Fig. 26.1
a Clear cell papillary RCC showing an aggregate of branching tubules, some cystically dilated and containing small papillary structures. The tubules are lined by cells with clear cytoplasm and low-grade nuclei. b Clear cell papillary RCC showing linear arrangement of nuclei away from the basal aspect of the cells, imparting a “piano keys” appearance
Immunoprofile
CCP-RCCs have an unusual immunoprofile which is distinct from both classic clear cell type RCC as well as papillary type RCC. These tumors are diffusely and strongly positive with cytokeratin 7 (CK7) in both the solid and cystic areas and negative with alpha-methylacyl CoA racemase (AMACR). This staining pattern is distinct from both classic clear cell type RCC which are commonly negative with CK7 and papillary type RCC which are typically AMACR positive.
Although diffuse membranous positivity with carbonic anhydrase IX (CA-IX) is well described in clear cell type RCCs, CCP-RCCs have been shown to be positive with CA-IX, showing a characteristic “cup-like” reactivity with the absence of staining on the luminal aspect [6]. These tumors are usually negative or show only focal expression of CD10 and are commonly diffusely positive with high-molecular weight cytokeratin (HMWCK, 34beta E12).
Molecular Profile
So far, these tumors have not been shown to have the von Hippel–Lindau (VHL) gene mutations or 3p deletions commonly observed in classic clear cell type RCCs. They also lack trisomy of chromosome 7 or loss of Y chromosome, cytogenetic changes commonly observed in papillary RCCs (PRCCs) [3, 7]. Recent studies have shown relative overexpression of VHL mRNA in CCP-RCCs when compared with clear-cell RCCs. CCP-RCCs have also been shown to express HIF-1α and Glucose-transporter 1 (GLUT-1). The co-expression CAIX, GLUT-1 and HIF-1α in the absence of VHL gene alterations suggest activation of the HIF pathway by non-VHL dependent mechanisms [8]. Low copy number gains of chromosomes 7 and 17 have also been reported in some cases [4].
Prognosis
CCP-RCCs are typically small, biologically indolent tumors. No lymph node or distant metastasis of these tumors has been reported to date in the literature.
Recently a distinct tumorous entity named renal angiomyoadenomatous tumor (RAT) has been described in the literature [9], composed of an intimate mixture of epithelial cells associated with a variably prominent smooth muscle stroma often forming abortive vascular structures. The epithelium, described in these tumors as having a predominant tubular architecture lined by cells with low-grade nuclei and clear cytoplasm, is similar to some of the cases illustrated in the description of CCP-RCC. The epithelial component of RAT is positive with CK7, CA-IX and negative with CD10: an immunoprofile identical to CCP-RCC. The smooth muscle stroma marks with common muscle markers. No VHL mutations have been identified in these tumors. The current perspective is that both CCP-RCC and RAT have orphologic and immunohistochemical similarities and probably represent the spectrum of a single entity [4, 5, 14]
Differential Diagnosis
The most critical differential diagnoses include classic clear cell type RCC and PRCC . Additionally, translocation-associated RCC may enter the differential diagnosis.
Clear cell RCCs, the most common subtype of RCC, are typically golden yellow with a variegated appearance including the presence of hemorrhage and necrosis. These RCCs show a variable cystic appearance ranging from focal to extensively multicystic on gross examination. The tumor cells have clear cytoplasm are arranged in nests, alveoli or solid sheets separated by a characteristic intricate delicate sinusoidal (racemose) vascular network, seen in the vast majority of cases. Some tumors may show prominent areas with granular/eosinophilic cytoplasm, usually associated with a high nuclear grade. While clear cell RCCs may show focal papillary/pseudopapillary areas, a prominent papillary architecture is uncommon as is the characteristic linear arrangement of nuclei seen in CCP-RCC. Most clear cell RCCs are negative with CK7, although focal CK7 expression can be seen in and around cystic areas . Diffuse CK7 expression in the majority of the tumor is not a characteristic of clear cell type RCC [11]. Clear cell RCCs typically show diffuse membranous reactivity with CA-IX [12] and CD10 [7, 13] and commonly lack reactivity to AMACR [14] and HMWCK [15].
