Chapter 14 Cardiovascular disease
Essential anatomy, physiology and embryology of the heart
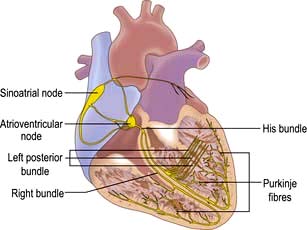
The conduction system of the heart
The sinus node
The sinus node is a complex spindle-shaped structure that lies in the lateral and epicardial aspects of the junction between the superior vena cava and the right atrium (Fig. 14.1). Physiologically, it generates impulses automatically by spontaneous depolarization of its membrane at a rate quicker than any other cardiac cell type. It is therefore the natural pacemaker of the heart.
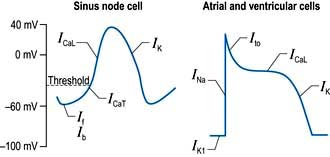
A number of factors are responsible for the spontaneous decay of the sinus node cell membrane potential (‘the pacemaker potential’), the most significant of which is a small influx of sodium ions into the cells. This small sodium current has two components: the background inward current (Ib) and the ‘funny’ (If) current (or pacemaker current) (Fig. 14.2). The term ‘funny’ current denotes ionic flow through channels activated in hyperpolarized cells (−60 mV or greater), unlike other time- and voltage-dependent channels activated by depolarization. The rate of depolarization of the sinus node membrane potential is modulated by autonomic tone (i.e. sympathetic and parasympathetic input), stretch, temperature, hypoxia, blood pH and in response to other hormonal influences (e.g. tri-iodothyronine and serotonin).
Atrial and ventricular myocyte action potentials
Action potentials in the sinus node trigger depolarization of the atrial and subsequently the ventricular myocytes. These cells have a different action potential from that of sinus node cells (Fig. 14.2). Their resting membrane potential is a consequence of a small flow of potassium ions into the cells through open ‘inward rectifier’ channels (IKl); at this stage sodium and calcium channels are closed. The arrival of adjacent action potentials triggers the opening of voltage-gated, fast, self-inactivating sodium channels, resulting in a sharp depolarization spike. This is followed by a partial repolarization of the membrane due to activation of ‘transient outward’ potassium channels.
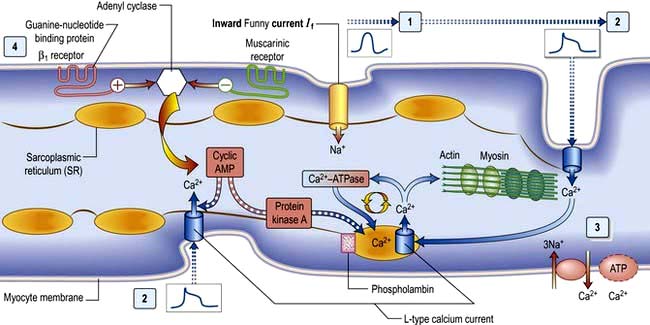
The plateau phase which follows is unique to myocytes and results from a small, but sustained inward calcium current through L-type calcium channels (ICaL) lasting 200–400 ms. This calcium influx is caused by a combined increase in permeability of the cell, especially the sarcolemmal membranes to calcium (Fig. 14.3). This plateau (or refractory) phase in myocyte action potential prevents early reactivation of the myocytes and directly determines the strength of contraction. The gradual inactivation of the calcium channels activates delayed rectifier potassium channels (IK) repolarizing the membrane. Atrial tissue is activated like a ‘forest fire’, but the activation peters out when the insulating layer between the atrium and the ventricle – the annulus fibrosus – is reached. Controversy exists about whether impulses from the sinoatrial (SA) node travel over specialized conducting ‘pathways’ or over ordinary atrial myocardium.
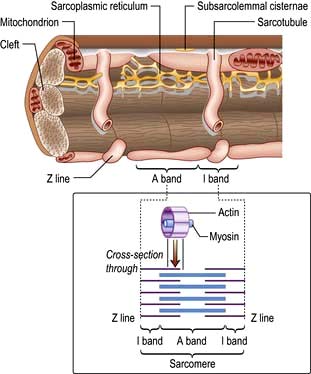
The cellular basis of myocardial contraction – excitation-contraction coupling
Each myocyte, approximately 100 µm long, branches and interdigitates with adjacent cells. An intercalated disc permits electrical conduction to adjacent cells. Myocytes contain bundles of parallel myofibrils. Each myofibril is made up of a series of sarcomeres (Fig. 14.4). A sarcomere (which is the basic unit of contraction) is bound by two transverse Z lines, to each of which is attached a perpendicular filament of the protein actin. The actin filaments from each of the two Z bands overlap with thicker parallel protein filaments known as myosin. Actin and myosin filaments are attached to each other by cross-bridges that contain ATPase, which breaks down adenosine triphosphate (ATP) to provide the energy for contraction.
