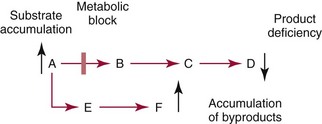
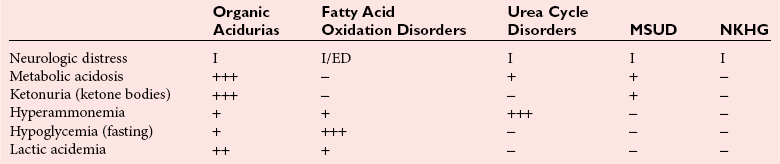
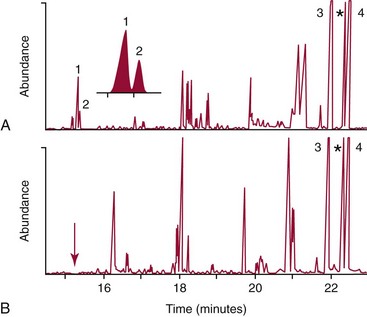
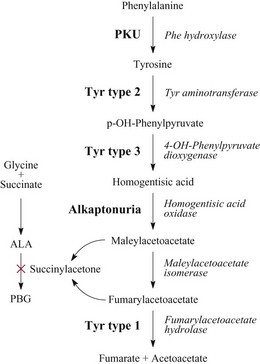
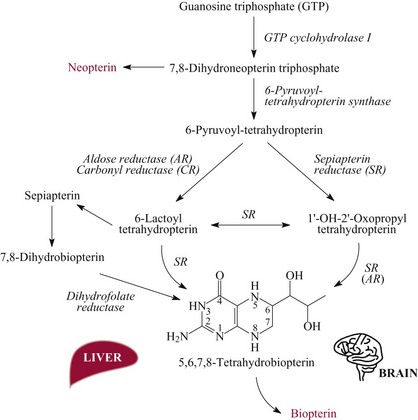
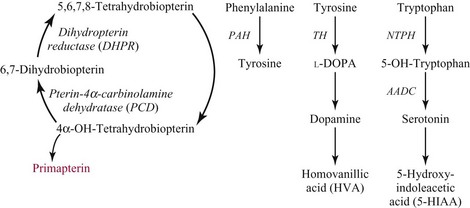
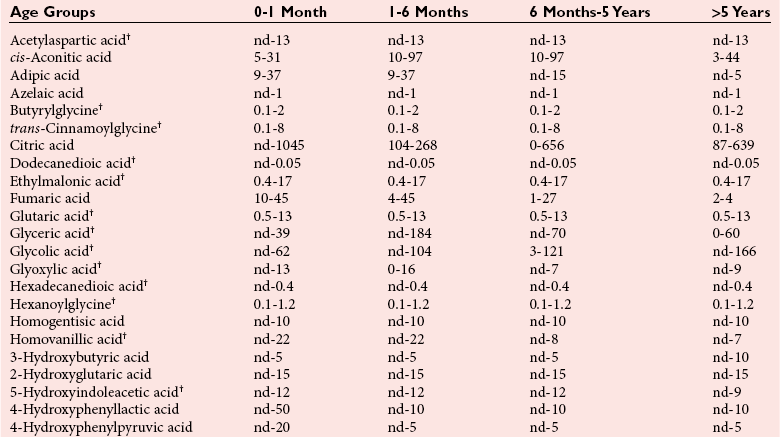
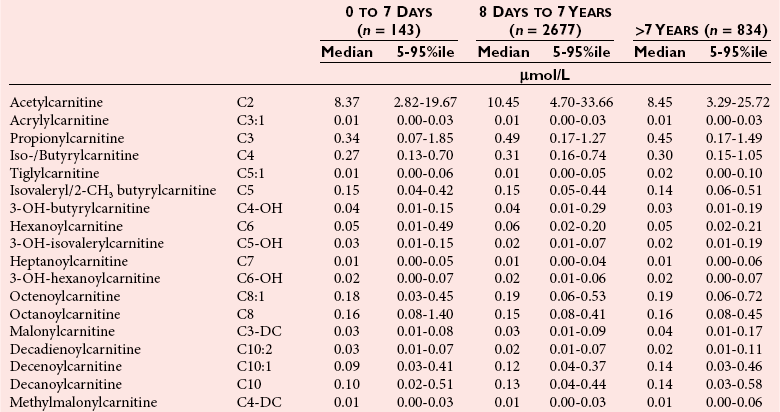
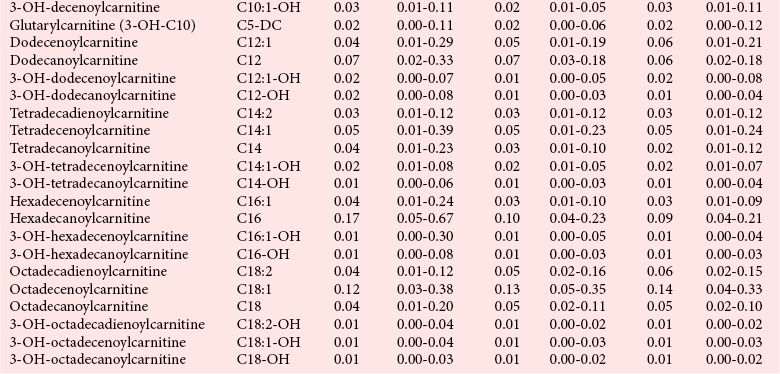
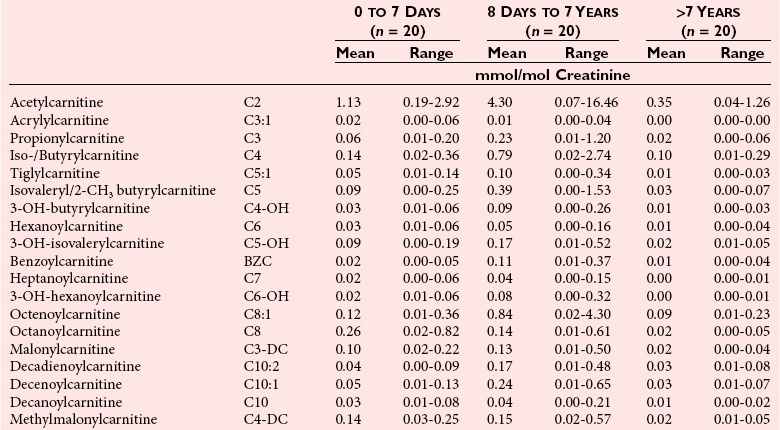
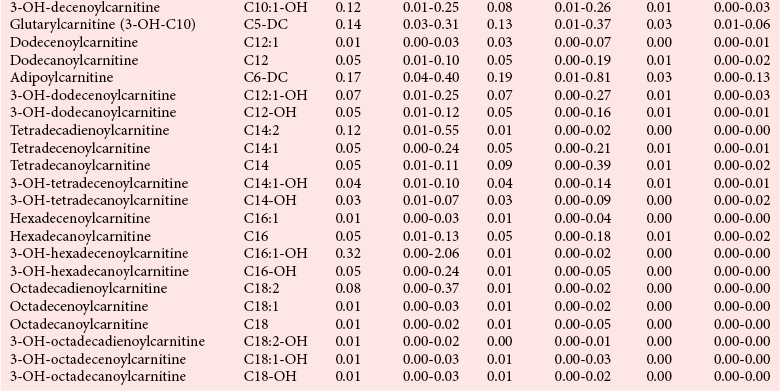
Chapter 58 Inborn errors of metabolism, which are due to impaired activity of enzymes, transporters, or cofactors, result in accumulation of abnormal metabolites (substrates) proximal to the metabolic block, or lack of necessary products. Figure 58-1 shows a hypothetical metabolic pathway in which the substrate A needs to be converted into the product D, with arrows representing individual enzymes. If an enzyme is defective (vertical rectangle), the concentration of substrate A will increase and the concentration of product D will decrease. It is then possible that high concentrations of substrate A will accumulate and become a source of substrate for enzymes not usually involved in its metabolism, thereby producing abnormal byproducts (E and F) through alternative pathways. Accumulation of specific metabolites and their byproducts within organs and tissues and/or the lack of reaction products are the chemical bases of the pathology observed in different inborn errors of metabolism. At the same time, measurement of some of these metabolites or their byproducts serves as the basis of biochemical diagnostic testing for inborn errors of metabolism and early detection by newborn screening programs. Symptoms of inborn errors of metabolism usually appear early in infancy, although several types become symptomatic in late childhood or adulthood. Signs and symptoms include (1) failure to thrive, (2) seizures, (3) mental retardation, (4) organ failure, and even (5) death. Inborn errors of metabolism have been divided into three broad categories, based on the effects of their metabolic derangement55: 1. Intoxication effect: IEMs in this category are the result of metabolites accumulating in the body that produce a toxic effect on different organs. The patient may become acutely ill after a symptom-free period, usually 24 to 72 hours, and concomitantly with ingestion of metabolites that are not metabolized, such as proteins or sugars. Types of IEMs that belong in this group include (1) aminoacidopathies (e.g., phenylketonuria, maple syrup urine disease, homocystinuria), (2) urea cycle defects (e.g., citrullinemia, argininosuccinic aciduria, ornithine transcarbamylase deficiency), (3) organic acidemias (e.g., propionic acidemia, methylmalonic acidemia, glutaric acidemia type I), and (4) disorders of sugar metabolism (galactosemia, hereditary fructose intolerance). Some of these disorders, such as phenylketonuria, affect primarily the brain, causing severe mental retardation but without acute decompensation. In other disorders (e.g., organic acidemias), symptoms appear shortly after protein intake (usually after the first few feedings) and include vomiting, lethargy, seizures, and coma leading rapidly to death if not recognized and treated appropriately. 2. Energy deficiency: Symptoms of disorders in this category are due to impaired energy production. In some cases, patients may be asymptomatic for a long time, until energy requirements are increased as the result of involuntary fasting, illness, infection, or strenuous exercise. A classic example of these disorders involves fatty acid oxidation (e.g., medium chain acyl–coenzyme A [CoA] dehydrogenase deficiency, very long chain acyl-CoA dehydrogenase deficiency, carnitine uptake defect, carnitine palmitoyl transferase deficiency I and II), although in this case, the block in the metabolic pathway, in addition to impairment in energy production, leads to the accumulation of intermediates that cause an intoxication effect.52 Other diseases in this group include glycogen storage disorders, in