, these studies suggest that 719Arg carriers have an increased risk of coronary heart disease, and may derive greater benefit from statin therapy.
Clinical Implications
The KIF6 findings from large clinical cohorts spurred much excitement about the potential use of KIF6Trp719Arg genotyping in cardiovascular disease risk assessment and the prediction of relative statin efficacy.89,92–97 Along these lines, a KIF6 genetic test was developed and marketed to health care practitioners. It is estimated that over 150,000 KIF6 genetic tests have been performed to date.98,99 However, recent studies have begun to refute the initial associations between KIF6 genotype and coronary event risk. These studies have found no increased risk of coronary events in carriers of the 719Arg allele, and no interaction between KIF6 genotype and statin risk reduction.100–103 For example, a meta-analysis of 17,000 cases and over 39,000 controls found that the KIF6 Trp719Arg polymorphism was not associated with the risk of clinical coronary artery disease.100 The reasons for the discrepancies in the literature are unclear, but false-positive results in the early studies may be a factor.104 Another concerning factor is that the biological relationship between KIF6 and cardiovascular disease and statin response has not been fully elucidated.104 For example, KIF6 genotype does not appear to mediate the LDL-lowering effects of statin therapy.92,95,102 In light of the conflicting reports and the unclear biological plausibility of this gene, many experts have begun to question the clinical utility of KIF6 genetic testing. Furthermore, some experts have put forth that KIF6 genotyping is not warranted at this time.99,100,102,104 Additional analyses of large-scale clinical trials, and the execution of genome-wide association studies, will be needed to ascertain the definitive role of KIF6Trp719Arg genotyping in cardiovascular disease risk assessment and statin efficacy.
Other Pharmacokinetic Genes of Interest
The CYP3A family plays a major role in the phase 1 metabolism of atorvastatin, lovastatin, and simvastatin (Figure 11–2). A few studies have investigated the impact of CYP3A4 and CYP3A5 gene polymorphisms on statin pharmacokinetics and pharmacodynamics. In this regard, the CYP3A4*1B (–392A>G) polymorphism, which results in increased CYP3A4 expression, was associated with higher LDL cholesterol levels following atorvastatin treatment. This finding was hypothesized to be due to increased metabolism and lower systemic atorvastatin concentrations.105 The CYP3A5*3/*3 genotype, which results in no CYP3A5 protein expression (i.e., nonexpressor phenotype), was associated with increased simvastatin plasma exposure, and enhanced lipid-lowering effects of atorvastatin, lovastatin, and simvastatin in clinical studies.106,107 Along these lines, the CYP3A5*3/*3 genotype was associated with more severe muscle damage compared with CYP3A5*1/*3 heterozygotes (i.e., expressor phenotype) in atorvastatin-treated patients.108 Additional studies are needed to confirm these above findings, and to determine whether CYP3A4 and CYP3A5 polymorphisms affect clinical outcomes in patients receiving statin therapy.
P-glycoprotein (encoded by ABCB1) and breast cancer resistance protein (encoded by ABCG2) are important efflux transporters that play a role in statin disposition (Figure 11–2). Three polymorphisms in the ABCB1 gene, c.1236C>T, c.2677G>A/T, and c.3435 C>T, have been studied in relation to statin pharmacokinetics and lipid-lowering effects. For example, the ABCB1 1236T/2677T/3435T diplotype was associated with higher plasma exposure of atorvastatin and simvastatin compared with the wild-type 1236C/2677G/3435C diplotype.109 In another study, carriers of the c.1236T allele experienced a greater reduction in total and LDL cholesterol following treatment with simvastatin, as compared with wild-type homozygotes.110 Of note, the effects of ABCB1 polymorphisms on statin pharmacokinetics may be statin-specific, as one study showed that ABCB1 1236/2677/3435 haplotypes were not associated with the pharmacokinetics of fluvastatin, pravastatin, lovastatin, and rosuvastatin.111 In terms of ABCG2, the c.421C>A has been shown to influence statin pharmacokinetics and pharmacodynamics. Specifically, the c.421A allele was associated with increased plasma concentrations of atorvastatin, fluvastatin, rosuvastatin, and simvastatin lactone.112–114 Interestingly, the effect of the c.421A allele on plasma exposure appears to be greatest for rosuvastatin.113–115 The c.421 A/A genotype has been associated with enhanced reductions in LDL cholesterol compared with the C/C genotype following rosuvastatin treatment in Chinese and Caucasian populations.116–118 Taken together, ABCB1 and ABCG2 polymorphisms affect statin pharmacokinetics and pharmacodynamics, and the effects appear to be statin-specific. Additional studies are needed to determine whether these drug transporter polymorphisms translate into differences in clinical outcomes and adverse effects in patients treated with statins.