CCP-RCC have also been reported in patients with VHL disease 16, 17], an autosomal dominant disorder associated with mutations in the VHL tumor suppressor gene located on short arm of chromosome 3. One recent study [16] reported that these tumors noted in 3 patients with VHL disease had the characteristic morphology and immunoprofile of sporadic CCP-RCC and also lacked 3p deletion. However, another study [17] has reported that the majority of these CCP-RCClike tumors (12/14) arising in patients with VHL disease lack the characteristic immunoprofile of sporadic CCP- RCC and frequently demonstrate chromosome 3p deletion. Overall, based on the current available data, it appears that that CCPRCC may occur in VHL disease and should be included in the differential diagnosis when working up cystic and/or bilateral renal tumors with clear cell features in this clinical setting.
PRCCs are the second most common subtype of RCCs and account for 10–15 % of all RCCs. These tumors have a unique genotype characterized by trisomy of chromosomes 7 and 17 and loss of chromosome Y. These tumors commonly show a predominant papillary or tubulo-papillary architecture mimicking CCP-RCC; however, they typically lack the homogenous optically clear cytoplasm seen in CCP-RCC. Delahunt and Eble [16] proposed a morphologic subdivision of PRCCs into type 1 and type 2 PRCCs for prognostic purposes. This subdivision is now included in the current WHO classification of renal carcinomas. Type 1 PRCCs, which are more common, are characterized by papillae lined by small cells with low nuclear grade and scant amphophilic cytoplasm arranged in a single layer. They frequently show aggregates of foamy macrophages within fibrovascular cores, cholesterol clefts, and foci of necrosis, all of which are typically absent in CCP-RCC. Although type 1 PRCCs are characteristically positive with AMACR, CK7, and CD10, they are usually negative or only focally positive with CA-IX [1, 11, 17]. Type 2 PRCCs are composed of tumor cells with high Fuhrman nuclear grade, abundant eosinophilic (oncocytic) cytoplasm and pseudostratification of nuclei on papillary cores. Type 2 PRCCs are unlikely to be confused be with the low-grade CCP-RCC .
Translocation-associated RCCs are a rare subtype of RCCs. They are more frequent in pediatric and young adults [1, 17] although a few cases have been described in adults [18]. These tumors typically show prominent papillary and/or solid alveolar growth patterns and are composed of cells with high nuclear grade and clear to granular, eosinophilic cytoplasm. Cells lined exclusively by clear cells are rare. Psammoma bodies are typically present [17]. These carcinomas are usually negative or only focally positive with epithelial markers (CK cocktail, CK7, epithelial membrane antigen, EMA) and vimentin. Transcription factor E3 (TFE3) and transcription factor EB (TFEB) are highly sensitive and specific markers for translocation-associated RCC, which are negative in CCP-RCC. These tumors are described in detail subsequently in this chapter .
Tubulocystic Carcinoma
Tubulocystic carcinoma was originally described in 1956 and classified as low-grade collecting duct carcinoma [19, 20]. Recently, it has become generally accepted that tubulocystic and collecting duct carcinomas are separate tumors from a clinical and molecular perspective, and the current name of tubulocystic carcinoma of the kidney was proposed in 2004 [19, 21]. While some argue that tubulocystic carcinoma is a distinct entity which deserves a separate designation [19], there are some studies that have found compelling evidence of a link between tubulocystic carcinoma and PRCC; first, tubulocystic carcinoma is more often multicentric, similar to papillary carcinoma. Second, tubulocystic carcinoma and papillary carcinoma often occur concurrently and can be intimately admixed. Finally, tubulocystic carcinomas have a similar immunohistochemical and molecular phenotype to PRCC [22]. Whether or not tubulocystic carcinoma is a distinct entity or a tumor closely related to PRCC has not been currently resolved.
Tubulocystic carcinomas are rare; in one study this subtype accounted for < 1 % of all RCCs [23]. All reported cases have occurred in adults (ranging from 30 to 94), with a male to female ratio of 3–7:1.