which hypoglycemia can occur in the presence or absence of any stress, mitochondrial disorders, and congenital lactic acidosis, in which the clinical course is progressive even in the absence of triggering conditions.43 3. Disorders of complex molecules: These disorders result from defects in the synthesis or catabolism of complex molecules. They are progressive, are independent of intercurrent events, and are not related to food intake. The metabolism of complex molecules is altered in all (1) lysosomal disorders, (2) peroxisomal disorders, and (3) disorders of intracellular trafficking and processing. For some of these disorders, a therapy is now available that is usually more effective if initiated before irreversible organ damage has occurred. For this reason, some of these conditions are also being considered for newborn screening. With few exceptions, the diagnosis of IEM is primarily a laboratory process. For example, routine laboratory tests in the symptomatic IEM patient and a high index of suspicion often point the clinician toward a specific IEM. For example, (1) hyperammonemia without metabolic acidosis suggests a defect of the urea cycle; (2) hypoketotic hypoglycemia usually with hyperammonemia to various degrees suggests fatty acid oxidation impairment; and (3) hyperammonemia with metabolic acidosis and ketosis is suggestive of an organic acidemia (Table 58-1). The diagnosis of IEMs requires specific tests that usually are performed in Biochemical Genetics Laboratories. The core group of tests necessary for the diagnosis of IEMs includes (1) amino acid analysis in plasma, urine (in few cases), and cerebrospinal fluid (in even fewer cases), (2) organic acid analysis in urine, and (3) carnitine and acylcarnitine profile in plasma. Despite constant progress in medical treatment, several IEMs result in severe morbidity and inevitable mortality early in life. Most of these disorders are inherited as autosomal recessive traits (Table 58-2); therefore the recurrence risk in subsequent pregnancies of the same couple is 25%. Genetic counseling of parents consists of a balanced assessment of (1) familial risk factors (parental consanguinity and ethnic origin), (2) risk of pregnancy loss as a consequence of the sampling procedure [0.5 to 1% by chorionic villus sampling (CVS), 0.5% by amniocentesis], (3) risk of maternal complications,53 (4) clinical validity of the prenatal test, (5) the burden of the disease, and (6) variable phenotypic expression of the disease even within the same family. TABLE 58-2 Clinical and Laboratory Characteristics of Disorders of Amino Acid Metabolism *OMIM, Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM/accessed September 29, 2011). Methods used for prenatal diagnosis of IEMs have different requirements in terms of timing, sample collection, and options for independent confirmation. CVS is performed at 10 to 13 weeks’ gestational age, has a higher risk of fetal loss as compared with classic amniocentesis, and might not provide accurate results because of possible contamination with maternal tissue. Alternatively, certain enzymes, such as those of the glycine cleavage pathway defective in glycine encephalopathy, are expressed only in cells of chorionic villi, rendering this procedure the only possibility when DNA testing is not possible.4 Amniocentesis is performed later in pregnancy (14 to 20 weeks) and provides both amniocytes and amniotic fluid to be used for independent and complementary diagnostic methods. Reliance on separate tests based on independent methods performed by laboratories with adequate prior experience is strongly encouraged to avoid incorrect or inconclusive results. In some IEMs (e.g., organic acidemias), amniotic fluid is tested for the presence or absence of specific metabolites, in addition to providing amniocytes for enzyme assay, DNA analysis, or both. The combination of at least two independent tests (e.g., enzyme assay + DNA; metabolite analysis + enzyme or DNA) enhances confidence in establishing a prenatal