Calcium is made available during the plateau phase of the action potential by calcium ions entering the cell and by being mobilized from the sarcoplasmic reticulum through the ryanodine receptor (RyR2) calcium-release channel. RyR2 activity is regulated by the protein calstabin 2 (see p. 770) and nitric oxide. The force of cardiac muscle contraction (‘inotropic state’) is thus regulated by the influx of calcium ions into the cell through calcium channels (Fig. 14.3). T (transient) calcium channels open when the muscle is more depolarized, whereas L (long-lasting) calcium channels require less depolarization. The extent to which the sarcomere can shorten determines the stroke volume of the ventricle. It is maximally shortened in response to powerful inotropic drugs or severe exercise.
Starling’s law of the heart
The contractile function of an isolated strip of cardiac tissue can be described by the relationship between the velocity of muscle contraction, the load that is moved by the contracting muscle, and the extent to which the muscle is stretched before contracting. As with all other types of muscle, the velocity of contraction of myocardial tissue is reduced by increasing the load against which the tissue must contract. However, in the non-failing heart, pre-stretching of cardiac muscle improves the relationship between the force and velocity of contraction (Fig. 14.5).
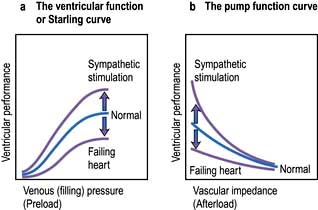
This phenomenon was described in the intact heart as an increase of stroke volume (ventricular performance) with an enlargement of the diastolic volume (preload), and is known as ‘Starling’s law of the heart’ or the ‘Frank–Starling relationship’. It has been transcribed into more clinically relevant indices. Thus, stroke work (aortic pressure × stroke volume) is increased as ventricular end-diastolic volume is raised. Alternatively, within certain limits, cardiac output rises as pulmonary capillary wedge pressure increases. This clinical relationship is described by the ventricular function curve (Fig. 14.5), which also shows the effect of sympathetic stimulation.
Nerve supply of the myocardium
Adrenergic stimulation and cellular signalling
β1-Adrenergic stimulation enhances Ca2+ flux in the myocyte and thereby strengthens the force of contraction (Fig. 14.3). Binding of catecholamines, e.g. norepinephrine (noradrenaline), to the myocyte β1-adrenergic receptor stimulates membrane-bound adenylate kinases. These enzymes enhance production of cyclic adenosine monophosphate (cAMP) that activates intracellular protein kinases, which in turn phosphorylate cellular proteins, including L-type calcium channels within the cell membrane. β1-Adrenergic stimulation of the myocyte also enhances myocyte relaxation.
The return of calcium from the cytosol to the sarcoplasmic reticulum (SR) is regulated by phospholamban (PL), a low-molecular-weight protein in the SR membrane. In its dephosphorylated state, PL inhibits Ca2+ uptake by the SR ATPase pump (Fig. 14.3). However, β1-adrenergic activation of protein kinase phosphorylates PL, and blunts its inhibitory effect. The subsequently greater uptake of Ca2+ ions by the SR hastens Ca2+ removal from the cytosol and promotes myocyte relaxation.
The cardiac cycle
The cardiac cycle (Fig. 14.6) consists of precisely timed rhythmic electrical and mechanical events that propel blood into the systemic and pulmonary circulations. The first event in the cardiac cycle is atrial depolarization (a P wave on the surface ECG) followed by right atrial and then left atrial contraction. Ventricular activation (the QRS complex on the ECG) follows after a short interval (the PR interval). Left ventricular contraction starts and shortly thereafter right ventricular contraction begins. The increased ventricular pressures exceed the atrial pressures, and close first the mitral and then the tricuspid valves.
