Chapter 22 Neurological disease
The impact of neurological disease
Neurology is a large and diverse subject which covers many conditions that require long-term coordinated care and have serious effects on the daily lives of patients and their families. Neurology includes conditions as diverse as cognitive disorders involving higher level mental functioning through to disorders of peripheral nerve and skeletal muscle. It is a specialty requiring good clinical skills and examination technique which cannot be replaced with investigations or imaging techniques alone (Table 22.1).
Table 22.1 UK incidence of common neurological conditions
| Conditions | Events per 100 000/year |
|---|---|
Cerebrovascular events | 210 |
Shingles (herpes zoster) and postherpetic neuralgia | 150 |
Diabetic and other neuropathies | 105 |
Epilepsy | 46 |
Parkinson’s disease | 19 |
Severe brain injury and subdural haematoma | 13 |
All CNS tumours | 9 |
Trigeminal neuralgia | 8 |
Meningitis | 7 |
Multiple sclerosis | 7 |
Presenile dementia (below 65 years) | 4 |
Myasthenia, all muscle and motor neurone disease | 5 |
Common symptoms and signs



Difficulty walking and falls
Change in walking pattern is a common complaint (Box 22.1). Arthritis and muscle pain make walking painful and slow (antalgic gait). The pattern of gait is valuable diagnostically.
 Box 22.1
Box 22.1






Spasticity and hemiparesis
Spasticity (p. 1082), more pronounced in extensor muscles, with or without weakness, causes stiff and jerky walking. Toes of shoes become scuffed, catching level ground. Pace shortens; a narrow base is maintained. Clonus – involuntary extensor rhythmic leg jerking – may occur.
Parkinson’s disease: shuffling gait
There is muscular rigidity (p. 1118) throughout extensors and flexors. Power is preserved; the pace shortens, and slows to a shuffle; its base remains narrow. A stoop and diminished arm swinging become apparent. Gait becomes festinant (hurried) with short rapid steps. There is difficulty turning quickly and initiating movement, sometimes with falls. Retropulsion means small backward steps, taken involuntarily when a patient is halted.
Cerebellar ataxia: broad-based gait
In lateral cerebellar lobe disease (p. 1083) stance becomes broad-based, unstable and tremulous. Ataxia describes this incoordination. When walking, the person tends to veer to the side of the affected cerebellar lobe.
Sensory ataxia: stamping gait
Peripheral sensory lesions (e.g. polyneuropathy, p. 1145) cause ataxia because of loss of proprioception (position sense). Broad-based, high-stepping, stamping gait develops. This form of ataxia is exacerbated by removal of sensory input (e.g. vision) and worse in the dark. Romberg’s test, first described in sensory ataxia of tabes dorsalis (p. 1129), becomes positive.
Lower limb weakness: slapping and waddling gaits
When weakness is distal, each foot must be lifted over obstacles. When ankle dorsiflexors are weak, e.g. in a common peroneal nerve palsy (p. 1144), the sole returns to the ground with an audible slap.
Dizziness, vertigo and blackouts
Vertigo (p. 1078) means the illusion of movement, a sensation of rotation or tipping. The patient feels the surroundings are spinning or moving. This is distressing and often accompanied by nausea or vomiting.
Blackout, like dizziness, is simply descriptive, implying either altered consciousness, visual disturbance or falling. Epilepsy (p. 1112) and syncope are mentioned in detail (p. 1116); hypoglycaemia and anaemia must be considered. Commonly no sinister cause is found. A careful history is essential.
Collapse is a vague term, but often used. Avoid it.
Examination and formulation
Following a short or detailed examination, relevant findings are summarized in a brief formulation – the basis for investigation, transfer of information and management (Practical Boxes 22.1, 22.2 and Table 22.2).
 Practical Box 22.1
Practical Box 22.1


















 Practical Box 22.2
Practical Box 22.2
10-part neurological examination
1. State of consciousness, arousal, appearance
2. Mental state, attitude, insight (see Box 22.5, p. 1069)
3. Cognitive function – orientation, recall, level of intellect, language, other cortical problems, e.g. apraxia
6. Neck – stiffness, palpation and auscultation of carotids
7. Cranial nerves (Table 22.3)