diagnosis. Before entertaining a prenatal diagnosis, one should ensure that the proband (individual first brought to medical attention in whom the diagnosis was established) related to the index case has a diagnosis confirmed by traditional methods, including enzymology when appropriate. If DNA analysis is considered, mutations of the index case should be known and confirmed as causative of the disease. Major advantages of direct metabolite analysis in amniotic fluid include independence from tissue expression and rapid turnaround time. However, analysis of direct metabolites in amniotic fluid has been reported for only a very limited number of diseases. Newborn screening is a public health activity aimed at early identification of conditions for which timely intervention is expected to result in elimination or reduction of morbidity, mortality, and disabilities. It is an important and effective component of preventive medicine. Originally instituted in the 1960s for the early detection of phenylketonuria (PKU), the number of diseases screened for in the newborn period has dramatically increased with the introduction of MS/MS multiplex analyses of acylcarnitine and amino acid profiles.13 Inclusion of IEMs as a whole in newborn screening panels has been controversial, because with few exceptions, their incidence, natural history, and prospective screening experience, as well as the effectiveness of treatment, have not yet been defined.10 However, implementation of this expanded screening allows collection of these data leading to a better understanding of these diseases. The complexity of the interpretation of MS/MS newborn screening results has prompted the development of algorithms for proper confirmatory testing and differential diagnosis of all detectable IEMs (http://www.acmg.net/resources/policies/ACT/condition-analyte-links.htm/accessed September 29, 2011). The most informative samples are collected from patients during acute metabolic decompensation. When possible, urine and blood should be collected at the same time. In several diseases, especially in fatty acid oxidation disorders, diagnostic abnormalities may not be detected when the patient has recovered from the acute episode. Urine and plasma/serum samples are stored at −20 °C until the need for specialized tests has been determined. Quantitative profiling of amino acids, carnitine, acylcarnitines, and fatty acids in plasma, and organic acids and acylglycines in urine, is the biochemical investigation necessary to diagnose these disorders. Alternatively, a blood spot on filter paper may provide enough material for one or more of the investigations described in this chapter. In case of death, collection of body fluid and tissue should be secured according to available protocols.12,54 Among IEMs, fatty acid oxidation (FAO) disorders are those recognized more often after the diagnosis of an affected sibling or as a cause of sudden death.5 Early reports attributed up to 5% of cases of sudden death in children younger than 5 years to FAO,7 and mounting evidence indicates that some of these disorders can cause mortality in adults as well.38 The postmortem evaluation of unexpected death, independent of age, especially when evidence of acute illness or infection is found, should consider FAO as a cause. This is accomplished by analysis of acylcarnitines in blood and bile spots (Figure 58-2).52 Reference intervals for acylcarnitines in postmortem blood and bile spots are listed in Table 58-3. TABLE 58-3 Acylcarnitine Reference Intervals in Postmortem Blood and Bile Dried Spots From Rinaldo P, Hahn S, Matern D. Inborn errors of amino acid, organic acid, and fatty acid metabolism. In: Burtis CA, Ashwood ER, Bruns DE, eds. Tietz textbook of clinical chemistry and molecular diagnostics, 4th edition. St. Louis, Elsevier Saunders, 2006:2241. Blood and bile are collected on filter paper identical to the cards used for newborn screening; once properly dried they are shipped to the laboratory at room temperature. In cases with a