Gross
Microscopy
Microscopically, tubulocystic carcinomas are well circumscribed and composed primarily of variably dilated cysts which are relatively evenly spaced in a fibrotic, hyalinized stroma. No background racemose type vascularity is typically present. The cysts are lined by a single layer of flat to columnar cells with granular oncocytic cytoplasm and nuclei with prominent nucleoli, similar to Fuhrman nuclear grade 3 (Fig. 26.2a). A characteristic feature is the presence of incomplete septae which are free-floating within cystic spaces (Fig. 26.2b). Occasional cysts can be quite dilated up to 1 cm, and prominent hobnailing of the cells can be present. Tumors can occasionally have focal clearing in the cytoplasm of the cells. Very focal cellular stratification and very focal papillae within the cysts have been described; however, prominent or extensive papillary architecture is not a typical feature and should prompt consideration of an admixed component of PRCC. Increased mitoses, necrosis, and angiolymphatic invasion are not typically present. Desmoplastic stroma and cellular ovarian type stroma are absent [19, 22, 24, 25]. Exceptional cases of tubulocystic carcinoma with poorly differentiated areas, some of which resemble collecting duct carcinoma, have been described [26].
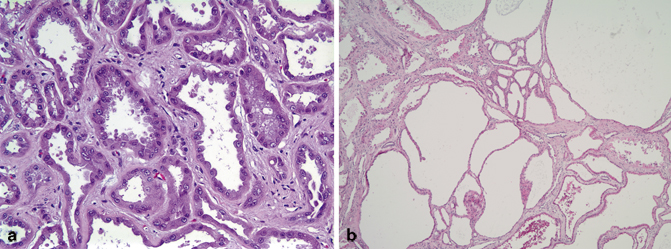
Fig. 26.2
a Tubulocystic carcinoma contains cells with abundant granular cytoplasm and hobnailing nuclei with prominent nucleoli. b Cystic spaces containing incomplete septae in tubulocystic carcinoma
In some studies, tubulocystic carcinomas have been found to be more often multicentric (up to 20 % in one series), similar to the rate of multicentricity in PRCC, and higher than the rates of multicentricity seen in other subtypes of RCC [22]. In other series, multicentricity rates in tubulocystic carcinoma were low (6 %) [19]. Also, in several series, concurrent or admixed papillary renal neoplasms were common (50 % in one larger series); the associated papillary neoplasms included papillary adenomas and type 1 and type 2 PRCCs [22, 25]. In those cases in which the PRCC was intimately admixed with the tubulocystic carcinoma, most had similar cellular morphology in both the tubulocystic and papillary areas [22]. This association with papillary renal neoplasms has not, however, been reported in other series [19].
The appropriate way to classify carcinomas which have both a tubulocystic and papillary component is somewhat controversial; some have recommended classifying such tumors as “renal cell carcinoma, unclassified type, with tubulocystic features” [22]. This would require close communication with the clinical team, however, because some clinicians consider “unclassified type” to necessarily indicate a high-grade aggressive RCC.
Immunoprofile
Tubulocystic carcinomas are typically positive for CK8, CK18, and CK19; most are negative for CK34betaE12, with a few rare cases exhibiting very focal positivity. Tumors are variably positive for CK7, with some series reporting heterogeneous staining for CK7 in most of their cases and others reporting only very focal or weak staining in the majority of their cases. AMACR, CD10, and kidney specific cadherin are frequently strongly positive in tubulocystic carcinomas. PAX-2 is described as being positive in only a subset of cases. Tubulocystic carcinomas are positive for PAX-8, vimentin, and RCC Ma [19, 22–25]. In a few cases in which a papillary RCC component was admixed with the tubulocystic carcinoma and for which immunohistochemical stains were performed, the papillary and tubulocystic component had a similar immunohistochemical profile [22].
Molecular Profile
One study has shown that the gene expression profiles of tubulocystic carcinoma and collecting duct carcinomas are distinct, with the majority of tubulocystic carcinomas showing a statistically significant relative overexpression of vimentin, p53, and AMACR, compared to collecting duct carcinomas [21]. In a few studies, the majority of tubulocystic carcinomas had a molecular profile similar to papillary carcinomas (gains of chromosomes 7 and 17 and loss of chromosome Y). In a few tumors with both a papillary carcinoma and tubulocystic carcinoma component which were analyzed, the molecular profile was found to be similar in both components. Notably, however, there is a subset of tubulocystic carcinomas which does not harbor the characteristic molecular changes of PRCC [22].