Other Pharmacodynamic Genes of Interest
Although the mechanism of action of the statins is to directly inhibit the HMG-CoA reducase enzyme, their subsequent effects on lipid homeostasis are complex. As such, identifying polymorphisms that predict statin pharmacodynamics is a challenging task. Traditionally, a candidate gene approach has been used to elucidate statin drug target pharmacogenomics. As a result, over 40 genes have been implicated in statin response.119 However, many of these initial genetic associations have a small effect on lipid homeostasis, and many examples have failed to be replicated in subsequent studies.
One of the genes most commonly associated with statin response in candidate gene analyses is apolipoprotein E (APOE). APOE is a component of chylomicrons and very-low-density lipoprotein cholesterol, and mediates chylomicron remnant removal. The three alleles most commonly studied in APOE include ε2, ε3 (wild-type), and ε4. The ε2 allele is associated with enhanced HMG-CoA activity, while the ε4 allele is associated with decreased HMG-CoA activity.119 Some, but not all, studies suggest that LDL-lowering response following statin treatment is greatest for the ε2 allele and smallest for the ε4 allele.119 For example, LDL-lowering following atorvastatin therapy was 3.5% greater in carriers of the ε2 allele compared with wild-type.120 Another candidate gene that has been variably associated with LDL response following statin therapy is HMGCR, which encodes HMG-CoA reductase (the target of statin action). Two polymorphisms in HMGCR have been associated with smaller reductions in total and LDL cholesterol following pravastatin therapy.121 However, this finding has not been consistently replicated in the literature. As previously mentioned, a large number of polymorphisms in other genes (e.g., CETP, PCSK9, ADAMTS1) have been implicated in statin response, but the effect sizes tend to be small, and the associations have not been consistently replicated between studies.
Recently, two genome-wide association studies have been conducted in the field of statin pharmacogenomics. The genome-wide approach allows for a comprehensive interrogation of the human genome, and potential identification of novel proteins involved in drug response.122 Participants from the Treating to New Targets (TNT) trial, who had received atorvastatin 10 mg daily for 8 weeks, were genotyped using whole genome and candidate gene approaches.123 Over 290,000 polymorphisms were evaluated; however, none were significantly associated with atorvastatin-induced changes in LDL, HDL, or triglyceride at the whole genome level. In a subsequent candidate gene analysis of this clinical population, only the APOE rs7412 polymorphism, which is contained in the ε2 allele, was associated with LDL cholesterol response after correction for multiple statistical tests.123 This is consistent with previous studies showing associations between the APOE ε2 variant and greater LDL-lowering following statin therapy. A recent genome-wide association study evaluated 3,936 patients who had participated in three different statin trials.124 The polymorphism that was most significantly associated with total cholesterol response at the whole genome level was located in the calmin (CLMN) gene. The function of CLMN, and its relationship to lipid homeostasis, is not known. This association remains to be replicated in additional cohorts.
Given the state of research in the field, polymorphisms related to statin pharmacodynamics are not ready for use in the clinical setting. In the future, genome-wide association studies will likely shed more light on genetic predictors of statin response and cardiovascular disease risk. Since many polymorphisms have a small effect on lipid response, it may be that a combination of polymorphisms (e.g., a genetic risk score) will be used to help guide statin therapy. Studies assessing the impact of genetics on cardiovascular outcomes will also be needed to move statin pharmacogenomics closer to the clinic.