higher index of suspicion, an effort should be made to collect and freeze a specimen of liver7 and to collect a skin biopsy for establishing the fibroblast culture to be used, if needed, to confirm a diagnosis. Although fatty infiltration of the liver and/or other organs (e.g., heart, muscle, kidneys) is a common observation in FAO disorders, the finding of macroscopic steatosis should not be used as the only criterion in deciding whether to investigate a possible underlying FAO disorder during postmortem evaluation of a case of sudden death. Sudden death from cardiac arrhythmia can be the only finding in fatty acid oxidation defects, and the absence of obvious physical findings on autopsy does not exclude this, especially in adults. In cases of sudden infant death, if parental permission to perform an autopsy is not granted, any leftover specimens or unused portions of blood spots collected for newborn screening, if still available, could be useful samples in obtaining a diagnosis. Several methods have been used in the analysis of amino acids in biological fluids such as plasma, urine, and cerebrospinal fluid. All involve chromatographic separation of amino acids with precolumn [high-performance liquid chromatography (HPLC), GC methods] or postcolumn (ion-exchange chromatography) derivatization, followed by detection by ultraviolet (UV), fluorescence, or mass spectrometry.1,35,61,65,77,78 The gold standard for analysis of amino acids remains ion-exchange chromatography (IEC), even though recent work using tandem mass spectrometry is showing promising results. The challenge with amino acid analysis includes the need (1) to cover a wide dynamic range, (2) to have a very low detection limit, and (3) to have a high upper limit of linearity. In addition to these analytical requirements, isomers need to be separated and quantified. With ion-exchange chromatography, the sample (plasma/urine/CSF) is deproteinized and injected onto an ion-exchange column (typically a lithium cation-exchange column). Amino acids are separated on the basis of their pKa by changing the pH and ionic strength of eluting buffers and the temperature of the column. Acidic amino acids are eluted first, followed by neutral then basic amino acids. After their elution from the column, amino acids are mixed with ninhydrin at 135 °C to form a colored adduct. The intensity of their absorbance is proportional to the concentration of the amino acid. Absorbance is read at two different wavelengths: 570 nm (maximum absorbance for amino acids) and 440 nm (maximum absorbance for imino acids, such as proline and hydroxyproline). The concentration of amino acids is calculated using an internal standard and external calibration. Identification of individual amino acids relies on retention time, ratio of absorbance at the two wavelengths (440/570 nm), and, in case of doubt, spiking of the sample with a standard. The label organic acids includes metabolites of almost all pathways of intermediary metabolism and exogenous compounds. Organic acids analyzed by gas chromatography/mass spectrometry (GC/MS) are separated on the basis of their volatility and solubility in the stationary nonpolar liquid phase of the capillary gas chromatography column. Before GC/MS analysis is performed, organic acids must be (1) extracted, usually with an organic solvent; (2) converted to volatile trimethylsilyl (TMS) derivatives; and (3) dissolved in organic solvents.40,78 With this technique, the mass spectrometer is employed as a detector, thus allowing positive identification of organic acids both by retention time and by their characteristic fragmentation spectrum. A random urine specimen is routinely used for this analysis; however, the most informative samples for the diagnosis of IEM are collected during acute metabolic decompensation. Organic acid analysis in blood or CSF usually is not informative to establish a diagnosis. Their interpretation is challenging because hundreds of compounds will be present in a specimen. Key factors in