Prognosis
The vast majority of reported cases of pure tubulocystic carcinoma presented at low stage (pT1), and the majority of patients were disease-free at the follow-up after resection. However, rare cases of pure tubulocystic carcinomas have been reported to present at high stage (pT3 or with pelvic lymph node metastases) , and occasional patients develop either local recurrence or distant metastasis. Interestingly, while all of the described tubulocystic carcinomas have had high nuclear grade features, the majority of tumors had a good prognosis, indicating that Fuhrman nuclear grade may not be predictive of prognosis in this tumor [19, 23–25]. Tubulocystic carcinomas with admixed high-grade PRCC or with poorly differentiated areas may, as expected, have worse prognoses. In one study, a patient with admixed tubulocystic carcinoma and high-grade PRCC developed metastases attributed to the high-grade PRCC [25]. In another study of tubulocystic carcinomas with poorly differentiated areas, two of the three patients had follow up; one had a local recurrence, and the other patient died of distant metastases [26].
Differential Diagnoses
The main items in the differential diagnosis for tubulocystic carcinoma primarily include cystic entities of the kidney , such as benign renal cysts, oncocytoma with prominent cystic change, multilocular cystic RCC, and cystic nephroma/mixed epithelial and stromal tumors (MESTs) of the kidney. On core biopsies (when the entire lesion is not available for examination), it may even be difficult to distinguish tubulocystic carcinoma from dilated nonneoplastic renal tubules. Typically, benign cysts and dilated nonneoplastic tubules are lined by unremarkable attenuated tubular epithelium; hobnailing cells and high nuclear grade features with prominent nucleoli should not be present. The presence of a distinct mass seen grossly or radiographically may also be helpful in distinguishing dilated nonneoplastic tubules from a tubulocystic carcinoma. While tubulocystic carcinomas have high grade, irregular nuclei with chromatin alteration, oncocytomas with prominent cystic change retain relatively round, uniform low-grade appearing nuclei with even chromatin and occasional nucleoli; prominent hobnailing is not typically present, and cystic areas may merge with areas of more conventional nests of oncocytoma. Multilocular cystic RCCs are lined by low-grade cells with optically clear cytoplasm, identical to those seen in low-grade clear cell RCCs ; cells with granular eosinophilic cytoplasm, high-grade nuclei, and prominent hobnailing should not be present. Cystic nephromas are composed of multiple cysts with lining epithelium that can range from attenuated to cuboidal or columnar to hobnailing; MESTs, which some believe to be closely related to cystic nephromas, can have similar cysts or may have more variably sized cysts, with some small branching tubules or cysts which can resemble glandular epithelium; ciliated epithelium and epithelium with clear cytoplasm may also be present. In both cystic nephromas and MESTs, the nuclei of the lining epithelium are typically low grade, without prominent nucleoli. In addition, cystic nephromas and MESTs are characterized by intervening cellular spindled “ovarian type” stroma which is estrogen receptor (ER) and progesterone receptor (PR) positive and can occasionally show cellular condensation surrounding the cysts, in contrast to the fibrotic, hyalinized stroma seen in tubulocystic carcinomas. Cystic nephromas and MESTs also often have thick walled or proliferating dilated blood vessels within the stroma, and in addition MESTs can have stromal smooth muscle or adipose tissue. The cysts in cystic nephromas and MESTs can be evenly distributed, as in tubulocystic carcinomas; however, in many cystic nephromas and MESTs, the cysts are clustered, with prominent intervening stroma, which would be unusual in a tubulocystic carcinoma [19, 22].
The characteristic incomplete septae which are free-floating within cystic spaces can be helpful, as these are unusual in the other cystic entities mentioned above. In difficult cases, immunostains may be helpful; tubulocystic carcinomas lack an ER, PR positive stroma and are AMACR positive.
Acquired-Cystic-Disease-Associated Renal Cell Carcinoma
ESRD has been known to be associated with an increased risk of developing RCC, with an overall incidence of renal carcinomas in end-stage kidneys of about 3–7 % [1]. A large clinicopathologic series published in 2006 by Tickoo et al. [2] proposed acquired-cystic-disease-associated renal cell carcinoma (ACD-associated RCC) as a specific subtype of renal cancer arising in end-stage renal disease. These tumors are commonly found incidentally on imaging for surveillance of chronic renal disease.