Clinical Case: Statins
DT is a 55-year-old white male of normal weight who is admitted to the coronary care unit for a STEMI. The patient has a past medical history significant for hypertension and atrial fibrillation. DT has no known drug allergies, and does not smoke or consume alcohol. Prior to admission he was taking metoprolol succinate 150 mg twice daily, warfarin 5 mg daily, and lisinopril 20 mg daily. After receiving medical management, the patient was discharged 7 days later on the following medications: aspirin 325 mg daily, sotalol 160 mg twice daily, metoprolol succinate 25 mg daily, dabigatran 150 mg twice daily, lisinopril 40 mg daily, simvastatin 40 mg at bedtime, clopidogrel 75 mg daily, and sublingual nitroglycerin as needed. Three months after discharge, DT began passing dark colored urine with complaints of severe muscular thigh pain, confusion, nausea and vomiting, and significant dehydration. DT was readmitted to the hospital where his serum creatinine was 3.4 mg/dL (baseline, 1 mg/dL), serum potassium was 5.9 mEq/L (baseline, 3.5 mEq/L), and creatine kinase was 100,000 U/L (baseline, 60 U/L). DT is diagnosed with rhabdomyolysis. In order to address potential causes of his rhabdomyolysis, SLCO1B1 genetic testing is performed. DT is found to have the SLCO1B1*5/*15 genetic makeup.
Question: Based on the case and his genetic results, what is the potential cause of DT’s rhabdomyolysis?
Answer: After ruling out potential medical causes for rhabdomyolysis (e.g., extreme physical exertion, muscle crush from a car accident, alcohol intoxication, metabolic abnormalities, hyper-thermia, or infection), drug-induced etiologies should be evaluated. A drug class that is often associated with rhabdomyolysis is the statins. Rhabdomyolysis associated with statin therapy has an incidence of 3.4 per 100,000 patient-years of treatment. Unfortunately, the etiology of statin-induced rhabdomyolysis is poorly understood. Theoretically, decreased mevalonate production may disrupt small regulatory proteins required for myocyte cell membrane maintenance leading to the disorder. The risk of statin-induced myotoxicity is multifactorial, and varies with the pharmacokinetic properties of the individual statins and with the clinical factors of the individual patient. Transport of statins into the liver is an active process via OATP1B1, which is located on the basolateral side of hepatocytes. The majority of statins, such as simvastatin, are metabolized in the liver by the CYP enzymes, particularly CYP3A4. In the myocytes, the transport is dependent on passive diffusion; thus, statin concentrations and dose have been shown to increase the risk of rhabdomyolysis, although in a nonlinear fashion. Predisposing risks for statin-induced rhabdomyolysis consist of any factors affecting statin volume of distribution or metabolism (e.g., advanced age or renal and hepatic dysfunction), recent surgery, heavy exercise, and certain comorbidities (e.g., hypothyroidism and diabetes). Medically, DT has no obvious predisposing medical risk factors. From the standpoint of drug-drug interactions that could increase concentrations of simvastatin, none exist. While multiple medications are known to significantly increase concentrations of simvastatin (e.g., amiodarone, dronedarone, verapamil, diltiazem, amlodipine, ranolazine, cyclosporine, gemfibrozil, macrolides, azole antifungals, and protease inhibitors), none of DT’s medications are metabolized by CYP3A4. Therefore, this patient remains a clinical conundrum, thus warranting genetic testing.
In this case, DT has the SLCO1B1*5/*15 diplotype. Recent studies have identified common variants in SLCO1B1, such as c.521 T>C, to alter OATP1B1 activity. The *5/*15 diplotype, which contains two variant c.521C alleles, is associated with decreased OATP1B1 transporter function. Such a variant could significantly increase simvastatin systemic concentrations and possibly lead to rhabdomyolysis. As this patient is post-MI, he warrants a statin for overall reduction in morbidity and mortality. After an episode of potential statin-induced rhabdomyolysis, many clinicians would dismiss the idea of reconsidering statin administration. However, in the case of DT, the benefits of a statin may outweigh the risks. Using the genetic data provided, this can steer potential therapeutic recommendations. For DT, potential treatment options include a more water-soluble statin with less myopathy (e.g., pravastatin or low-dose rosuvastatin), a statin that does not appear to be affected by SLCO1B1 polymorphisms (e.g., fluvastatin), or an alternative lipid-lowering agent such as niacin or ezetimibe. However, it is important to note that ezetimibe may lack the pleiotropic cardiovascular effects that statins and niacin seem to possess. Taken together, SLCO1B1 genotyping may be useful in this case to elucidate potential etiologies of statin-induced rhabdomyolysis.
HYPERTENSION
Hypertension is a highly prevalent disease, affecting 34% of adults in the United States.125 It results in numerous adverse clinical consequences such as cardiovascular morbidity and mortality, stroke, and chronic kidney disease. A variety of pharmacologic agents exist for the treatment of hypertension, and current guidelines recommend the following drug classes as potential first-line treatment options: angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, β-blockers, calcium channel blockers, and thiazide diuretics.126
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