9. Coordination and fine movements







Table 22.2 Six grades of muscle power
| Grade | Definition |
|---|---|
5 | Normal power |
4 | Active movement against gravity and resistance |
3 | Active movement against gravity |
2 | Active movement with gravity eliminated |
1 | Flicker of contraction |
0 | No contraction |
Functional neuroanatomy
The neurone and synapse
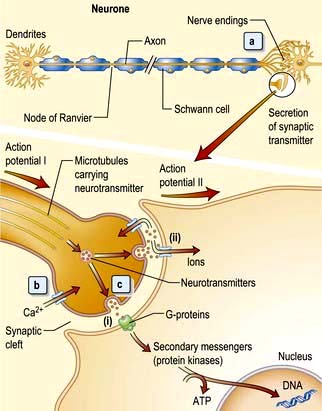
The neurone is the functional unit of the entire nervous system (Fig. 22.1). Its cell body and axon terminate in a synapse. Size and type of each group of neurones vary. A thoracic spinal cord α-motor neurone has an axonal length of >1 metre and innervates between several hundred and 2000 muscle fibres in one leg – a motor unit. By contrast, some spinal or intracerebral interneurones have axons under 100 µm long, terminating on one neuronal cell body.
Clinical features of focal brain lesions: general mechanisms
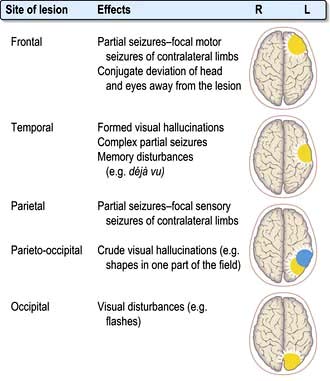
 Suppression or destruction of neurones and surrounding structures (Fig. 22.2). This is the commonest process – part of the system simply fails to work.
Suppression or destruction of neurones and surrounding structures (Fig. 22.2). This is the commonest process – part of the system simply fails to work.
 Synchronous discharge of neurones by irritative lesions (Fig. 22.3), e.g. cortical lesions, causes epilepsy, either partial or generalized.
Synchronous discharge of neurones by irritative lesions (Fig. 22.3), e.g. cortical lesions, causes epilepsy, either partial or generalized.
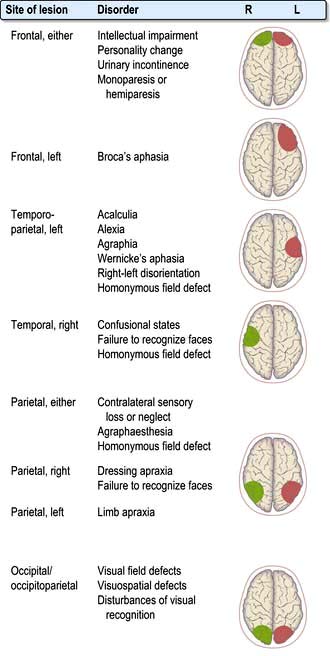
Localization within the cerebral cortex



Aphasia
Dysarthria
Dysarthria is disordered articulation – slurred speech. Language is intact. Paralysis, slowing or incoordination of muscles of articulation or local discomfort causes various patterns of dysarthria. Examples are the gravelly speech of pseudobulbar palsy (p. 1080), the jerky, ataxic speech of cerebellar lesions, the monotone of Parkinson’s, and speech in myasthenia that fatigues and dies away. Many aphasic patients are also dysarthric.
Memory and its disorders
Memory loss (the amnestic syndrome) is part of dementia (p. 1137) but also occurs as an isolated entity (Box 22.2).
 Box 22.2
Box 22.2















Cranial nerves (Table 22.3)
I: Olfactory nerve
| Number | Name | Main clinical action |
|---|---|---|
I | Olfactory | Smell |
II | Optic | Vision, fields, afferent light reflex |
III | Oculomotor | Eyelid elevation, eye elevation, ADduction, depression in ABduction, efferent (pupil) |
IV | Trochlear | Eye intorsion, depression in ADduction |
V | Trigeminal | Facial (and corneal) sensation, mastication muscles |
VI | Abducens | Eye ABduction |
VII | Facial | Facial movement, taste fibres |
VIII | Vestibular | Balance and hearing |
| Cochlear |
|
IX | Glossopharyngeal | Sensation – soft palate, taste fibres |
X | Vagus | Cough, palatal and vocal cord movements |
XI | Accessory | Head turning, shoulder shrugging |
XII | Hypoglossal | Tongue movement |
II: Optic nerve and visual system (Fig. 22.4)
Light regulated by the pupillary aperture is converted into action potentials by retinal rod, cone and ganglion cells (see page 1055). The lens, under control of the ciliary muscle, produces the image (inverted) on the retina. Axons in the optic nerve (1) decussate at the optic chiasm (2), fibres from the nasal retina cross and join with uncrossed fibres originating in the temporal retina to form the optic tract (3). Each optic tract thus carries information from the contralateral visual hemifield.
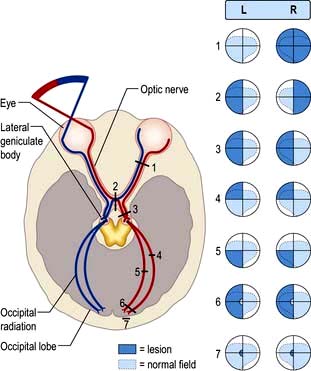
From the lateral geniculate body, fibres pass in the optic radiation through the parietal and temporal lobes (4 and 5) to reach the visual cortex of the occipital lobe (6 and 7), which is somatotopically organized with macular vision located at the occipital pole (see Fig. 22.4).
Visual field defects
Optic nerve lesions
 Reduced acuity in affected eye
Reduced acuity in affected eye