Acquired cystic kidney disease develops in approximately half of patients undergoing dialysis. While cysts are present in 8 % of all patients beginning dialysis, both the number and size of cysts progressively increase as the duration of dialysis increases (> 90 % incidence after 10 years or more of dialysis). The type of dialysis (peritoneal vs. hemodialysis) does not appear to be significant. These patients are at increased risk of developing RCC, with a risk 100 times that of the general population. ACD-associated RCC is becoming a well-recognized subtype of RCC and is diagnosed based on the characteristic histologic appearance and the background cystic disease. These RCCs are the most common subtype of RCC noted in end-stage kidneys and are almost always seen in patients on dialysis. Although more common in end-stage kidneys with ACD, where ACD-associated RCCs account for 46 % of dominant masses, ACD-associated RCCs can also occur in noncystic end-stage kidneys.
It is important to remember that other RCC subtypes can be seen in both acquired cystic and noncystic end-stage kidneys. They include well-documented subtypes such as PRCC, clear cell RCC and chromophobe RCC which account for approximately 40 % of all RCCs arising in end-stage kidneys. Several causes have been proposed for the increased incidence of RCCs in ESRD including depressed cellular and humoral immunity in renal failure, impaired antioxidant defense, chronic infections and inflammation with release of free radicals causing deoxyribonucleic acid (DNA) damage and mutations, use of immunosuppressive medications, and proliferative activity induced by the oxalate crystals [1].
Gross
ACD-associated RCCs are commonly multifocal and bilateral. They can have a thick fibrous capsule and can appear to have arisen in cysts. They are usually well circumscribed and frequently show foci of hemorrhage, necrosis, and calcification. The background kidney can be normal in size or small and scarred. In acquired cystic end-stage kidneys, numerous cortical and medullary cysts are noted, ranging in size from 0.5 to 3 cm. The cysts initially form in the cortex, but in advanced cases medullary cysts can occur.
Microscopy
Cells in these RCCs are arranged in a variety of architectural patterns including acinar, papillary, solid, and cystic. The tumor cells are large with abundant eosinophilic/oncocytic cytoplasm and large nuclei with prominent nucleoli, reminiscent of Fuhrman grade 3 nuclei. Frequent cytoplasmic lumina confer the characteristic “cribriform” or “sieve-like” architecture to this tumor. Another characteristic feature seen in the vast majority of these tumors is the presence of abundant intratumoral oxalate crystals [2, 27, 28]. These crystals are seen within the tumor and are not associated with foci of necrosis or inflammation (Fig. 26.3). Focal areas composed of cells with clear cytoplasm are not uncommon; such foci can mimic clear cell RCC. Sarcomatoid and rhabdoid features may be seen in a subset of cases. In the background kidney, especially in the setting of acquired cystic kidney disease, there are numerous cysts lined by a similar population of eosinophilic cells. While these cysts are usually distributed throughout the kidney, occasionally they can be clustered together.
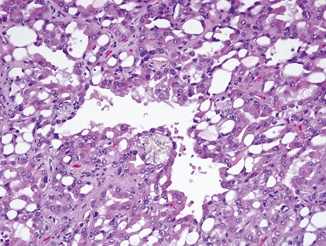
Fig. 26.3
Numerous vacuoles present within acquired cystic disease-associated carcinoma. Associated oxalate crystals are evident
Immunoprofile
Tumor cells are diffusely positive with AMACR and usually negative or only focally positive with CK7. CK AE1/AE3, CD10, and RCC marker are typically positive, and lack of high-molecular weight CK expression has been reported [1, 29, 30]. In our anecdotal, unpublished experience, these tumors are diffusely positive with PAX-8. The cysts in the background kidney are also usually diffusely positive with AMACR and negative or only focally positive with CK7.
Molecular Profile
ACD-associated RCCs lack the characteristic changes seen in clear cell RCCs (VHL gene mutation or 3p deletion) or PRCCs (trisomy of chromosomes 7 and 17 as well as loss of chromosome Y). Comparative genomic hybridization (CGH) and fluorescence in situ hybridization (FISH) studies have shown chromosomal gains on multiple chromosomes including chromosomes 3, 7, 16, 17, and Y. Chromosomal losses are uncommon while frequent gains on chromosomes 3 and Y have been reported [30, 31].