correct interpretation of results include (1) recognition of abnormal patterns and possible interferences due to dietary or medication artifacts, (2) knowledge about metabolic disorders and their presentation, and (3) information about the clinical status of patients. Acylcarnitines derive from conjugation of carnitine with acyl-CoA. Carnitine [3-hydroxy-4-(trimethylazaniumyl)butanoate] is a water-soluble molecule that is essential in the transfer of long-chain fatty acids inside mitochondria for beta-oxidation. In addition, carnitine binds acyl residues accumulating in several organic acidemias and in fatty acid oxidation disorders, to facilitate their excretion. In the presence of a metabolic block (organic acidemia or fatty acid oxidation disorder), specific acylcarnitines, derived from conjugation of carnitine with acyl-CoA upstream of the metabolic block, accumulate, producing a pattern that is characteristic for each disease or group of diseases, so acylcarnitine analysis plays an essential role in the diagnosis of metabolic disorders (see Figure 58-2). Such an analysis is usually performed by tandem mass spectrometry (MS/MS) with or without liquid chromatographic separation prior to MS/MS detection.39,41 Plasma/serum is the biological fluid of choice, and whole blood spotted on filter paper is used for screening of newborns. Concentrations of acylcarnitines in plasma differ from concentrations in whole blood, especially for long-chain acylcarnitines. This is thought to be due to binding of long-chain acylcarnitines to the membranes of blood cells, resulting in reduced long-chain species in plasma. Plasma for analysis of acylcarnitines should be separated immediately after collection and kept frozen until analysis. Hemolysis has been known to result in elevated long-chain acylcarnitines, misleading the diagnosis. Storage of the sample at room temperature or even refrigerated may result in hydrolysis and, consequently, reduced concentrations of acylcarnitines. Urine acylcarnitine analysis is performed only in the diagnostic work-up of specific disorders, such as glutaric acidemia type I, and only if equivocal results are obtained with other tests. Quantification of acylcarnitines is typically performed using stable isotope dilution. However, some deuterated internal standards are not available for all identified acylcarnitine species. Caution should be taken when acylcarnitine results from different laboratories are compared, because the values may change depending on the internal standards used.14 Age-appropriate reference intervals should be used in the interpretation of biochemical genetics tests. Reference intervals for urine organic acids, urine acylglycines, plasma, and urine acylcarnitines are listed in Tables 58-4, 58-5, and 58-6. TABLE 58-4 Reference Intervals of Selected Organic Acids and Acylglycines in Urine, mmol/mol Creatinine* *TIC detection limit: 0.1 mmol/mol creatinine; SIM detection limit (with stable isotope labeled internal standard): 0.01 mmol/mol creatinine. †Measured using a stable isotope-labeled internal standard. From Rinaldo P, Hahn S, Matern D. Inborn errors of amino acid, organic acid, and fatty acid metabolism. In: Burtis CA, Ashwood ER, Bruns DE, eds. Tietz textbook of clinical chemistry and molecular diagnostics, 4th edition. St. Louis, Elsevier Saunders, 2006:2238. TABLE 58-5 Acylcarnitine Reference Intervals in Plasma From Rinaldo P, Hahn S, Matern D. Inborn errors of amino acid, organic acid, and fatty acid metabolism. In: Burtis CA, Ashwood ER, Bruns DE, eds. Tietz textbook of clinical chemistry and molecular diagnostics, 4th edition. St. Louis, Elsevier Saunders, 2006:2239. TABLE 58-6 Acylcarnitine Reference Intervals in Urine From Rinaldo P, Hahn S, Matern D. Inborn errors of amino acid, organic acid, and fatty acid metabolism. In: Burtis CA, Ashwood ER, Bruns DE, eds. Tietz textbook of clinical chemistry and molecular diagnostics, 4th edition. St. Louis, Elsevier Saunders, 2006:2240. Table 58-2 provides a summary of the most common