 Impaired colour vision (assess with Ishihara plates)
Impaired colour vision (assess with Ishihara plates)
 An afferent pupillary defect (see below)
An afferent pupillary defect (see below)

Causes are listed in Box 22.3.
 Box 22.3
Box 22.3
Causes of optic neuropathy
 Inflammatory (optic neuritis), e.g. demyelination, sarcoidosis, vasculitis
Inflammatory (optic neuritis), e.g. demyelination, sarcoidosis, vasculitis
 Optic nerve trauma or compression, e.g. glioma, meningioma, aneurysm, bone disorders affecting orbit
Optic nerve trauma or compression, e.g. glioma, meningioma, aneurysm, bone disorders affecting orbit
 Toxic, e.g. tobacco-alcohol, ethambutol, methyl alcohol, quinine, hydroxy chloroquine, radiation
Toxic, e.g. tobacco-alcohol, ethambutol, methyl alcohol, quinine, hydroxy chloroquine, radiation
 Ischaemic optic neuropathy, e.g. giant cell arteritis
Ischaemic optic neuropathy, e.g. giant cell arteritis
 Hereditary optic neuropathies, e.g. Leber’s
Hereditary optic neuropathies, e.g. Leber’s
 Nutritional deficiency, e.g. vitamin B1 and B12
Nutritional deficiency, e.g. vitamin B1 and B12
 Infection, e.g. orbital cellulitis, syphilis, TB
Infection, e.g. orbital cellulitis, syphilis, TB
 Neurodegenerative disorders, e.g. leucodystrophies
Neurodegenerative disorders, e.g. leucodystrophies

Papilloedema
Papilloedema means swelling of the optic disc. Causes are shown in Box 22.4. The earliest signs of swelling are disc pinkness, with blurring and heaping up of disc margins, nasal first. There is loss of spontaneous pulsation of retinal veins within the disc. The physiological cup becomes obliterated, the disc engorged with dilated vessels. Small haemorrhages often surround the disc.
 Box 22.4
Box 22.4
Causes of optic disc swelling
 Raised intracranial pressure (papilloedema)
Raised intracranial pressure (papilloedema)
 Brain tumour, abscess, or haemorrhage. Idiopathic intracranial hypertension, hydrocephalus
Brain tumour, abscess, or haemorrhage. Idiopathic intracranial hypertension, hydrocephalus

 Optic neuritis, e.g. multiple sclerosis
Optic neuritis, e.g. multiple sclerosis
 Ischaemic optic neuropathy, e.g. giant cell arteritis
Ischaemic optic neuropathy, e.g. giant cell arteritis
 Toxic optic neuropathy, e.g. methanol
Toxic optic neuropathy, e.g. methanol


 Central retinal vein thrombosis
Central retinal vein thrombosis



 Vasculitis, e.g. systemic lupus erythematosus
Vasculitis, e.g. systemic lupus erythematosus

 Hypercapnia, chronic hypoxia, hypocalcaemia
Hypercapnia, chronic hypoxia, hypocalcaemia


Various conditions simulate true disc swelling. Marked hypermetropic (long-sighted) refractive errors make a disc appear pink, distant and ill-defined. Myelinated nerve fibres at disc margins and hyaline bodies (drusen, p. 1064) can be mistaken for disc swelling.
Disc infiltration also causes a swollen disc with raised margins (e.g. in leukaemia).
Inflammatory optic neuropathy (optic neuritis)
A plaque of demyelination within the optic nerve is the most common cause in Western populations. Dedicated MRI imaging of the optic nerves may show the inflammatory plaque and imaging of the brain may show additional inflammatory lesions which confer a higher risk of developing multiple sclerosis (MS). Approximately 50% of patients go on to develop MS with prolonged follow-up (see p. 1123). Recovery of visual acuity to 6/9 or better occurs in 95% of cases over months, with recovery time improved by high-dose i.v. steroids given acutely.
Optic neuritis may be caused by infective or other inflammatory disorders, e.g. sarcoidosis or vasculitides (see Box 22.3).
Anterior ischaemic optic neuropathy
Individuals with small hypermetropic discs seem to be predisposed, and often there are relatively few vascular risk factors. Less commonly arteritis is the cause (see p. 536).