Prognosis
Most tumors are small and have a good prognosis. Metastasis is rare and when present is usually to regional lymph nodes. Rare cases have presented with extrarenal extension, renal vein extension (pT3 disease), and/or sarcomatoid and rhabdoid differentiation [32].
Differential Diagnosis
The most common differential diagnosis of ACD-associated RCC includes PRCC, clear cell RCC, and RCCs with oncocytic cytoplasm .
PRCCs as mentioned previously can be subdivided into type 1 and type 2 PRCCs for prognostic purposes [16]. Type 2 PRCCs may be confused with ACD-associated RCCs as both can have tubulopapillary architecture as well as cells with predominantly oncocytic cytoplasm and high-grade nuclei with prominent nucleoli; rarely type 1 PRCCs, which tend to show scant basophilic or focally clear cytoplasm with low-grade nuclei, may also be confused with ACD-associated RCC. While PRCCs can be seen in the setting of cystic end-stage kidneys, these tumors lack the characteristic intratumoral oxalate crystals as well as the cytoplasmic lumina imparting a cribriform appearance seen in ACD-associated RCCs. Also, while both tumors express diffuse AMACR expression, the vast majority of PRCCs are diffusely positive with CK7 unlike ACD-associated RCCs.
Classic clear cell RCCs can occasionally cause diagnostic confusion, as ACD-associated RCCs can show foci with clear cell morphology mimicking clear cell RCCs. However, ACD-associated RCCs lack the characteristic delicate sinusoidal “racemose” vasculature characteristically seen in the majority of clear cell RCCs. Although clear cell RCCs can frequently have eosinophilic granular cytoplasm with high-grade nuclei showing prominent nucleoli, clear cell RCCs lack the varied architecture including papillary and cribriform patterns as well as the oxalate crystals frequently seen in ACD-associated RCCs. While both these tumor types are usually negative with CK7, clear cell RCCs are also negative or show only focal AMACR expression. Strong, diffuse, membranous expression with CA-IX is commonly seen in clear cell RCCs. In our experience, CD10 is not very useful in distinguishing these tumors.
RCCs with oncocytic cytoplasm such as chromophobe RCCs or high-grade unclassified type RCCs with oncocytic cytoplasm can rarely enter into the differential diagnosis; careful attention to immunomorphologic features can usually resolve the diagnosis. ACD-associated RCCs lack the plant-like architecture, koilocytic atypia, and diffuse CK7 positivity commonly noted in chromophobe RCCs. Unclassified RCC is a diagnosis of exclusion; these tumors lack the characteristic morphology and oxalate crystals seen in ACD-associated RCCs.
Translocation-Associated Renal Cell Carcinomas
While renal carcinomas associated with Xp11.2 translocations/TFE3 gene fusions have been included in the most recent WHO classification [33], the spectrum of translocation-associated RCCs has greatly expanded. This group of closely related carcinomas is defined by a translocation involving one of the members of the microphthalmia-associated transcription factor (MiTF) family, which codes for basic helix-loop-helix/leucine zipper transcription factors. Members of this family include TFE3, TFEB, TFEC, and MiTF. They share homologous DNA binding and activation domains, and may have functional overlap; MiTF is important in melanogenesis. RCCs which harbor these translocations are collectively referred to as MiTF/TFE family translocation-associated carcinomas and include Xp11.2/TFE3 translocation-associated carcinoma and its purported subtype, melanotic Xp11 translocation tumor, as well as TFEB (t(6;11)) associated carcinoma [34–36].
Xp11.2 Translocation-Associated RCC
Xp11.2/TFE3 translocation-associated RCCs are rare, with reported incidences in large series of adult neoplasms of 1.6–4.2 %. This carcinoma constitutes a much higher proportion of RCC in children and young adults; reported incidences in this population range widely (20–76 %) depending on the study and the age cutoff. However, given the rarity of RCC in children and young adults, it is likely that the absolute number of Xp11.2 translocation associated carcinomas is higher in adult populations than in pediatric populations [17, 36]. A significant proportion (15 %) are associated with a history of chemotherapy [37].