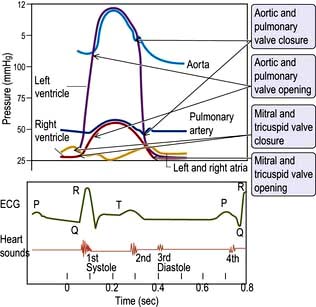
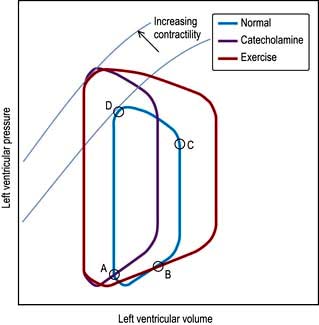
Until the aortic and pulmonary valves open, the ventricles contract with no change of volume (isovolumetric contraction). When ventricular pressures rise above the aortic and pulmonary artery pressures, the pulmonary valve and then the aortic valve open and ventricular ejection occurs. As the ventricles begin to relax, their pressures fall below the aortic and pulmonary arterial pressures, and aortic valve closure is followed by pulmonary valve closure. Isovolumetric relaxation then occurs. After the ventricular pressures have fallen below the right atrial and left atrial pressures, the tricuspid and mitral valves open. The cardiac cycle can be graphically depicted as the relationship between the pressure and volume of the ventricle. This is shown in Figure 14.7, which illustrates the changing pressure-volume relationships in response to increased contractility and to exercise.
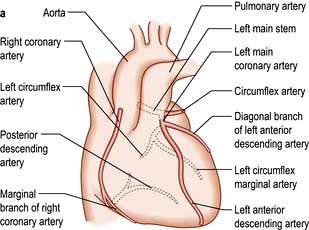
The coronary circulation
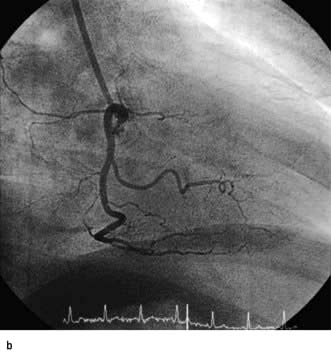
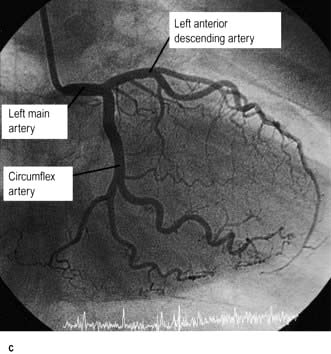
The coronary arterial system (Fig. 14.8) consists of the right and left coronary arteries. These arteries branch from the aorta, arising immediately above two cusps of the aortic valve. These arteries are unique in that they fill during diastole, when not occluded by valve cusps and when not squeezed by myocardial contraction. The right coronary artery arises from the right coronary sinus and courses through the right side of the AV groove, giving off vessels that supply the right atrium and the right ventricle. The vessel usually continues as the posterior descending coronary artery, which runs in the posterior interventricular groove and supplies the posterior part of the interventricular septum and the posterior left ventricular wall.
Blood vessel control and functions of the vascular endothelium


Vasomotor control
Nitric oxide is a diffusible gas with a very short half-life, produced in endothelial cells from the amino acid L-arginine via the action of the enzyme NO synthase (NOS), which is controlled by cytoplasmic calcium/calmodulin (Fig. 14.9). It is produced in response to various stimuli (Table 14.1), triggering vascular smooth muscle relaxation through activation of guanylate cyclase, leading to an increase in the intracellular levels of cyclic 3,5-guanine monophosphate (cGMP). Its cardiovascular effects protect against atherosclerosis, high blood pressure, heart failure and thrombosis. NO is also the neurotransmitter in various ‘nitrergic’ nerves in the central and peripheral nervous systems and may play a role in the central regulation of vascular tone. The class of drugs used to treat erectile dysfunction, the phosphodiesterase (PDE5) inhibitors, prevent the breakdown of cGMP and promote vasodilatation.
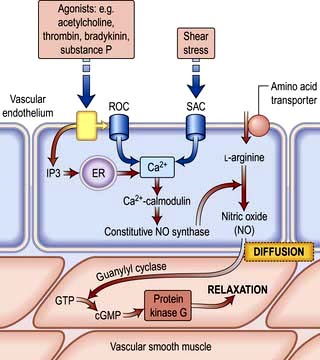
Table 14.1 Some of the products and functions of the vascular endothelium
| Endothelial product | Function(s) | Stimulus |
|---|---|---|
Nitric oxide | ||
Prostacyclin (PGI2) | ||
Prostanoids | ||
Endothelin | ||
Endothelial-derived hyperpolarizing factor | ||
Angiotensin-converting enzyme | ||
von Willebrand factor | ||
Adhesion molecules |
|
|
P, L, E selectins | ||
Vascular endothelial growth factor (VEGF) |
ICAM, intracellular adhesion molecule; VCAM, vascular cell adhesion molecule, PECAM, platelet/endothelial cell adhesion molecule; TNF, tumour necrosis factor; IL, interleukin.