disorders of amino acid metabolism and transport and their characteristics. Several of these disorders are discussed in the following sections. Hyperphenylalaninemias result from the impaired conversion of phenylalanine to tyrosine, leading to an increased concentration of phenylalanine in body fluids. They are caused by a primary deficiency of phenylalanine hydroxylase, the enzyme that converts phenylalanine to tyrosine (Figure 58-3), or, in rare cases (<2% of total cases in the United States), by a defect in synthesis (Figure 58-4) or recycling (Figure 58-5) of the essential cofactor tetrahydrobiopterin. The combined incidence of these conditions is about 1 : 10,000 to 1 : 20,000 live births. In PKU, accumulation of phenylalanine and other metabolites such as phenyllactate and phenylpyruvate (phenylketones) occurs. Elevated phenylalanine interferes with neurotransmitter synthesis and uptake, leading to clinical symptoms of phenylketonuria.63 In patients with PKU due to phenylalanine hydroxylase deficiency, dietary treatment with a special formula without phenylalanine should be started as soon as possible—ideally before 3 weeks of age.63 Diet needs to be continued for life. Phenylalanine concentrations are monitored periodically and should remain between 60 and 360 µmol/L (reference interval, 30 to 80 µmol/L) to ensure adequate brain development. If the concentration of phenylalanine is too low, growth of the child is compromised; if it is too high, executive functioning is impaired. High concentrations of phenylalanine in the first years of life lead to mental retardation. Phenylalanine at high concentrations is teratogenic and, depending on the concentration and the period of exposure during pregnancy, will cause (1) increased risk of spontaneous abortion, (2) congenital heart defects, (3) facial dysmorphism, (4) microcephaly, and (5) developmental delay (even in the absence of microcephaly) in the fetuses of women with PKU. Adverse pregnancy outcomes in pregnant women with PKU are minimized by maintaining phenylalanine concentrations less than 360 µmol/L.37 In approximately 2% of cases, hyperphenylalaninemia is due to a deficiency of biosynthesis (see Figure 58-4) or recycling (see Figure 58-5) of the cofactor tetrahydrobiopterin (BH4). BH4 is also a cofactor for tyrosine and tryptophan hydroxylases (see Figure 58-5).42 Infants with BH4 deficiencies show signs of neurologic involvement despite adequate dietary control of phenylalanine concentrations. Impairment of tyrosine and tryptophan hydroxylases reduces the synthesis of the neurotransmitters dopamine and serotonin, with severe neurologic consequences. BH4 is also a cofactor for nitric oxide synthase, which catalyzes the generation of nitric oxide from arginine, although the clinical consequences of this latter impairment are not known. Five enzyme deficiencies leading to BH4 deficiency have been reported (see Table 58-2). One of these, sepiapterin reductase deficiency, impairs BH4 synthesis only in the brain, because alternative pathways are available for its synthesis in the liver. As a result, patients with this latter condition have normal activity of phenylalanine hydroxylase in the liver and no hyperphenylalaninemia.21 Among patients with BH4 deficiencies and elevated phenylalanine, 50% of cases are due to 6-pyruvoyltetrahydropterin synthase (6-PTPS) deficiency. These patients are clinically indistinguishable from those with classic PKU (caused by phenylalanine hydroxylase deficiency), when diagnosed through newborn screening, but they progressively deteriorate with loss of head control, truncal hypotonia with hypertonia of the extremities, drooling, swallowing difficulties, and myoclonic seizures at between 2 and 6 months of age.42 Treatment of these patients requires BH4, which usually normalizes the concentration of phenylalanine (except in some cases of DHPR deficiency), and neurotransmitter precursors (L-dopa/carbidopa and 5-OH-tryptophan), which obviate the need for tyrosine and tryptophan hydroxylase.