Occipital cortex
Homonymous hemianopic defects are produced by unilateral posterior cerebral artery infarction (see p. 1101 stroke). The macular cortex (at each occipital pole) may be spared.
Widespread bilateral occipital lobe damage by infarction (‘top of the basilar’ syndrome), trauma or coning causes cortical blindness (Anton’s syndrome). The patient cannot see but characteristically lacks insight into this; he or she may even deny it. Pupillary responses remain normal (p. 1101).
The pupils
Pupillary reactions to light and accommodation may be tested (Fig. 22.5). A bright torch (not an ophthalmoscope light!) should be used to test the pupillary light reaction.
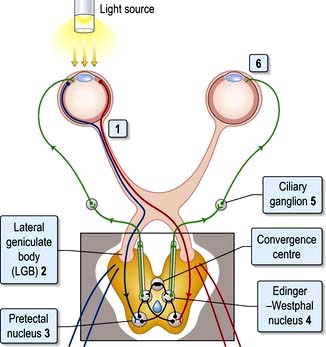




Horner’s syndrome (see Box 22.5)
 Box 22.5
Box 22.5
Causes of Horner’s syndrome













The clinical features of Horner’s syndrome are:





III, IV, VI: Oculomotor, trochlear and abducens nerves
Control of eye movements
 the ipsilateral occipital cortex – pathway concerned with tracking objects
the ipsilateral occipital cortex – pathway concerned with tracking objects
 the vestibular nuclei – pathways linking eye movements with position of the head and neck (vestibulo-ocular reflex, p. 1072).
the vestibular nuclei – pathways linking eye movements with position of the head and neck (vestibulo-ocular reflex, p. 1072).
Conjugate lateral eye movements are coordinated from each PPRF via the medial longitudinal fasciculus (MLF, Fig. 22.6). Fibres from the PPRF pass both to the ipsilateral VIth nerve nucleus (lateral rectus) and, having crossed the midline, to the opposite IIIrd nerve nucleus (medial rectus and other muscles) via the MLF, thus linking the eyes for lateral gaze.
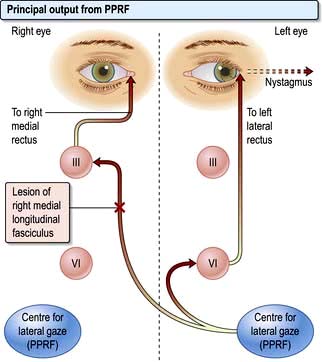
Abnormalities of conjugate lateral gaze
Internuclear ophthalmoplegia (INO)
Damage to one MLF causes internuclear ophthalmoplegia (INO), a common complex brainstem eye movement disorder seen frequently in MS. In a right INO there is a lesion of the right MLF (Fig. 22.6). On attempted left lateral gaze the right eye fails to ADduct. The left eye develops nystagmus in ABduction. The side of the lesion is on the side of impaired ADduction, not on the side of the (obvious, unilateral) nystagmus. When present bilaterally, INO is almost pathognomonic of MS.
Nystagmus
Jerk nystagmus
 Horizontal or rotary jerk nystagmus may be either of peripheral (vestibular) or central origin (VIIIth nerve, brainstem, cerebellum and connections).
Horizontal or rotary jerk nystagmus may be either of peripheral (vestibular) or central origin (VIIIth nerve, brainstem, cerebellum and connections).
 Vertical jerk nystagmus is caused typically by central lesions.
Vertical jerk nystagmus is caused typically by central lesions.

III: Oculomotor nerve
The nucleus of the IIIrd nerve lies ventral to the aqueduct in the midbrain. It supplies four external ocular muscles (superior, inferior and medial recti, and inferior oblique), levator palpebrae superioris (which lifts the eyelid) and parasympathetic constriction of the pupil. Causes of a IIIrd nerve lesion are listed in Box 22.6.
 Box 22.6
Box 22.6




Signs of a complete IIIrd nerve palsy:
 Unilateral complete ptosis (levator weakness)
Unilateral complete ptosis (levator weakness)
 Eye deviated down and out (unopposed lateral rectus and superior oblique)
Eye deviated down and out (unopposed lateral rectus and superior oblique)

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