PGI2 is synergistic to NO and also plays a role in the local regulation of vasomotor tone.
Angiotensin-converting enzyme located on the endothelial cell membrane converts circulating angiotensin I (synthesized by the action of renin on angiotensinogen) to angiotensin II which has vasoconstrictor properties and leads to aldosterone release (Fig. 12.5). Aldosterone promotes sodium absorption from the kidney and together with the angiotensin-induced vasoconstriction provides haemodynamic stability.
Anti- and pro-thrombotic mechanisms
In addition to their ability to prevent clotting, endothelial cells also aid thrombosis. They are responsible for the production of von Willebrand factor through a unique organelle called the Weibel–Palade body, which not only acts as a carrier for factor VIII but also promotes platelet adhesion by binding to exposed collagen (p. 414).
Modulation of immune responses
In response to various inflammatory mediators, the vascular endothelium expresses various so-called ‘adhesion molecules’ which promote leucocyte attraction, adhesion and infiltration into the blood vessel wall (Chapter 3).
Symptoms of heart disease
The following symptoms occur with heart disease:






The severity of cardiac symptoms or fatigue is classified according to the New York Heart Association (NYHA) grading of cardiac status (see Table 14.19). The differential diagnosis of chest pain is given in Table 14.2.
Table 14.2 Differential diagnosis of chest pain
| Central | Lateral/peripheral |
|---|---|
Cardiac | Pulmonary |
Ischaemic heart disease (infarction or angina) | Infarction |
Coronary artery spasm | Pneumonia |
Pericarditis/myocarditis | Pneumothorax |
Mitral valve prolapse | Lung cancer |
Aortic aneurysm/dissection | Mesothelioma |
Non-cardiac | Non-pulmonary |
Pulmonary embolism | Bornholm disease (epidemic myalgia) |
Oesophageal disease (see Box 6.3) | Herpes zoster |
Mediastinitis | Trauma (ribs/muscular) |
Costochondritis (Tietze disease) |
|
Trauma (soft tissue, rib) |
|
Central chest pain
 Retrosternal heavy or gripping sensation with radiation to the left arm or neck that is provoked by exertion and eased with rest or nitrates – angina (p. 729)
Retrosternal heavy or gripping sensation with radiation to the left arm or neck that is provoked by exertion and eased with rest or nitrates – angina (p. 729)
 Similar pain at rest – acute coronary syndrome (p. 734)
Similar pain at rest – acute coronary syndrome (p. 734)
 Severe tearing chest pain radiating through to the back – aortic dissection (p. 787)
Severe tearing chest pain radiating through to the back – aortic dissection (p. 787)
 Sharp central chest pain that is worse with movement or respiration but relieved with sitting forward – pericarditis pain (p. 774)
Sharp central chest pain that is worse with movement or respiration but relieved with sitting forward – pericarditis pain (p. 774)
 Sharp stabbing left submammary pain associated with anxiety – Da Costa’s syndrome.
Sharp stabbing left submammary pain associated with anxiety – Da Costa’s syndrome.
Dyspnoea
Central sleep apnoea syndrome (CSAS). If hypopnoea occurs rather than apnoea, the phenomenon is termed ‘periodic breathing’, but the two variations are known together as CSAS. This occurs due to malfunctioning of the respiratory centre in the brain, caused by poor cardiac output with concurrent cerebrovascular disease. The symptoms of CSAS, such as daytime somnolence and fatigue, are similar to those of obstructive sleep apnoea syndrome (OSAS, p. 818) and there is considerable overlap with the symptoms of heart failure. CSAS is believed to lead to myocardial hypertrophy and fibrosis, deterioration in cardiac function and complex arrhythmias, including non-sustained ventricular tachycardia, hypertension and stroke. Patients with CSAS have a worse prognosis compared to similar patients without CSAS.
Palpitations
 Premature beats (ectopics) are felt by the patient as a pause followed by a forceful beat. This is because premature beats are usually followed by a pause before the next normal beat, as the heart resets itself. The next beat is more forceful as the heart has had a longer diastolic period and therefore is filled with more blood before this beat.
Premature beats (ectopics) are felt by the patient as a pause followed by a forceful beat. This is because premature beats are usually followed by a pause before the next normal beat, as the heart resets itself. The next beat is more forceful as the heart has had a longer diastolic period and therefore is filled with more blood before this beat.
 Paroxysmal tachycardias (see p. 698) are felt as a sudden racing heart beat.
Paroxysmal tachycardias (see p. 698) are felt as a sudden racing heart beat.
 Bradycardias (p. 702) may be appreciated as slow, regular, heavy or forceful beats. Most often, however, they are simply not sensed. All palpitations can be graded by the NYHA cardiac status (see Table 14.19).
Bradycardias (p. 702) may be appreciated as slow, regular, heavy or forceful beats. Most often, however, they are simply not sensed. All palpitations can be graded by the NYHA cardiac status (see Table 14.19).
Syncope
Syncope is a transient loss of consciousness due to inadequate cerebral blood flow. The cardiovascular causes are listed in Table 14.3.
Table 14.3 Cardiovascular causes of syncope
 A vasovagal attack is a simple faint and is the most common cause of syncope. The mechanism begins with peripheral vasodilatation and venous pooling of blood, leading to a reduction in the amount of blood returned to the heart. The near-empty heart responds by contracting vigorously, which in turn stimulates mechanoreceptors (stretch receptors) in the inferoposterior wall of the left ventricle. These in turn trigger reflexes via the central nervous system, which act to reduce ventricular stretch (i.e. further vasodilatation and sometimes profound bradycardia), but this causes a drop in blood pressure and therefore syncope. These episodes are usually associated with a prodrome of dizziness, nausea, sweating, tinnitus, yawning and a sinking feeling. Recovery occurs within a few seconds, especially if the patient lies down.
A vasovagal attack is a simple faint and is the most common cause of syncope. The mechanism begins with peripheral vasodilatation and venous pooling of blood, leading to a reduction in the amount of blood returned to the heart. The near-empty heart responds by contracting vigorously, which in turn stimulates mechanoreceptors (stretch receptors) in the inferoposterior wall of the left ventricle. These in turn trigger reflexes via the central nervous system, which act to reduce ventricular stretch (i.e. further vasodilatation and sometimes profound bradycardia), but this causes a drop in blood pressure and therefore syncope. These episodes are usually associated with a prodrome of dizziness, nausea, sweating, tinnitus, yawning and a sinking feeling. Recovery occurs within a few seconds, especially if the patient lies down.


 Micturition syncope refers to loss of consciousness whilst passing urine.
Micturition syncope refers to loss of consciousness whilst passing urine.

Obstructive. The obstructive cardiac causes listed in Table 14.3 all lead to syncope due to restriction of blood flow from the heart into the rest of the circulation, or between the different chambers of the heart.
Arrhythmias. Stokes–Adams attacks (p. 700) are a sudden loss of consciousness unrelated to posture and due to intermittent high-grade AV block, profound bradycardia or ventricular standstill. The patient falls to the ground without warning, is pale and deeply unconscious. The pulse is usually very slow or absent. After a few seconds the patient flushes brightly and recovers consciousness as the pulse quickens. Often there are no sequelae, but patients may injure themselves during falls. Occasionally a generalized convulsion may occur if the period of cerebral hypoxia is prolonged, leading to a misdiagnosis of epilepsy.
FURTHER READING
Moya A, Sutton R, Ammirati F et al. Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS). Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J 2009; 30(21):2631–2671.
Examination of the cardiovascular system
General examination
 Clubbing (p. 799) is seen in congenital cyanotic heart disease, particularly Fallot’s tetralogy and also in 10% of patients with subacute infective endocarditis.
Clubbing (p. 799) is seen in congenital cyanotic heart disease, particularly Fallot’s tetralogy and also in 10% of patients with subacute infective endocarditis.

 Cyanosis is a dusky blue discoloration of the skin (particularly at the extremities) or of the mucous membranes when the capillary oxygen saturation is less than 85%. Central cyanosis (p. 799) is seen with shunting of deoxygenated venous blood into the systemic circulation, as in the presence of a right-to-left heart shunt. Peripheral cyanosis is seen in the hands and feet, which are cold. It occurs in conditions associated with peripheral vasoconstriction and stasis of blood in the extremities leading to increased peripheral oxygen extraction. Such conditions include congestive heart failure, circulatory shock, exposure to cold temperatures and abnormalities of the peripheral circulation, e.g. Raynaud’s, p. 788.
Cyanosis is a dusky blue discoloration of the skin (particularly at the extremities) or of the mucous membranes when the capillary oxygen saturation is less than 85%. Central cyanosis (p. 799) is seen with shunting of deoxygenated venous blood into the systemic circulation, as in the presence of a right-to-left heart shunt. Peripheral cyanosis is seen in the hands and feet, which are cold. It occurs in conditions associated with peripheral vasoconstriction and stasis of blood in the extremities leading to increased peripheral oxygen extraction. Such conditions include congestive heart failure, circulatory shock, exposure to cold temperatures and abnormalities of the peripheral circulation, e.g. Raynaud’s, p. 788.
The arterial pulse
Rhythm


Character of pulse
 Carotid pulsations are not normally apparent on inspection of the neck but may be visible (Corrigan’s sign) in conditions associated with a large-volume pulse, including high output states (such as thyrotoxicosis, anaemia or fever) and in aortic regurgitation.
Carotid pulsations are not normally apparent on inspection of the neck but may be visible (Corrigan’s sign) in conditions associated with a large-volume pulse, including high output states (such as thyrotoxicosis, anaemia or fever) and in aortic regurgitation.
 A ‘collapsing’ or ‘water hammer’ pulse (Fig. 14.10) is a large-volume pulse characterized by a short duration with a brisk rise and fall. This is best appreciated by palpating the radial artery with the palmer aspect of four fingers while elevating the patient’s arm above the level of the heart. A collapsing pulse is characteristic of aortic valvular regurgitation or a persistent ductus arteriosus.
A ‘collapsing’ or ‘water hammer’ pulse (Fig. 14.10) is a large-volume pulse characterized by a short duration with a brisk rise and fall. This is best appreciated by palpating the radial artery with the palmer aspect of four fingers while elevating the patient’s arm above the level of the heart. A collapsing pulse is characteristic of aortic valvular regurgitation or a persistent ductus arteriosus.

 A plateau pulse is small in volume and slow in rising to a peak due to aortic stenosis (Fig. 14.10).
A plateau pulse is small in volume and slow in rising to a peak due to aortic stenosis (Fig. 14.10).
 Alternating pulse (pulsus alternans). This is characterized by regular alternate beats that are weak and strong. It is a feature of severe myocardial failure and is due to the prolonged recovery time of damaged myocardium; it indicates a very poor prognosis. It is easily noticed when taking the blood pressure because the systolic pressure may vary from beat to beat by as much as 50 mmHg (Fig. 14.10).
Alternating pulse (pulsus alternans). This is characterized by regular alternate beats that are weak and strong. It is a feature of severe myocardial failure and is due to the prolonged recovery time of damaged myocardium; it indicates a very poor prognosis. It is easily noticed when taking the blood pressure because the systolic pressure may vary from beat to beat by as much as 50 mmHg (Fig. 14.10).
 Bigeminal pulse (pulsus bigeminus). This is due to a premature ectopic beat following every sinus beat. The rhythm is not regular (Fig. 14.10) because every weak pulse is premature.
Bigeminal pulse (pulsus bigeminus). This is due to a premature ectopic beat following every sinus beat. The rhythm is not regular (Fig. 14.10) because every weak pulse is premature.
 Pulsus bisferiens (Fig. 14.10). This is a pulse that is found in hypertrophic cardiomyopathy and in mixed aortic valve disease (regurgitation combined with stenosis). The first systolic wave is the ‘percussion’ wave produced by the transmission of the left ventricular pressure in early systole. The second peak is the ‘tidal’ wave caused by recoil of the vascular bed. This normally happens in diastole (the dicrotic wave), but when the left ventricle empties slowly or is obstructed from emptying completely, the tidal wave occurs in late systole. The result is a palpable double pulse.
Pulsus bisferiens (Fig. 14.10). This is a pulse that is found in hypertrophic cardiomyopathy and in mixed aortic valve disease (regurgitation combined with stenosis). The first systolic wave is the ‘percussion’ wave produced by the transmission of the left ventricular pressure in early systole. The second peak is the ‘tidal’ wave caused by recoil of the vascular bed. This normally happens in diastole (the dicrotic wave), but when the left ventricle empties slowly or is obstructed from emptying completely, the tidal wave occurs in late systole. The result is a palpable double pulse.
 Dicrotic pulse (Fig. 14.10) results from an accentuated dicrotic wave. It occurs in sepsis, hypovolaemic shock and after aortic valve replacement.
Dicrotic pulse (Fig. 14.10) results from an accentuated dicrotic wave. It occurs in sepsis, hypovolaemic shock and after aortic valve replacement.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