Newborn Screening and Inborn Errors of Metabolism
Clinical Presentation
Diagnosis
Prenatal Diagnosis
Newborn Screening
Evaluation of Symptomatic Patients
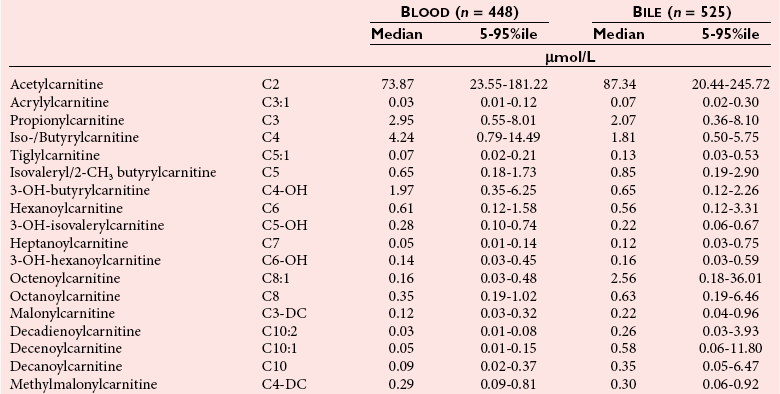
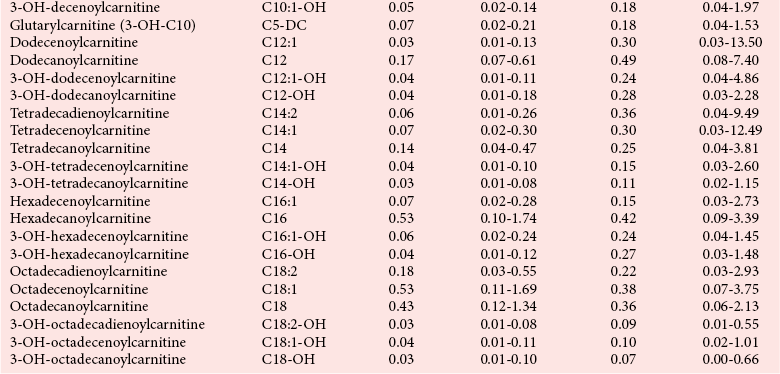
Postmortem Screening
Biochemical Genetics Tests: Analytical Considerations
Amino Acid Analysis by Ion-Exchange Chromatography