Microscopy
The most characteristic morphology of Xp11.2 translocation-associated carcinomas is architectural heterogeneity; within any given tumor, cells can be variably arranged in sheets, nests, trabeculae, true papillae, or pseudopapillae. The cells typically have voluminous cytoplasm which can range from eosinophilic and granular to clear and can have bulging cell borders. Within any tumor the nuclear grade can vary, but these are almost uniformly at least focally of high Fuhrman nuclear grade. Clear cells arranged in some areas around true papillae and in other areas in solid sheets and nests with prominent cell borders is a relatively specific morphologic feature seen in many of these tumors (Fig. 26.4a). Many of these tumors exhibit, at least focally, a pseudoalveolar pattern in which cells are arranged in alveoli with central cellular discohesion (Fig. 26.4b). In some areas, the discohesion can lead to the formation of pseudopapillae. Prominent psammoma bodies and scattered xanthoma cells have been described in some tumors (Fig. 26.4c). Different gene fusions may lead to differing morphologic features; those with the ASPL-TFE3 typically are composed of large polygonal cells with abundant cytoplasm and high-grade nuclei arranged in an alveolar or pseudopapillary pattern. Psammoma bodies can be extensive. In contrast, those with the PRCC-TFE3 gene fusion typically have tumor cells with less abundant cytoplasm arranged in nests; psammoma bodies are rare or absent [34, 36, 38, 39]. Recently, newly described Xp11 translocation-associated RCCs have expanded the morphologic spectrum; tumors with a dual population of cells, some with voluminous abundant cytoplasm and prominent cell borders and some with less abundant cytoplasm arranged around hyaline material, similar to that seen in t(6;11) associated RCCs; tumors with pleomorphic neoplastic giant cells; tumors with hobnailing cells arranged in tubules and cysts; tumors with low-grade spindled areas; tumors with prominent cystic change; and tumors resembling infiltrative urothelial carcinoma have been described [18, 40, 41].
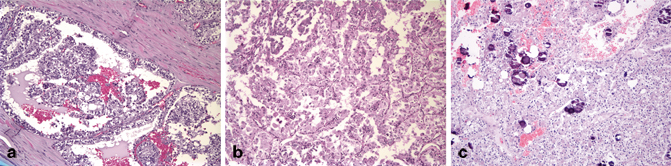
Fig. 26.4
Xp11 translocation-associated carcinoma showing a clear cells arranged around true papillae, b areas with an alveolar architecture, and c numerous psammoma bodies
Immunohistochemistry
The majority of Xp11.2 translocation-associated carcinomas are either negative for or only very focally positive for epithelial markers such as CK cocktail, CK CAM5.2, CK7, and EMA. The other characteristic immunostain is for the mutant (chimeric) TFE3 protein, which is overexpressed relative to native TFE3; the immunostain utilizes an antibody directed against the C-terminal portion of TFE3, which is preserved across all described gene fusions . Reported sensitivity (82–97.5 %) and specificity (79–99.6 %) for this immunostain vary widely [42–44]. This is likely due to differences in methodology. In addition, the immunostain can be technically challenging, and depending on fixation of the tissue and the methodology of the stain, native TFE3 can pick up the stain; typically Xp11.2 translocation carcinomas exhibit moderate to strong, diffuse nuclear staining that can be appreciated at low power. Therefore, appropriate caution should be used in interpreting this immunostain; comparison with the staining pattern in the adjacent normal kidney is helpful, and it should be interpreted in the context of the overall morphology as well as with any supporting molecular findings [36, 42]. These carcinomas frequently label for PAX-2 and PAX-8. Vimentin is usually negative or only very focally positive. The tumors are typically positive for CD10, RCC, and AMACR [38, 39, 45, 46]. CA-IX is either negative or only very focally positive, usually around areas of necrosis [46]. One relatively new marker that shows promise is cathepsin-K, a protease whose expression is driven by MiTF in osteoclasts; cytoplasmic expression of cathepsin-K can help distinguish MiTF/TFE RCCs from other RCCs. It has been found to be relatively specific but not particularly sensitive for Xp11 translocation carcinomas. Interestingly, in one study cathepsin-K was found to be positive in RCCs harboring the PRCC-TFE3 gene fusion but not in those harboring ASPSCR1-TFE3 gene fusion, suggesting that expression of this marker may be dependent on the particular gene fusion expressed [47, 48].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