Urine Organic Acid Analysis by Gas Chromatography/Mass Spectrometry
Plasma Acylcarnitine Profile
Reference Intervals

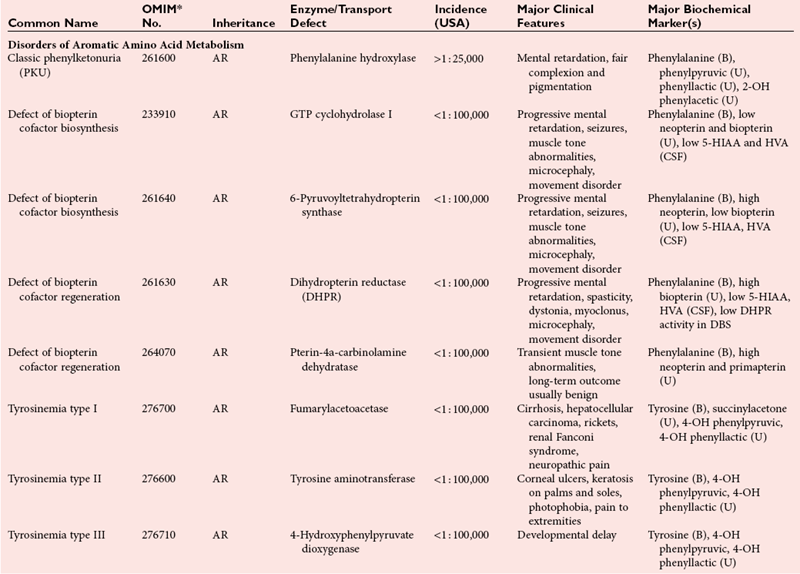
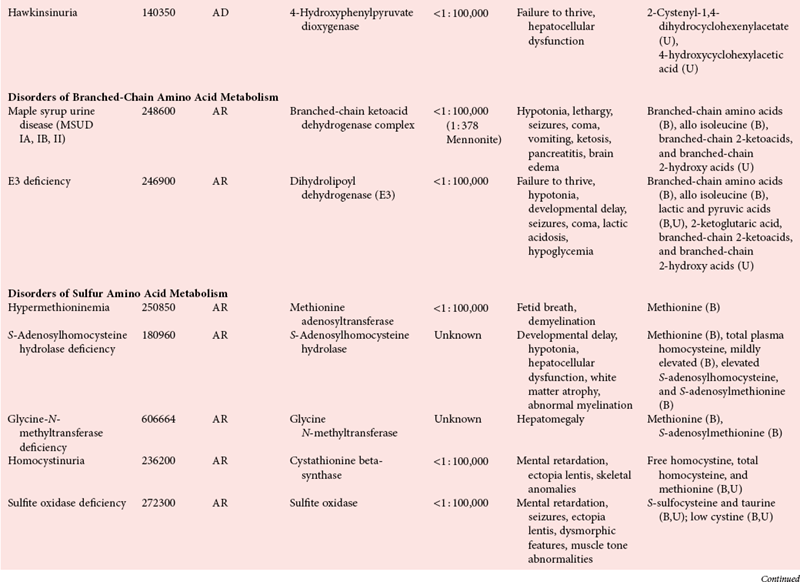
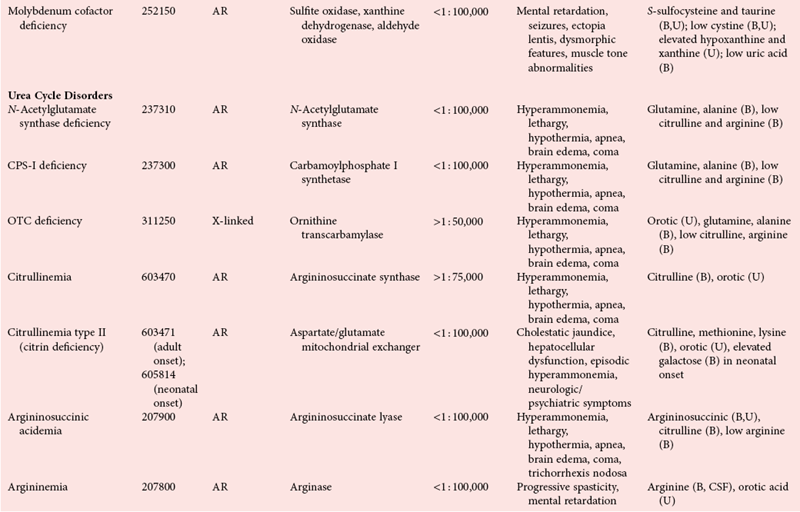
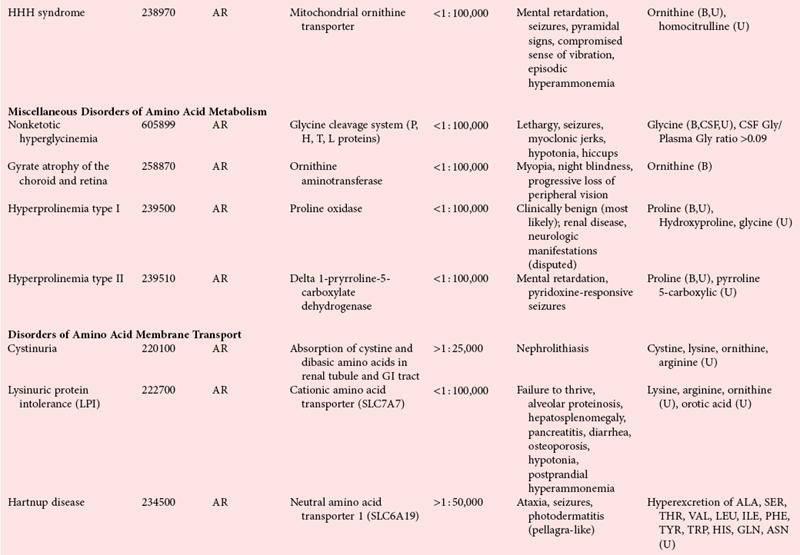
Disorders of Amino Acid Metabolism
Classic Phenylketonuria and Other Hyperphenylalaninemias
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine