The nervous system coordinates and organizes the functions of all body systems. This intricate network of interlocking receptors and transmitters is a dynamic system that controls and regulates every mental and physical function. It has three main divisions:
♦ central nervous system (CNS)—the brain and spinal cord (See Reviewing the central nervous system, page 250.)
♦ peripheral nervous system—the motor and sensory nerves, which carry messages between the CNS and remote parts of the body (See Reviewing the peripheral nervous system,pages 251 and 252.)
♦ autonomic nervous system—actually part of the peripheral nervous system, regulates involuntary functions of the internal organs.
The fundamental unit that participates in all nervous system activity is the neuron, a highly specialized cell that receives and transmits electrochemical nerve impulses through delicate, threadlike fibers that extend from the central cell body. Axons carry impulses away from the cell body; dendrites carry impulses to it. Most neurons have several dendrites but only one axon.
♦ Sensory (or afferent) neurons transmit impulses from receptors to the spinal cord or the brain.
♦ Motor (or efferent) neurons, or motoneurons, transmit impulses from the CNS to regulate the activity of muscles or glands.
♦ Interneurons, also known as connecting or association neurons, carry signals through complex pathways between sensory and motoneurons. Interneurons account for 99% of all the neurons in the nervous system.
From birth to death, the nervous system efficiently organizes and controls the smallest actions, thoughts, or feelings; monitors communication and the instinct for survival; and allows introspection, wonder, abstract thought, and self-awareness. Together, the CNS and peripheral nervous system keep a person alert, awake, oriented, and able to move about freely without discomfort and with all body systems working to maintain homeostasis.
Thus, any disorder affecting the nervous system can cause signs and symptoms in any and all body systems. Patients with nervous system disorders commonly have signs and symptoms that are elusive, subtle, and sometimes latent.
Pathophysiologic changes
Typically, disorders of the nervous system involve some alteration in arousal, cognition, movement, muscle tone, homeostatic mechanisms, or pain. Most disorders cause more than one alteration, and the close intercommunication between the CNS and peripheral nervous system means that one alteration may lead to another.
AROUSAL
Arousal refers to the level of consciousness or state of awareness. A person who is aware of himself and the environment and can respond in specific ways is said to be fully conscious. Full consciousness requires that the reticular activating system (RAS), higher systems in the cerebral cortex, and thalamic connections are intact and functioning properly. Several mechanisms can alter arousal:
Reviewing the central nervous system
The central nervous system includes the brain and spinal cord. The brain consists of the cerebrum, cerebellum, brain stem, and primitive structures that lie below the cerebrum: the diencephalon, limbic system, and reticular activating system (RAS). The spinal cord is the primary pathway for messages between peripheral areas of the body and the brain. It also mediates spinal reflexes.
Cerebrum
The left and right cerebral hemispheres are joined by the corpus callosum, a mass of nerve fibers that allows communication between corresponding centers in the right and left hemispheres. Each hemisphere is divided into four lobes, based on anatomic landmarks and functional differences. The lobes are named for the cranial bones that lie over them (frontal, temporal, parietal, and occipital):
♦ frontal lobe — influences personality, judgment, abstract reasoning, social behavior, language expression, and movement (in the motor portion)
♦ temporal lobe — controls hearing, language comprehension, and storage and recall of memories (although memories are stored throughout the brain)
♦ parietal lobe — interprets and integrates sensations, including pain, temperature, and touch; also interprets size, shape, distance, and texture (The parietal lobe of the nondominant hemisphere, usually the right, is especially important for awareness of body schema [shape].)
♦ occipital lobe—functions primarily in interpreting visual stimuli.
The cerebral cortex, the thin surface layer of the cerebrum, is composed of gray matter (unmyelinated cell bodies). The surface of the cerebrum has convolutions (gyri) and creases or fissures (sulci).
Cerebellum
The cerebellum, which also has two hemispheres, maintains muscle tone, coordinates muscle movement, and controls balance.
Brain stem
Composed of the pons, midbrain, and medulla oblongata, the brain stem relays messages between upper and lower levels of the nervous system. The cranial nerves originate from the pons, midbrain, and medulla oblongata:
♦ pons—connects the cerebellum with the cerebrum and the midbrain to the medulla oblongata, and contains one of the respiratory centers
♦ midbrain—mediates the auditory and visual reflexes
♦ medulla oblongata—regulates respiratory, vasomotor, and cardiac function.
Primitive structures
The diencephalon contains the thalamus and hypothalamus, which lie beneath the cerebral hemispheres. The thalamus relays all sensory stimuli (except olfactory) as they ascend to the cerebral cortex. Thalamic functions include primitive awareness of pain, screening of incoming stimuli, and focusing of attention. The hypothalamus controls or affects body temperature, appetite, water balance, pituitary secretions, emotions, and autonomic functions, including sleep and wake cycles.
The limbic system lies deep within the temporal lobe. It initiates primitive drives (hunger, aggression, and sexual and emotional arousal) and screens all sensory messages traveling to the cerebral cortex.
The RAS, a diffuse network of hyperexcitable neurons fanning out from the brain stem through the cerebral cortex, screens all incoming sensory information and channels it to appropriate areas of the brain for interpretation. RAS activity also stimulates wakefulness.
Spinal cord
The spinal cord joins the brain stem at the level of the foramen magnum and terminates near the second lumbar vertebra.
A cross section of the spinal cord reveals a central H-shaped mass of gray matter divided into dorsal (posterior) and ventral (anterior) horns. Gray matter in the dorsal horns relays sensory (afferent) impulses; in the ventral horns, motor (efferent) impulses. White matter (myelinated axons of sensory and motor nerves) surrounds these horns and forms the ascending and descending tracts.
Reviewing the peripheral nervous system
The peripheral nervous system consists of the cranial nerves (CN), the spinal nerves, and the autonomic nervous system (ANS).
Cranial nerves
The 12 pairs of cranial nerves transmit motor or sensory messages, or both, primarily between the brain or brain stem and the head and neck. All cranial nerves, except for the olfactory and optic nerves, originate from the midbrain, pons, or medulla oblongata. The cranial nerves are sensory, motor, or mixed (both sensory and motor) as follows:
♦ olfactory (CN I)—Sensory: smell
♦ optic (CN II)—Sensory: vision
♦ oculomotor (CN III)—Motor: extraocular eye movement (superior, medial, and inferior lateral), pupillary constriction, and upper eyelid elevation
♦ trochlear (CN IV)—Motor: extraocular eye movement (inferior medial)
♦ trigeminal (CN V)—Sensory: transmitting stimuli from face and head, corneal reflex; Motor: chewing, biting, and lateral jaw movements
♦ abducens (CN VI)—Motor: extraocular eye movement (lateral)
♦ facial (CN VII)—Sensory: taste receptors (anterior two-thirds of tongue); Motor: facial muscle movement, including muscles of expression (those in the forehead and around the eyes and mouth)
♦ acoustic (CN VIII)—Sensory: hearing, sense of balance
♦ glossopharyngeal (CN IX)—Motor: swallowing movements; Sensory: sensations of throat; taste receptors (posterior one-third of tongue)
♦ vagus (CN X)—Motor: movement of palate, swallowing, gag reflex; activity of the thoracic and abdominal viscera, such as heart rate and peristalsis; Sensory: sensations of throat, larynx, and thoracic and abdominal viscera (heart, lungs, bronchi, and GI tract)
♦ spinal accessory (CN XI)—Motor: shoulder movement, head rotation
♦ hypoglossal (CN XII)—Motor: tongue movement.
Spinal nerves
The 31 pairs of spinal nerves are named according to the vertebra immediately below their exit point from the spinal cord. Each spinal nerve consists of afferent (sensory) and efferent (motor) neurons, which carry messages to and from particular body regions, called dermatomes.
Autonomic nervous system
The ANS innervates all internal organs. Sometimes known as the visceral efferent nerves, autonomic nerves carry messages to the viscera from the brain stem and neuroendocrine system. The ANS has two major divisions: the sympathetic (thoracolumbar) nervous system and the parasympathetic (craniosacral) nervous system.
Sympathetic nervous system
Sympathetic nerves exit the spinal cord between the levels of the 1st thoracic and 2nd lumbar vertebrae—hence the name thoracolumbar. These preganglionic neurons enter small relay stations (ganglia) near the cord. The ganglia form a chain that disseminates the impulse to postganglionic neurons, which reach many organs and glands, and can produce widespread, generalized responses.
The physiologic effects of sympathetic activity include:
♦ vasoconstriction
♦ elevated blood pressure
♦ enhanced blood flow to skeletal muscles
♦ increased heart rate and contractility
♦ heightened respiratory rate
♦ smooth-muscle relaxation of the bronchioles, GI tract, and urinary tract
♦ sphincter contraction
♦ pupillary dilation and ciliary muscle relaxation
♦ increased sweat gland secretion
reduced pancreatic secretion.
Parasympathetic nervous system
The fibers of the parasympathetic, or craniosacral, nervous system leave the central nervous system (CNS) by way of the cranial nerves from the midbrain and medulla and with the spinal nerves between the 2nd and 4th sacral vertebrae (S2 to S4).
After leaving the CNS, the long preganglionic fiber of each parasympathetic nerve travels to a ganglion near a particular organ or gland, and the short postganglionic fiber enters the organ or gland. Parasympathetic nerves have a specific response involving only one organ or gland.
The physiologic effects of parasympathetic system activity include:
♦ reduced heart rate, contractility, and conduction velocity
♦ bronchial smooth-muscle constriction
♦ increased GI tract tone and peristalsis with sphincter relaxation
♦ urinary system sphincter relaxation and increased bladder tone
♦ vasodilation of external genitalia, causing erection
♦ pupillary constriction
♦ increased pancreatic, salivary, and lacrimal secretions.
The parasympathetic system has little effect on mental or metabolic activity.
♦ direct destruction of the reticular activating system and its pathways
♦ destruction of the entire brain stem, either directly by invasion or indirectly by impairment of its blood supply
♦ compression of the RAS by disease, either from direct pressure or compression as structures expand or herniate.
Those mechanisms may result from structural, metabolic, or psychogenic disturbances:
♦ Structural changes include infections, vascular problems, neoplasms, trauma, and developmental and degenerative conditions. They usually are identified by their location relative to the tentorial plate, the double fold of dura that supports the temporal and occipital lobes and separates the cerebral hemispheres from the brain stem and cerebellum. Those above the tentorial plate are called supratentorial, whereas those below are called infratentorial.
♦ Metabolic changes that affect the nervous system include hypoxia, electrolyte disturbances, hypoglycemia, drugs, and toxins (endogenous and exogenous). Essentially, any systemic disease can affect the nervous system.
♦ Psychogenic changes are commonly associated with mental and psychiatric illnesses. Ongoing research has linked neuroanatomy and neurophysiology of the CNS and supporting structures, including neurotransmitters, with certain psychiatric illnesses. For example, dysfunction of the limbic system has been associated with schizophrenia, depression, and anxiety disorders.
Decreased arousal may be a result of diffuse or localized dysfunction in supratentorial areas:
♦ Diffuse dysfunction reflects damage to the cerebral cortex or underlying subcortical white matter. Disease is the most common cause of diffuse dysfunction; other causes include neoplasms, closed trauma with subsequent bleeding, and pus accumulation.
♦ Localized dysfunction reflects mechanical forces on the thalamus or hypothalamus. Masses (such as bleeding, infarction, emboli, and tumors) may directly impinge on the deep diencephalic structures or herniation may compress them.
Stages of altered arousal
An alteration in arousal usually begins with some interruption or disruption in the diencephalon. When this occurs, the patient shows evidence of dullness, confusion, lethargy, and stupor. Continued decreases in arousal result from midbrain dysfunction and are evidenced by a deepening of the stupor. Eventually, if the medulla and pons are affected, coma results.
A patient may move back and forth between stages or levels of arousal, depending on the cause of the altered arousal state, initiation of treatment, and response to the treatment. Typically, if the underlying problem isn’t or can’t be corrected, then the patient will progress through the various stages of decreased consciousness, termed rostral-caudal progression. Six levels of altered arousal or consciousness have been identified. (See Stages of altered arousal.)
Typically, five areas of neurologic function are evaluated to help identify the cause of altered arousal:
♦ level of consciousness (includes awareness and cognitive functioning, which reflect cerebral status)
♦ pattern of breathing (helps to localize the cause to the cerebral hemisphere or brain stem)
♦ pupillary changes (reflects the level of brain stem function; the brain stem areas that control arousal are anatomically next to the areas that control the pupils)
♦ eye movement and reflex responses (help identify the level of brain stem dysfunction and its mechanism, such as destruction or compression)
♦ motor responses (help identify the level, side, and severity of brain dysfunction).
COGNITION
Cognition is the ability to be aware, to use intuition, and to perceive, reason, judge, and remember. It reflects higher functioning of the cerebral cortex, including the frontal, parietal, and temporal lobes, and portions of the brain stem. Typically, an alteration in cognition results from direct destruction by ischemia and hypoxia or from indirect destruction by compression or the effects of toxins and chemicals.
Stages of altered arousal
This chart highlights the six levels or stages of altered arousal and their manifestations.
Stage
Manifestations
Confusion
♦ Loss of ability to think rapidly and clearly
♦ Impaired judgment and decision making
Disorientation
♦ Beginning of loss of consciousness
♦ Disorientation to time progresses to include disorientation to place
♦ Impaired memory
♦ Lack of recognition of self (last to go)
Lethargy
♦ Limited spontaneous movement or speech
♦ Easy to arouse by normal speech or touch
♦ Possible disorientation to time, place, or person
Obtundation
♦ Mild to moderate reduction in arousal
♦ Limited responsiveness to environment
♦ Ability to fall asleep easily without verbal or tactile stimulation from others
♦ Ability to answer questions with minimum response
Stupor
♦ State of deep sleep or unresponsiveness
♦ Arousable (motor or verbal response only to vigorous and repeated stimulation)
♦ Withdrawal or grabbing response to stimulation
Coma
♦ Lack of motor or verbal response to external environment or any stimuli
♦ No response to noxious stimuli, such as deep pain
♦ Unable to be aroused by any stimulus
Altered cognition may manifest as agnosia, aphasia, or dysphasia:
♦ Agnosia is a defect in the ability to recognize the form or nature of objects. Usually, agnosia involves only one sense—hearing, vision, or touch.
♦ Aphasia is loss of the ability to comprehend or produce language.
♦ Dysphasia is impairment of the ability to comprehend or use symbols in either verbal or written language, or to produce language.
Dysphasia typically arises from the left cerebral hemisphere, usually the frontotemporal region. However, different types of dysphasia occur, depending on the specific area of the brain involved. For example, a dysfunction in the posteroinferior frontal lobe (Broca’s area) causes a motor dysphasia in which the patient can’t find the words to speak and has difficulty writing and repeating words. Dysfunction in the pathways connecting the primary auditory area to the auditory association areas in the middle third of the left superior temporal gyrus causes a form of dysphasia called word deafness: The patient has fluent speech, but comprehension of the spoken word and ability to repeat speech are impaired. Rather than words, the patient hears only noise that has no meaning, yet reading comprehension and writing ability are intact.
Dementia
Dementia is the loss of more than one intellectual or cognitive function, which interferes with the ability to function in daily life. The patient may experience a problem with orientation, general knowledge and information, vigilance (attentiveness, alertness, and watchfulness), recent memory, remote memory, concept formulation, abstraction (ability to generalize about nonconcrete thoughts and ideas), reasoning, or language use.
The underlying mechanism is a defect in the neuronal circuitry of the brain. The extent of dysfunction reflects the total quantity of neurons lost and the area where this loss occurred.
Factors that have been associated with dementia include:
♦ degeneration
♦ cerebrovascular disorders
♦ compression
♦ effects of toxins
♦ metabolic conditions
♦ biochemical imbalances
♦ demyelinization
♦ infection.
Three major types of dementia have been identified: amnestic, intentional, and cognitive. Each type affects a specific area of the brain, resulting in characteristic impairments:
♦ Amnestic dementia typically results from defective neuronal circuitry in the temporal lobe. Characteristically, the patient exhibits difficulty in naming things, loss of recent memory, and loss of language comprehension.
♦ Intentional dementia results from a defect in the frontal lobe. The patient is easily distracted and, although able to follow simple commands, can’t carry out such sequential functions as planning, initiating, and regulating behavior or achieving specific goals. The patient may exhibit personality changes and a flat affect. Possibly appearing accident prone, he may lose motor function, as evidenced by a wide shuffling gait, small steps, muscle rigidity, abnormal reflexes, incontinence of bowel and bladder and, possibly, total immobility.
♦ Cognitive dementia reflects dysfunctional neuronal circuitry in the cerebral cortex. Typically, the patient loses remote memory, language comprehension, and mathematical skills, and has difficulty with visual-spatial relationships.
MOVEMENT
Movement involves a complex array of activities controlled by the cerebral cortex, the pyramidal system, the extrapyramidal system, and the motor units (the axon of the lower motoneuron from the anterior horn cell of the spinal cord and the muscles innervated by it). A problem in any one of these areas can affect movement. (See Reviewing motor impulse transmission.)
For movement to occur, the muscles must change their state from one of contraction to relaxation or vice versa. A change in muscle innervation anywhere along the motor pathway affects movement. Certain neurotransmitters, such as dopamine, play a role in altered movement.
Alterations in movement typically include excessive movement (hyperkinesia) or decreased movement (hypokinesia). Hyperkinesia is a broad category that includes many different types of abnormal movements. Each type of hyperkinesia is associated with a specific underlying pathophysiologic mechanism affecting the brain or motor pathway. (See Types of hyperkinesia,pages 256 and 257.) Hypokinesia usually involves loss of voluntary control, even though peripheral nerve and muscle functions are intact. The types of hypokinesia include paresis, akinesia, bradykinesia, and loss of associated neurons.
Paresis
Paresis is a partial loss of motor function (paralysis) and muscle power, which the patient will commonly describe as weakness. Paresis can result from the dysfunction of the:
♦ upper motoneurons in the cerebral cortex, subcortical white matter, internal capsule, brain stem, or spinal cord
♦ lower motoneurons in the brain stem motor nuclei and anterior horn of the spinal cord, or problems with their axons as they travel to the skeletal muscle
♦ motor units affecting the muscle fibers or the neuromuscular junction.
Upper motoneurons
Upper motoneuron dysfunction reflects an interruption in the pyramidal tract and consequent decreased activation of the lower motoneurons innervating one or more areas of the body. Upper motoneuron dysfunction usually affects more than one muscle group and generally affects distal muscle groups more severely than proximal groups. Onset of spastic muscle tone over several days to weeks commonly accompanies upper motoneuron paresis, unless the dysfunction is acute. In acute dysfunction, flaccid tone and loss of deep tendon reflexes indicates spinal shock, caused by a severe, acute lesion below the foramen magnum. Incoordination associated with upper motoneuron paresis manifests as slow, coarse movement with abnormal rhythm.
Lower motoneurons
Lower motoneurons are of two basic types: large (alpha) and small (gamma). Dysfunction of the large motoneurons of the anterior horn of the spinal cord, the motor nuclei of the brain stem, and their axons causes impairment of voluntary and involuntary movement. The extent of paresis is directly correlated to the number of large lower motoneurons affected. If only a small portion of the large motoneurons are involved, paresis occurs; if all motor units are affected, paralysis.
The small motoneurons play two necessary roles in movement: maintaining muscle tone and protecting the muscle from injury. Usually, when the large motoneurons are affected, dysfunction of the small motoneurons causes reduced or absent muscle tone, flaccid paresis, and paralysis.
Reviewing motor impulse transmission
Motor impulses that originate in the motor cortex of the frontal lobe travel through upper motoneurons of the pyramidal or extrapyramidal tract to the lower motoneurons of the peripheral nervous system.
In the pyramidal tract, most impulses from the motor cortex travel through the internal capsule to the medulla, where they cross (decussate) to the opposite side and continue down the spinal cord as the lateral corticospinal tract, ending in the anterior (or ventral) horn of the gray matter at a specific spinal cord level. Some fibers don’t cross in the medulla but continue down the anterior corticospinal tract and cross near the level of termination in the anterior horn. The fibers of the pyramidal tract are considered upper motoneurons. In the anterior horn of the spinal cord, upper motoneurons relay impulses to the lower motoneurons, which carry them via the spinal and peripheral nerves to the muscles, producing a motor response.
Motor impulses that regulate involuntary muscle tone and muscle control travel along the extrapyramidal tract from the premotor area of the frontal lobe to the pons of the brain stem, where they cross to the opposite side. The impulses then travel down the spinal cord to the anterior horn, where they’re relayed to lower motoneurons for ultimate delivery to the muscles.
Types of hyperkinesia
This chart summarizes some of the most common types of hyperkinesia, their manifestations, and the underlying mechanisms involved in their development.
Type
Manifestations
Mechanisms
Akathisia
♦ Ranges from mildly compulsive movement (usually the legs) to severely frenzied motion
♦ Partly voluntary, with ability to suppress for short periods
♦ Relief obtained by performing motion
Possibly associated with impaired dopaminergic transmission
Asterixis
♦ Irregular flapping-hand movement
♦ More prominent when arms are outstretched
Believed to result from buildup of toxins not broken down by the liver (such as ammonia)
Athetosis
♦ Slow, sinuous, irregular movements in the distal extremities
♦ Characteristic hand posture
♦ Slow, flucutating grimaces
Believed to result from injury to the putamen of the basal ganglion
Injury to subthalamus nucleus, causing inhibition of the nucleus
Chorea
♦ Random, irregular, involuntary, rapid contractions of muscle groups
♦ Nonrepetetive
♦ Diminishes with rest; disappears during sleep
♦ Increases during emotional stress or attempts at voluntary movement
Excess concentration or heightened sensitivity to dopamine in the basal ganglia
Hyperactivity
♦ Prolonged, generalized, increased activity
♦ Mainly involuntary but possibly subject to voluntary control
♦ Continual changes in body posture or excessive performance of a simple activity at inappropriate times
Possibly due to injury to frontal lobe and reticular activating system (RAS)
Intentional cerebellar tremor
♦ Tremor secondary to movement
♦ Most severe when nearing end of the movement
Errors in the feedback from the periphery and goal-directed movement due to disease of dentate nucleus and superior cerebellar peduncle
Myoclonus
♦ Shocklike contractions
♦ Throwing limb movements
♦ Random occurrence
♦ Triggered by startle
♦ Present even during sleep
Irritability of nervous system and spontaneous discharge of neurons in the cerebral cortex, cerebellum, RAS, and spinal cord
Parkinsonian tremor
♦ Regular, rhythmic, slow flexion and extension contraction
♦ Primarily affects metacarpophalangeal and wrist joints
♦ Disappears with voluntary movement
Loss of inhibitory effects of dopamine in basal ganglia
Wandering
♦ Moving about without attention to environment
Possibly due to bilateral injury to globus pallidus or putamen
Motor units
The muscles innervated by motoneurons in the anterior horn of the spinal cord may also be affected. Paresis results from a decrease in the number or force of activated muscle fibers in the motor unit. The action potential of each motor unit decreases so that additional motor units are needed more quickly to produce the power necessary to move the muscle. Dysfunction of the neuromuscular junction causes paresis in a similar fashion; however, the capability of the motor units to function is lost, not the actual number of units.
Akinesia
Akinesia is a partial or complete loss of voluntary and associated movements, as well as a disturbance in the time needed to perform a movement. Commonly caused by dysfunction of the extrapyramidal tract, akinesia is associated with dopamine deficiency at the synapse or a defect in the postsynaptic receptors for dopamine.
Bradykinesia
Bradykinesia refers to slow voluntary movements that are labored, deliberate, and hard to initiate. The patient has difficulty performing movements consecutively and at the same time. Like akinesia, bradykinesia involves a disturbance in the time needed to perform a movement.
Loss of associated neurons
Movement involves not only the innervation of specific muscles to accomplish an action but also the work of other innervated muscles that enhance the action. Loss of associated neurons involves alterations in movement that accompany the usual habitual voluntary movements for skill, grace, and balance. For example, when a person expresses emotion, the muscles of the face change and the posture relaxes. Loss of associated neurons involving emotional expression results in a flat, blank expression and a stiff posture. Loss of associated neurons necessary for locomotion results in a decrease in arm and shoulder movement, hip swinging, and rotation of the cervical spine.
MUSCLE TONE
Like movement, muscle tone involves complex activities controlled by the cerebral cortex, pyramidal system, extrapyramidal system, and motor units. Normal muscle tone is the slight resistance that occurs in response to passive movement. When one muscle contracts, reciprocal muscles relax to permit movement with only minimal resistance. For example, when the elbow is flexed, the biceps contracts and feels firm and the triceps is somewhat relaxed and soft; with continued flexion, the biceps relaxes and the triceps contracts. Thus, when a joint is moved through range of motion, the resistance is normally smooth, even, and constant.
The two major types of altered muscle tone are hypotonia (decreased muscle tone) and hypertonia (increased muscle tone).
Hypotonia
Hypotonia (also referred to as muscle flaccidity) typically reflects cerebellar damage, but rarely it may result from pure pyramidal tract damage.
Hypotonia is thought to involve a decrease in muscle spindle activity as a result of a decrease in neuron excitability. Flaccidity generally occurs with loss of nerve impulses from the motor unit responsible for maintaining muscle tone.
It may be localized to a limb or muscle group, or it may be generalized, affecting the entire body. Flaccid muscles can be moved rapidly with little or no resistance; eventually they become limp and atrophy.
Hypertonia
Hypertonia is increased resistance to passive movement. There are four types of hypertonia:
♦ Spasticity is hyperexcitability of stretch reflexes caused by damage to the lateral corticospinal tract and the motor, premotor, and supplementary motor areas. (See How spasticity develops.)
♦ Paratonia (gegenhalten) is variance in resistance to passive movement in direct proportion to the force applied; the cause is frontal lobe injury.
♦ Dystonia is sustained, involuntary twisting movements resulting from slow muscle contraction; the cause is lack of appropriate inhibition of reciprocal muscles.
♦ Rigidity, or constant, involuntary muscle contraction, is resistance in both flexion and extension; the causes are damage to basal ganglion (cogwheel or lead-pipe rigidity) or loss of cerebral cortex inhibition or cerebellar control (gamma and alpha rigidity).
Hypertonia usually leads to atrophy of unused muscles. However, in some cases, if the motor reflex arc remains functional but isn’t inhibited by the higher centers, the overstimulated muscles may hypertrophy.
HOMEOSTATIC MECHANISMS
For proper function, the brain must maintain and regulate pressure inside the skull (intracranial pressure [ICP]) as it also maintains the flow of oxygen (O2) and nutrients to its tissues. Both of these are accomplished by balancing changes in blood flow and cerebrospinal fluid (CSF) volume.
Constriction and dilation of the cerebral blood vessels help to regulate ICP and delivery of nutrients to the brain. These vessels respond to changes in concentrations of carbon dioxide (CO2), O2, and hydrogen ions (H+). For example, if the CO2 concentration in blood increases, the gas combines with body fluids to form carbonic acid, which eventually releases H+. An increase in H+ concentration causes the cerebral vessels to dilate, increasing blood flow to the brain, increasing cerebral perfusion and, subsequently, causing a drop in H+ concentration. A decrease in O2 concentration also stimulates cerebral vasodilation, increasing blood flow and O2 delivery to the brain.
Should these normal autoregulatory mechanisms fail, the abnormal blood chemistry stimulates the sympathetic nervous system to cause vasoconstriction of the large and medium-sized cerebral arteries. This helps to prevent increases in blood pressure from reaching the smaller cerebral vessels.
CSF volume remains relatively constant. However, if ICP rises—even as little as 5 mm Hg—the arachnoid villi open and excess CSF drains into the venous system.
The blood-brain barrier also helps to maintain homeostasis in the brain. This barrier is composed of tight junctions between the endothelial cells of the cerebral vessels and neuroglial cells and is relatively impermeable to most substances. However, some substances required for metabolism pass through the blood-brain barrier, depending on their size, solubility, and electrical charge. This barrier also regulates water flow from the blood, thereby helping to maintain the volume within the skull.
Increased ICP
ICP is the pressure that the brain tissue, CSF, and cerebral blood (intracranial components) exert against the skull. The skull is a rigid structure; therefore, a change in the volume of the intracranial contents triggers a reciprocal change in one or more of the intracranial components to maintain a consistent pressure. Any condition that alters the normal balance of the intracranial components—including increased brain volume, increased blood volume, or increased CSF volume—can increase ICP.
Initially, the body uses its compensatory mechanisms (described above) to attempt to maintain homeostasis and lower ICP. However, if these mechanisms become overwhelmed and are no longer effective, ICP continues to rise. Cerebral perfusion pressure falls and cerebral blood flow decreases. Ischemia leads to cellular hypoxia, which initiates vasodilation of cerebral blood vessels in an attempt to increase cerebral blood flow. Unfortunately, this only causes the ICP to increase further. As the pressure continues to rise, compression of brain tissue and cerebral vessels further impairs cerebral blood flow.
If ICP continues to rise, the brain begins to shift under the extreme pressure and may herniate to an area of lesser pressure. When the herniating brain tissue’s blood supply is compromised, cerebral ischemia and hypoxia worsen. The herniation increases pressure in the area where the pressure was lower, thus impairing its blood supply. As ICP approaches systemic blood pressure, cerebral perfusion slows even more, ceasing when ICP equals systemic blood pressure. (See What happens when ICP rises, page 260.)
How spasticity develops
Motor activity is controlled by pyramidal and extrapyramidal tracts that originate in the motor cortex, basal ganglia, brain stem, and spinal cord. Nerve fibers from the various tracts converge and synapse at the anterior (or ventral) horn of the spinal cord. Together they maintain segmental muscle tone by modulating the stretch reflex arc. This arc, shown in a simplified version below, is basically a negative feedback loop in which muscle stretch (stimulation) causes reflexive contraction (inhibition), thus maintaining muscle length and tone.
Damage to certain tracts results in loss of inhibition and disruption of the stretch reflex arc. Uninhibited muscle stretch produces exaggerated, uncontrolled muscle activity, accentuating the reflex arc, and eventually resulting in spasticity.
Cerebral edema
Cerebral edema is an increase in the fluid content of brain tissue that leads to an increase in the intracellular or extracellular fluid volume. Cerebral edema may result from an initial injury to the brain tissue, or it may develop in response to cerebral ischemia, hypoxia, and hypercapnia.
Cerebral edema is classified in four types— vasogenic, cytotoxic, ischemic, or interstitial— depending on the underlying mechanism responsible for the increased fluid content.
♦ Vasogenic—Injury to the vasculature increases capillary permeability and disruption of blood brain barrier; leakage of plasma proteins into the extracellular spaces pulls water into the brain parenchyma.
♦ Cytotoxic (metabolic)—Toxins cause failure of the active transport mechanisms; loss of intracellular potassium and influx of sodium (and water) cause cells in the brain to swell.
♦ Ischemic—It’s caused by cerebral infarction and initially confined to intracellular compartment; after several days, released lysosomes from necrosed cells disrupt blood brain barrier.
♦ Interstitial—Movement of CSF from ventricles to extracellular spaces increases brain volume.
Commonly, patients simultaneously experience more than one type of cerebral edema. Regardless of the type, blood vessels become distorted and brain tissue is displaced, ultimately leading to herniation.
PAIN
Pain is the result of a complex series of steps from a site of injury to the brain, which interprets the stimuli as pain. Pain that originates outside the nervous system is termed nociceptive pain; pain that originates from within the nervous system, neurogenic or neuropathic pain.
What happens when ICP rises
Intracranial pressure (ICP) is the pressure exerted within the intact skull by the intracranial volume—about 10% blood, 10% cerebrospinal fluid (CSF), and 80% brain tissue. The rigid skull has little space for expansion of these substances.
The brain compensates for increases in ICP by regulating the volume of the three substances in the following ways:
♦ limiting blood flow to the head
♦ displacing CSF into the spinal canal
♦ increasing absorption or decreasing production of CSF—withdrawing water from brain tissue and excreting it through the kidneys.
When compensatory mechanisms become overworked, small changes in volume lead to large changes in pressure. The following chart will help you to understand increased ICP’s pathophysiology.
Nociception
Nociception begins when noxious stimuli reach pain fibers. Sensory receptors called nociceptors—which are free nerve endings in the tissues—are stimulated by various agents, such as chemicals, temperature, or mechanical pressure. If a stimulus is sufficiently strong, impulses travel via the afferent nerve fibers along sensory pathways to the spinal cord, where they initiate autonomic and motor reflexes. The information also continues to travel to the brain, which perceives it as pain. Several theories have been developed to explain pain. (See Theories of pain, pages 262 and 263.)
Nociception consists of four steps: transduction, transmission, modulation, and perception.
Transduction
Transduction is the conversion of noxious stimuli into electrical impulses and subsequent depolarization of the nerve membrane. These electrical impulses are created by algesic substances that sensitize the nociceptors and are released at the site of injury or inflammation. Examples include H+ and potassium ions, serotonin, histamine, prostaglandins, bradykinin, and substance P.
Transmission
A delta fibers and C fibers transmit pain sensations from the tissues to the CNS.
A delta fibers are lightly myelinated fibers with a small diameter. Mechanical or thermal stimuli elicit a rapid or fast response. These fibers transmit localized, sharp, stinging, or pinprick-type pain sensations. A delta fibers connect with secondary neuron groupings on the dorsal horn of the spinal cord.
C fibers are smaller and unmyelinated. They connect with second order neurons in lamina I and II (the latter includes the sub-stantia gelatinosa, an area in which pain is modulated). C fibers respond to chemical stimuli, rather than heat or pressure, triggering a slow pain response, usually within 1 second. This dull ache or burning sensation isn’t localized and leads to two responses: an acute response transmitted immediately through fast pain pathways, which prompts the person to evade the stimulus, and lingering pain transmitted through slow pain pathways, which persists or worsens.
The A delta and C fibers carry the pain signal from the peripheral tissues to the dorsal horn of the spinal cord. Excitatory and inhibitory interneurons and projection cells (neurons that connect pathways in the cerebral cortex of the CNS and peripheral nervous system) carry the signal to the brain by way of crossed and uncrossed pathways. An example of a crossed pathway is the spinothalamic tract, which enters the brain stem and ends in the thalamus. Sensory impulses travel from the medial and lateral lemniscus (tract) to the thalamus and brain stem. From the thalamus, other neurons carry the information to the sensory cortex, where pain is perceived and understood.
Another example of a crossed pathway is the ascending spinoreticulothalamic tract, which is responsible for the psychological components of pain and arousal. At this site, neurons synapse with interneurons before they cross to the opposite side of the cord and make their way to the medulla and, eventually, the reticular activating system, mesencephalon, and thalamus. Impulses then are transmitted to the cerebral cortex, limbic system, and basal ganglia.
After stimuli are delivered, responses from the brain must be relayed back to the original site. Several pathways carry the information in the dorsolateral white columns to the dorsal horn of the spinal cord. Some corticospinal tract neurons end in the dorsal horn and allow the brain to pay selective attention to certain stimuli while ignoring others. This allows transmission of the primary signal while suppressing the tendency for signals to spread to adjacent neurons.
Modulation
Modulation refers to modifications in pain transmission. Some neurons from the cerebral cortex and brain stem activate inhibitory processes, thus modifying the transmission. Substances—such as serotonin from the mesencephalon, norepinephrine from the pons, and endorphins from the brain and spinal cord— inhibit pain transmission by decreasing the release of nociceptive neurotransmitters. Spinal reflexes involving motoneurons may initiate a protective action, such as withdrawal from a pinprick, or may enhance the pain, such as when trauma causes a muscle spasm in the injured area.
Perception
Perception is the end result of pain transduction, transmission, and modulation. It encompasses the emotional, sensory, and subjective aspects of the pain experience. Pain perception is thought to occur in the cortical structures of the somatosensory cortex and limbic system. Alertness, arousal, and motivation are believed to result from the action of the reticular activating system and limbic system. Cardiovascular responses and typical fight or flight responses are thought to involve the medulla and hypothalamus.
Theories of pain
Over the years numerous theories have attempted to explain the sensation of pain and describe how it occurs. This chart highlights some major theories about pain.
Theory
Major assumptions
Comment
Specificity
♦ Four types of cutaneous sensation (touch, warmth, cold, pain); each results from stimulation of specific skin receptor sites and neural pathways.
♦ Specific pain neurons transmit pain sensation along specific pain fibers.
♦ At synapses in the substantia gelatinosa, pain impulses cross to the opposite side of the cord and ascend the specific pain pathways of the spinothalamic tract to the thalamus and the pain receptor areas of the cerebral cortex.
♦ Focuses on the direct relationship between the pain stimulus and perception; doesn’t account for adaptation to pain and the psychosocial factors modulating it.
Intensity
♦ Pain results from excessive stimulation of sensory receptors. Disorders or processes causing pain create an intense summation of nonnoxious stimuli.
♦ Doesn’t explain existence of intense stimuli not perceived as pain.
Pattern
♦ Nonspecific receptors transmit specific patterns (characterized by the length of the pain sensation, the amount of involved tissue, and the summation of impulses) from the skin to the spinal cord, leading to pain perception.
♦ Includes some components of the intensity theory; pain possibly a response to intense stimulation of the sensory receptors regardless of receptor type or pathway.
Neuromatrix
♦ A pattern theory. Sensations imprinted in the brain. Sensory inputs may trigger a pattern of sensation from the neuromatrix (a proposed network of neurons looping between the thalamus and the cortex, and the cortex and the limbic system).
♦ Sensation pattern is possible without the sensory trigger.
♦ Explains existence of phantom pain.
Gate control
♦ Pain is transmitted from skin via the small diameter A delta and C fibers to cells of the substantia gelatinosa in the dorsal horn, where interconnections between other sensory pathways exist. Stimulation of the large-diameter fast, myelinated A beta and A alpha fibers closes gate, which restricts transmission of the impulse to the CNS and diminishes pain perception.
♦ Large fiber stimulation is possible through massage, scratching or rubbing the skin, or through electrical stimulation. Concurrent firing of pain and touch paths reduces transmission and perception of the pain impulses but not of touch impulses.
♦ An increase in small-fiber activity inhibits the substantia gelatinosa cells, “opening the gate” and increasing pain transmission and perception.
♦ Substantia gelatinosa acts as a gate-control system to inhibit the flow of nerve impulses from peripheral fibers to the CNS.
♦ Central T cells act as a CNS control to stimulate selective brain processes that influence the gate-control system. Inhibition of T cells closes the gate, pain impulses aren’t transmitted to the brain.
♦ T-cell activation of neural mechanisms in the brain is responsible for pain perception and response; transmitters partly regulate the release of substance P, the peptide that conveys pain information. Pain modulation is also partly controlled by the neurotransmitters, enkephalin and serotonin.
♦ Persistent pain initiates a gradual decline in the fraction of impulses that pass through the various gates.
♦ Descending efferent impulses from the brain may be responsible for closing, partially opening, or completely opening the gate.
♦ Provides the basis for use of massage and electrical stimulation in pain management; being used to develop additional theories and models.
Melzack-Casey Conceptual Model
♦ Three major psychological dimensions of pain: sensory-discriminative from thalamus and somatosensory cortex, motivational-affective from the reticular formation, and cognitive-evaluative.
♦ Interactions among the three produce descending inhibitory influences that alter pain input to the dorsal horn and ultimately modify the sensory pain experience and motivational-affective dimensions.
♦ Pain is localized and identified by its characteristics, evaluated by past experiences, and undergoes further cognitive processing. The complex sensory, motivational, and cognitive interactions determine motor activities and behaviors associated with the pain experience.
♦ Takes into account the powerful role of psychological functioning in determining the quality and intensity of pain.
The following three variables contribute to the wide variety of individual pain experiences:
♦ pain threshold—level of intensity at which a stimulus is perceived as pain
♦ perceptual dominance—existence of pain at another location that’s given more attention
♦ pain tolerance—duration or intensity of pain to be endured before a response is initiated.
Neurogenic pain
Neurogenic pain is associated with neural injury. Pain results from spontaneous discharges from the damaged nerves, spontaneous dorsal root activity, or degeneration of modulating mechanisms. Neurogenic pain doesn’t activate nociceptors, and there’s no typical pathway for transmission.
Disorders
This section discusses disorders of the nervous system, some of which can have far-reaching effects in all body systems. They include Alzheimer’s disease, amyotrophic lateral sclerosis, arteriovenous malformations, Bell’s palsy, cerebral palsy, complex regional pain syndrome, Creutzfeldt-Jakob disease, Guillain-Barré syndrome, headache, head trauma, Huntington’s disease, hydrocephalus, intracranial aneurysm, meningitis, multiple sclerosis, myasthenia gravis, Parkinson’s disease, Reye’s syndrome, seizure disorder, spinal cord trauma, and stroke.
ALZHEIMER’s DISEASE
Alzheimer’s disease is a degenerative disorder of the cerebral cortex, especially the frontal lobe, which accounts for more than half of all cases of dementia.
Although primarily found in the elderly population, 1% to 10% of cases have their onset in middle age.
Because this is a primary progressive dementia, the prognosis for a patient with this disease is poor.
Causes
The exact cause of Alzheimer’s disease is unknown. Factors that have been associated with its development include:
♦ neurochemical factors, such as deficiencies in the neurotransmitters acetylcholine, somatostatin, substance P, and norepinephrine
♦ environmental factors, such as repeated head trauma or exposure to aluminum or manganese
♦ genetic factors.
Genetic studies show that an autosomal dominant form of Alzheimer’s disease is associated with early onset and early death. A family history of Alzheimer’s disease and the presence of Down syndrome are two established risk factors.
Pathophysiology
The brain tissue of patients with Alzheimer’s disease has three distinct and characteristic features:
♦ neurofibrillatory tangles (fibrous proteins)
♦ beta-amyloid plaques (composed of degenerating axons and dendrites)
♦ granulovascular degeneration of neurons.
Additional structural changes include cortical atrophy, ventricular dilation, deposition of amyloid (a glycoprotein) around the cortical blood vessels, and reduced brain volume. Also found is a selective loss of cholinergic neurons in the pathways to the frontal lobes and hippocampus, areas that are important for memory and cognitive functions. Examination of the brain after death commonly reveals an atrophic brain, in many cases weighing less than 1,000 g (normal: 1,380 g). (See Abnormal cellular structures in Alzheimer’s disease, pages 266 and 267.)
Signs and symptoms
The typical signs and symptoms reflect neurologic abnormalities associated with pathophysiologic changes of the disease:
♦ gradual loss of recent and remote memory, loss of sense of smell, and flattening of affect and personality
♦ difficulty with learning new information
♦ deterioration in personal hygiene
♦ inability to concentrate
♦ increasing difficulty with abstraction and judgment
♦ impaired communication
♦ severe deterioration in memory, language, and motor function
♦ loss of coordination
♦ inability to write or speak
♦ personality changes
♦ tendency to wander
♦ nocturnal awakenings
♦ loss of eye contact and fearful look
♦ signs of anxiety, such as wringing of hands
♦ acute confusion, agitation, compulsiveness, or fearfulness when overwhelmed with anxiety
♦ disorientation and emotional lability
♦ progressive deterioration of physical and intellectual ability.
Complications
♦ Injury secondary to violent behavior or wandering
♦ Pneumonia and other infections
♦ Malnutrition
♦ Dehydration
♦ Aspiration
♦ Death
Diagnosis
Alzheimer’s disease is diagnosed by exclusion— that is, by ruling out other disorders as the cause for the patient’s signs and symptoms. The only true way to confirm Alzheimer’s disease is by finding pathologic changes in the brain at autopsy. However, these diagnostic tests may be useful:
♦ Positron emission tomography shows changes in the metabolism of the cerebral cortex.
♦ Computed tomography scan shows evidence of early brain atrophy in excess of that which occurs in normal aging.
♦ Magnetic resonance imaging shows no lesion as the cause of the dementia.
♦ EEG shows evidence of slowed brain waves in the later stages of the disease.
♦ Cerebral blood flow studies show abnormalities in blood flow.
Treatment
No cure or definitive treatment exists for Alzheimer’s disease. Therapy may include:
♦ a cholinesterase inhibitor, such as donepezil, rivastigmine, and galantamine, to prevent the breakdown of acetylcholine (involved in the brain’s memory and thinking skills)
♦ memantine, an uncompetitive low-to-moderate affinity N-methyl-D-aspartate receptor antagonist, to regulate the activity of glutamate (involved in the brain’s information processing, storing, and retrieval)
♦ a vitamin E supplement to help defend the brain against damage.
Many clinical trials are under way for the treatment of Alzheimer’s disease, including a vaccine that would stimulate the immune system to recognize and attack the beta-amyloid plaques that occur with the disease.
Special considerations
Overall care is focused on supporting the patient’s remaining abilities and compensating for those he has lost.
♦ Provide the patient with a safe environment. Encourage him to exercise, as ordered, to help maintain mobility.
♦ Establish an effective communication system with the patient and family to help them adjust to the patient’s altered cognitive abilities.
♦ Anxiety may cause the patient to become agitated or fearful. Intervene by helping him focus on another activity.
♦ Establish a routine for the patient to help him maintain a sense of control over his environment.
♦ Offer emotional support to the patient and family members. Behavior problems may be worsened by excess stimulation or change in established routine. Teach them about the disease, and refer them to social service and community resources for legal and financial advice and support.
AMYOTROPHIC LATERAL SCLEROSIS
Commonly called Lou Gehrig disease, after the New York Yankees first baseman that died of this disorder, amyotrophic lateral sclerosis (ALS) is the most common of the motoneuron diseases causing muscular atrophy. Other motoneuron diseases include progressive muscular atrophy and progressive bulbar palsy. Onset usually occurs between ages 40 and 70. A chronic, progressively debilitating disease, ALS may be fatal in less than 1 year or continue for 10 years or more, depending on the muscles affected. More than 30,000 Americans have ALS; about 5,000 new cases are diagnosed each year; and the disease affects three times as many men as women.
Causes
The exact cause of ALS is unknown, but 5% to 10% of cases have a genetic component—an autosomal dominant trait that affects men and women equally.
The first and most important breakthrough so far in ALS genetic research was the discovery of mutations in the SOD1 gene. This mutation is seen in about 20% of patients with familial ALS. However, this mutation is quite complex. Researchers understand how the gene functions, but the mutation is an acquisition of a toxic property, meaning that the gene gains a function, rather than losing its normal function. Researchers are perplexed about what that gain of function is, so genetic studies continue.
Several mechanisms have been considered, including:
♦ a slow-acting virus
♦ a nutritional deficiency related to a disturbance in enzyme metabolism
♦ a metabolic interference in nucleic acid production by the nerve fibers
♦ an autoimmune disorder that affects immune complexes in the renal glomerulus and basement membrane.
Precipitating factors for acute deterioration include any severe stress, such as a myocardial infarction, trauma, a viral infection, and physical exhaustion.
Pathophysiology
ALS progressively destroys the upper and lower motoneurons. It doesn’t affect cranial nerves III, IV, and VI and, therefore, some facial movements, such as blinking, persist. Intellectual and sensory functions aren’t affected.
Some believe that glutamate—the primary excitatory neurotransmitter of the CNS— accumulates to toxic levels at the synapses. The affected motor units are no longer innervated, and progressive degeneration of axons causes loss of myelin. Some nearby motor nerves may sprout axons in an attempt to maintain function but, ultimately, nonfunctional scar tissue replaces normal neuronal tissue.
Signs and symptoms
Typical signs and symptoms of ALS include:
♦ fasciculations accompanied by spasticity, atrophy, and weakness due to degeneration of the upper and lower motoneurons, and loss of functioning motor units, especially in the muscles of the forearms and the hands
♦ impaired speech, difficulty chewing and swallowing, choking, and excessive drooling from degeneration of cranial nerves V, IX, X, and XII
♦ difficulty breathing, especially if the brain stem is affected
♦ muscle atrophy due to loss of innervation.
Mental deterioration doesn’t usually occur, but patients may become depressed because of the disease. Progressive bulbar palsy may cause crying spells or inappropriate laughter.
Complications
♦ Respiratory tract infections
♦ Respiratory failure
♦ Aspiration
Diagnosis
Although no diagnostic tests are specific to ALS, the following may aid in the diagnosis:
♦ Electromyography shows abnormalities of electrical activity in involved muscles.
♦ Muscle biopsy shows atrophic fibers interspersed between normal fibers.
♦ Nerve conduction studies show normal results.
♦ Computed tomography scan and EEG show normal results and thus rule out multiple sclerosis, spinal cord neoplasm, polyarteritis, syringomyelia, myasthenia gravis, progressive muscular dystrophy, and progressive stroke.
Abnormal cellular structures in Alzheimer’s disease
How and why neurons die in Alzheimer’s disease is largely unknown. However, several characteristic abnormal cellular structures, which scientists believe cause cell malfunction or cell death, are found in the brain of every patient with Alzheimer’s disease. These structures include excessive granulovacuoles, neurofibrillary tangles, and amyloid plaques.
Granulovacuolar degeneration
Granulovacuolar degeneration is found inside the neurons of the hippocampus. An abnormally high number of fluid-filled spaces, called vacuoles, enlarge the cell’s body, possibly causing the cell to malfunction or die.
Neurofibrillary tangles
Neurofibrillary tangles are bundles of filaments inside the neuron that abnormally twist around one another. Numerous neurofibrillary tangles are found in areas of the brain associated with memory and learning (hippocampus), fear and aggression (amygdala), and thinking (cerebral cortex). Scientists believe the neurofibrillary tangles play a role in the memory loss and personality changes that the patient with Alzheimer’s disease suffers.
Amyloid plaques
Amyloid plaques (senile plaques) are found outside neurons in the extracellular space of the cerebral cortex and hippocampus. They contain a core of beta amyloid protein that’s surrounded by abnormal nerve endings, or neurites. Amyloid plaques also occur in the walls of cerebral blood vessels, causing the condition called amyloid angiopathy.
Neurotransmitters: The messengers
Alzheimer’s disease also destroys the way some neurons talk to each other. Neurons communicate via a chemical message that passes between two cells across a tiny gap called a synapse. A neuron receives messages from its dendrites and passes the information to the cell body, where the message is received. The message can then be sent to the end of the axon, where sacs containing chemicals called neurotransmitters are released. The sacs empty the neurotransmitter into the synapse between the two cells. The chemical message is picked up by the other cell and the process continues.
Groups of neurons that use the same neurotransmitter form specialized network systems. Several of these systems are damaged in Alzheimer’s disease. One in particular, the cholinergic system, uses the neurotransmitter acetylcholine to send its messages. The relay center for this system, the nucleus basalis of Meynert, suffers severe neuron loss and decreased production of acetylcholine. This is believed to play a role in memory loss.
Treatment
ALS has no cure. Treatment is supportive and may include:
♦ riluzole, a neuroprotector, to modulate glutamate activity and slow disease progression
♦ diazepam, dantrolene, or baclofen to decrease spasticity
♦ quinidine to relieve painful muscle cramps
♦ an anticholinergic, such as glycopyrolate or transdermal scopalamine, to decrease oral secretions
♦ thyrotropin-releasing hormone (I.V. or intrathecally) to temporarily improve motor function (successful only in some patients)
♦ mechanical ventilation to support pulmonary function
♦ respiratory, speech, and physical therapy to maintain function as much as possible
♦ psychological support to assist with coping with this progressive, fatal illness.
Special considerations
Remember that because mental status remains intact while progressive physical degeneration takes place, the patient acutely perceives every change. This threatens the patient’s relationships, career, income, muscle coordination, sexuality, and energy.
♦ Implement a rehabilitation program designed to maintain independence as long as possible.
♦ Elevate the head of the bed to relieve orthopnea.
♦ Help the patient obtain assistive equipment, such as a walker and a wheelchair. Arrange for a visiting nurse to oversee the patient’s status, provide support, and teach the family about the illness.
♦ Depending on the patient’s muscular capacity, assist with bathing, personal hygiene, and transfers from wheelchair to bed. Help establish a regular bowel and bladder routine.
♦ To help the patient handle an increased accumulation of secretions and dysphagia, teach him to suction himself. He should have a suction machine handy at home to reduce the fear of choking.
♦ To prevent skin breakdown, provide good skin care when the patient is bedridden. Turn him often, keep his skin clean and dry, and use pressure-reducing devices, such as an alternating air mattress.
♦ If the patient has trouble swallowing, give him soft, solid foods and position him upright during meals. Gastrostomy, percutaneous jejunostomy, or nasogastric tube feedings may be necessary if he can no longer swallow. Teach the family (or the patient if he’s still able to feed himself) how to administer gastrostomy feedings.
♦ Provide emotional support. A discussion of directives regarding health care decisions should be instituted before the patient becomes unable to communicate his wishes. Prepare the patient and family members for his eventual death, and encourage the start of the grieving process. Patients with ALS may benefit from a hospice program or the local ALS support group chapter.
ARTERIOVENOUS MALFORMATIONS
Arteriovenous malformations (AVMs) are tangled masses of thin-walled, dilated blood vessels between arteries and veins that don’t connect by capillaries. AVMs are common in the brain, primarily in the posterior portion of the cerebral hemispheres. Abnormal channels between the arterial and venous system mix oxygenated and unoxygenated blood and, thereby, prevent adequate perfusion of brain tissue.
AVMs range in size from a few millimeters to large malformations extending from the cerebral cortex to the ventricles. Commonly, more than one AVM is present. Males and females are affected equally, and some evidence exists that AVMs are hereditary. Most AVMs are present at birth; however, symptoms typically don’t occur until the person is between ages 10 and 20.
Causes
♦ Acquired from penetrating injuries such as trauma
♦ Congenital, due to a hereditary defect
Pathophysiology
AVMs lack the typical structural characteristics of the blood vessels. The vessels of an AVM are thin; one or more arteries feed into the AVM, causing it to appear dilated and tortuous. The typically high-pressured arterial flow moves into the venous system through the connecting channels to increase venous pressure, engorging and dilating the venous structures. An aneurysm may develop. If the AVM is large enough, the shunting can deprive the surrounding tissue of adequate blood flow. Additionally, the thin-walled vessels may ooze small amounts of blood or actually rupture, causing hemorrhage into the brain or subarachnoid space.
Signs and symptoms
Typically, the patient experiences few, if any, signs and symptoms unless the AVM is large, leaks, or ruptures. Possible signs and symptoms include:
♦ chronic mild headache and confusion from AVM dilation, vessel engorgement, and increased intracranial pressure (ICP)
♦ seizures secondary to compression of the surrounding tissues by the engorged vessels
♦ systolic bruit over carotid artery, mastoid process, or orbit, indicating turbulent blood flow
♦ focal neurologic deficits (depending on the location of the AVM) resulting from compression and diminished perfusion
♦ signs and symptoms of intracranial (intracerebral, subarachnoid, or subdural) hemorrhage, including sudden severe headache, seizures, confusion, lethargy, and meningeal irritation from bleeding into the brain tissue or subarachnoid space
♦ hydrocephalus from AVM extension into the ventricular lining.
Complications
Complications depend on the severity (location and size) of the AVM and may include:
♦ aneurysm development and subsequent rupture
♦ hemorrhage (intracerebral, subarachnoid, or subdural, depending on the location of the AVM)
♦ hydrocephalus.
Diagnosis
♦ Cerebral arteriogram confirms the presence of AVMs and evaluates blood flow.
♦ Doppler ultrasonography of cerebrovascular system indicates abnormal, turbulent blood flow.
Treatment
Treatment can be supportive, corrective, or both, and includes:
♦ support measures, including aneurysm precautions to prevent possible rupture
♦ surgery—block dissection, laser, or ligation— to repair the communicating channels and remove the feeding vessels
♦ embolization or stereotactic radiosurgery if surgery isn’t possible, to close the communicating channels and feeder vessels and thus reduce the blood flow to the AVM.
Special considerations
♦ Monitor vital signs frequently.
♦ Control hypertension, seizure activity, and other activity or stress that could elevate the patient’s systemic blood pressure by administering drug therapy as ordered, conducting ongoing neurologic assessments, and maintaining a quiet, therapeutic environment.
♦ If the AVM has ruptured, intervene to control elevated ICP and intracranial hemorrhage. The patient with a ruptured AVM may have intracerebral or intraventricular bleeding; bleeding into the subarachnoid, subdural, or epidural space; or bleeding into the brain itself, usually causing a concurrent elevation in ICP.
BELL’s PALSY
Bell’s palsy is a disease of the seventh cranial nerve (facial) that produces unilateral or bilateral facial weakness or paralysis, although bilateral involvement occurs in less than 10% of cases. The onset of symptoms is rapid. Although it affects all age-groups, Bell’s palsy occurs most commonly in people younger than age 60. In 80% to 90% of patients, it subsides spontaneously, with complete recovery in 1 to 8 weeks; however, recovery may be delayed in elderly patients. If recovery is partial, contractures may develop on the paralyzed side of the face. Bell’s palsy may recur on the same or opposite side of the face.
Causes
♦ Hemorrhage
♦ Infection
♦ Local trauma
♦ Meningitis
♦ Tumor
Pathophysiology
Bell’s palsy reflects an inflammatory reaction around the seventh cranial nerve, usually at the internal auditory meatus where the nerve leaves bony tissue. This inflammatory reaction produces a conduction block that inhibits appropriate neural stimulation to the muscle by the motor fibers of the facial nerve, resulting in the characteristic unilateral or bilateral facial weakness.
Signs and symptoms
The signs and symptoms exhibited by the patient typically reflect interference in motor function associated with the seventh cranial nerve. Bell’s palsy usually produces unilateral facial weakness, occasionally with aching pain around the angle of the jaw or behind the ear. On the weak side, the mouth droops (causing the patient to drool saliva from the corner of his mouth), and taste perception is distorted over the affected anterior portion of the tongue. The forehead appears smooth, and the patient’s ability to close his eye on the weak side is markedly impaired. When he tries to close this eye, it rolls upward (Bell’s phenomenon) and shows excessive tearing. Although Bell’s phenomenon also occurs in people who are otherwise healthy, it isn’t apparent because the eyelids close completely and cover this eye motion. In Bell’s palsy, incomplete eye closure makes this upward motion obvious. Other symptoms include loss of taste and ringing in the ear.
Complications
♦ Corneal abrasion
♦ Infection (masked by steroid use)
♦ Poor functional recovery
Diagnosis
Diagnosis is based on clinical presentation: distorted facial appearance and inability to raise the eyebrow, close the eyelid, smile, show the teeth, or puff out the cheek on the affected side. After 10 days, electromyography helps predict the level of expected recovery by distinguishing temporary conduction defects from a pathologic interruption of nerve fibers.
Treatment
Treatment consists of prednisone, an oral corticosteroid that reduces facial nerve edema and improves nerve conduction and blood flow. After the 14th day of prednisone therapy, electrotherapy may help prevent atrophy of facial muscles. Persistent paralysis may require surgical treatment.
Special considerations
Patient care includes observation for adverse reactions to prednisone, pain relief, and emotional support.
♦ During treatment with prednisone, watch for adverse reactions, especially GI distress and fluid retention. If GI distress is troublesome, a concomitant antacid usually provides relief. If the patient has diabetes, prednisone must be used with caution, which necessitates frequent monitoring of the serum glucose level.
♦ To reduce pain, apply moist heat to the affected side of the face, taking care not to burn the skin.
♦ To help maintain muscle tone, massage the patient’s face with a gentle upward motion two or three times daily for 5 to 10 minutes, or have him massage his face himself. When he’s ready for active exercises, teach him to exercise by grimacing in front of a mirror.
♦ Advise the patient to protect his eye by covering it with an eye patch, especially when outdoors and at night (when sleeping). Tell him to keep warm and avoid exposure to dust and wind. When exposure is unavoidable, instruct him to cover his face.
♦ To prevent excessive weight loss, help the patient cope with difficulty in eating and drinking. Instruct him to chew on the unaffected side of his mouth. Provide a soft, nutritionally balanced diet, eliminating hot foods and fluids. Arrange for privacy at mealtimes to reduce embarrassment. Give the patient frequent and complete mouth care, being particularly careful to remove residual food that collects between the cheeks and gums.
♦ Offer psychological support.
CEREBRAL PALSY
The most common cause of crippling in children, cerebral palsy (CP) is a group of neuromuscular disorders caused by prenatal, perinatal, or postnatal damage to the upper motoneurons. Although nonprogressive, these disorders may become more obvious as an affected infant grows.
The three major types of CP—spastic, athetoid, and ataxic—may occur alone or in combination. Motor impairment may be minimal (sometimes apparent only during physical activities, such as running) or severely disabling. Common associated defects are seizures, speech disorders, and mental retardation.
Cerebral palsy occurs in an estimated 2 to 3 per 1,000 live births in the United States every year. Incidence is highest in premature infants (anoxia plays the greatest role in contributing to cerebral palsy) and in those who are small for gestational age. Almost one-half of the children with CP are mentally retarded, approximately one-fourth have seizure disorders, and more than three-fourths have impaired speech. Additionally, many children with CP have dental abnormalities, vision and hearing defects, and reading disabilities.
CP is more common in whites than in other ethnicities. The prognosis varies. Treatment may make a near-normal life possible for children with mild impairment. Those with severe impairment require special services and schooling.
Causes
The exact cause of CP is unknown; however, conditions resulting in cerebral anoxia, hemorrhage, or other CNS damage are probably responsible. Potential causes vary with time of damage.
Prenatal causes include:
♦ abnormal placental attachment
♦ anoxia
♦ exposure to radiation
♦ isoimmunization
♦ malnutrition
♦ maternal diabetes
♦ maternal infection (especially rubella)
♦ toxemia.
Perinatal and birth factors may include:
♦ abruptio placentae
♦ breech presentation
♦ depressed maternal vital signs from general or spinal anesthesia
♦ forceps delivery
♦ infection or trauma during infancy
♦ multiple births (especially infants born last)
♦ placenta previa
♦ premature birth
♦ prolapsed cord with delay in blood delivery to the head
♦ prolonged or unusually rapid labor. Postnatal causes include:
♦ kernicterus resulting from erythroblastosis fetalis
♦ prolonged anoxia
♦ systemic disease resulting in cerebral thrombosis or embolus.
Pathophysiology
In the early stages of brain development, a lesion or abnormality causes structural and functional defects that in turn cause impaired motor function or cognition. Even though the defects are present at birth, problems may not be apparent until months later, when the axons have become myelinated and the basal ganglia are mature.
Signs and symptoms
Shortly after birth, the infant with CP may exhibit some typical signs and symptoms, including:
♦ excessive lethargy or irritability
♦ high-pitched cry
♦ poor head control
♦ weak sucking reflex.
Additional physical findings that may suggest CP results from impaired development or damage to the motor areas of the brain and may include:
♦ delayed motor development (inability to meet major developmental milestones)
♦ abnormal head circumference, typically smaller than normal for age (because the head grows as the brain grows)
♦ abnormal postures, such as straightening the legs when the patient is lying on his back, toes down; holding the head higher than normal when in a prone position due to arching of the back
♦ abnormal reflexes (neonatal reflexes lasting longer than expected, extreme reflexes, or clonus)
♦ abnormal muscle tone and performance (scooting on the back to crawl, toe-first walking).
Each type of CP typically produces a distinctive set of clinical features, although some children display a mixed form of the disease. (See Assessing signs of cerebral palsy,page 272.)
Complications
Complications depend on the type of CP and the severity of the involvement. Possible complications include:
♦ contractures
♦ skin breakdown and ulcer formation
♦ muscle atrophy
♦ malnutrition
♦ seizure disorders
♦ speech, hearing, and vision problems
♦ language and perceptual deficits
♦ mental retardation
♦ dental problems
♦ respiratory difficulties, including aspiration from poor gag and swallowing reflexes.
Diagnosis
No diagnostic tests are specific to CP. However, neurologic screening will exclude other possible conditions, such as infection, spina bifida, or muscular dystrophy. Diagnostic tests that may be performed include:
♦ Developmental screening reveals delay in achieving milestones.
♦ Vision and hearing screening demonstrates degree of impairment.
♦ EEG identifies the source of seizure activity.
Suspect CP whenever an infant exhibits an alteration in neurologic function during clinical observation. This may include difficulty in sucking or moving voluntarily. Infants particularly at risk include those with a low birth weight, low Apgar score at 5 minutes, seizures, and metabolic disturbances. However, all infants should have a screening test for CP as a regular part of their 6-month checkup.
Treatment
CP can’t be cured, but treatment can help affected children reach their full potential within the limits set by this disorder. Such treatment requires a comprehensive and cooperative effort, involving physicians, nurses, teachers, psychologists, the child’s family, and occupational, physical, and speech therapists. Home care is usually possible. Treatment typically includes:
♦ braces, casts, or splints and special appliances, such as adapted eating utensils and a low toilet seat with arms, to help the child perform activities of daily living independently
♦ an artificial urinary sphincter for the incontinent child
Assessing signs of cerebral palsy
Each type of cerebral palsy (CP) is manifested by specific findings. This chart highlights the major signs and symptoms associated with each type of CP. The manifestations reflect impaired upper motoneuron function and disruption of the normal stretch reflex.
Type of CP
Signs and symptoms
Spastic CP (due to impairment of the pyramidal tract [most common type])
♦ Hyperactive deep tendon reflexes
♦ Increased stretch reflexes
♦ Rapid alternating muscle contraction and relaxation
♦ Muscle weakness
♦ Underdevelopment of affected limbs
♦ Muscle contraction in response to manipulation
♦ Tendency toward contractures
♦ Typical walking on toes with a scissors gait, crossing one foot in front of the other
Athetoid CP (due to impairment of the extrapyramidal tract)
♦ Involuntary movements usually affecting arms more severely than legs, including:
– grimacing
– wormlike writhing
– dystonia
– sharp jerks
♦ Difficulty with speech due to involuntary facial movements
♦ Increasing severity of movements during stress; decreased with relaxation and disappearing entirely during sleep
Ataxic CP (due to impairment of the extrapyramidal tract)
♦ Disturbed balance
♦ Incoordination (especially of the arms)
♦ Hypoactive reflexes
♦ Nystagmus
♦ Muscle weakness
♦ Tremor
♦ Lack of leg movement during infancy
♦ Wide gait as the child begins to walk
♦ Sudden or fine movements impossible (due to ataxia)
Mixed CP
♦ Spasticity and athetoid movements
♦ Ataxic and athetoid movements (resulting in severe impairment)
♦ range-of-motion exercises to minimize contractures
♦ an anticonvulsant to control seizures
♦ a muscle relaxant (sometimes) to reduce spasticity
♦ surgery to decrease spasticity or correct contractures
♦ muscle transfer or tendon lengthening surgery to improve function of joints
♦ rehabilitation, including occupational, physical, and speech therapy, to maintain or improve functional abilities.
Special considerations
A child with CP may be hospitalized for orthopedic surgery or for treatment of other complications.
♦ Speak slowly and distinctly. Encourage the child to ask for items he wants. Listen patiently and don’t rush him.
♦ Plan a high-calorie diet that’s adequate to meet the child’s high energy needs.
♦ During meals, maintain a quiet, unhurried atmosphere with as few distractions as possible. The child should be encouraged to feed himself and may need special utensils and a chair with a solid footrest. Teach him to place food far back in his mouth to facilitate swallowing.
Only gold members can continue reading. Log In or Register to continue
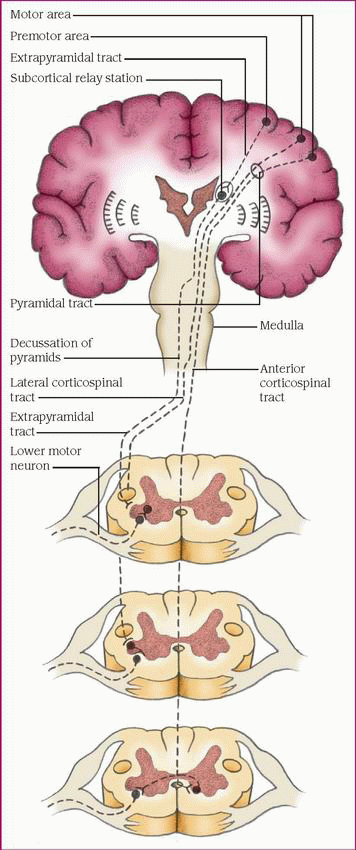
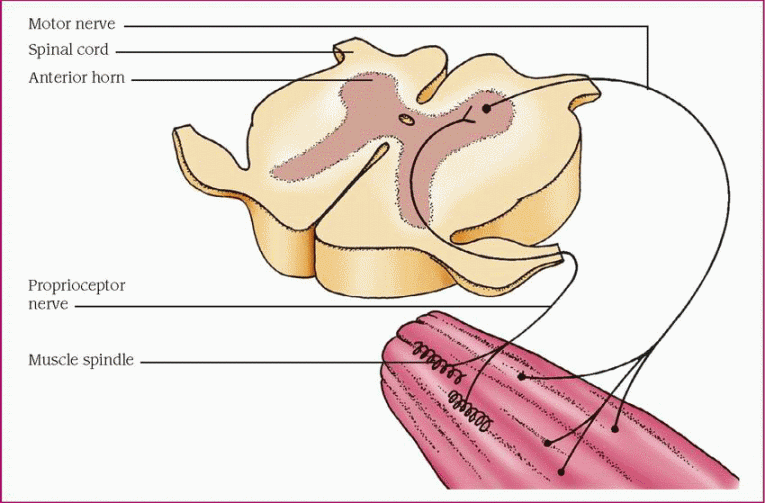
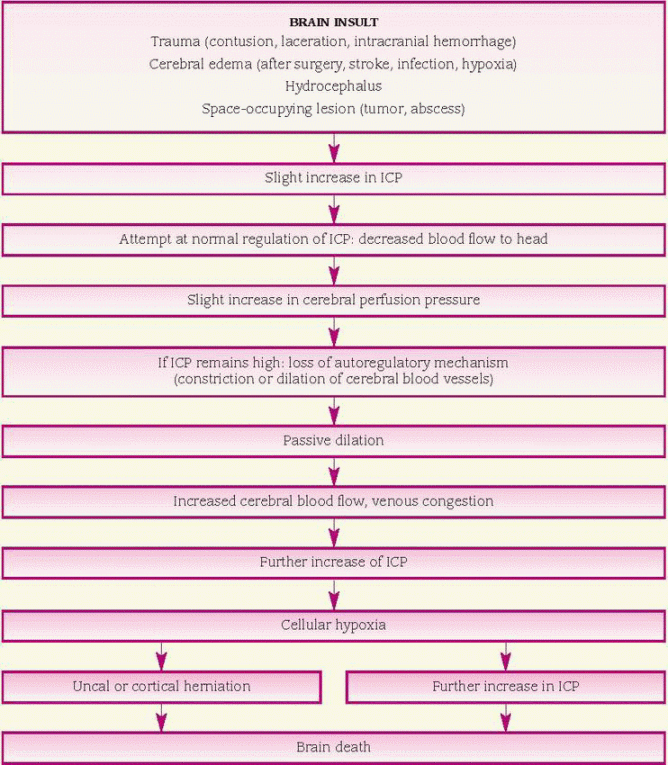
 Although primarily found in the elderly population, 1% to 10% of cases have their onset in middle age.
Although primarily found in the elderly population, 1% to 10% of cases have their onset in middle age. Genetic studies show that an autosomal dominant form of Alzheimer’s disease is associated with early onset and early death. A family history of Alzheimer’s disease and the presence of Down syndrome are two established risk factors.
Genetic studies show that an autosomal dominant form of Alzheimer’s disease is associated with early onset and early death. A family history of Alzheimer’s disease and the presence of Down syndrome are two established risk factors. The first and most important breakthrough so far in ALS genetic research was the discovery of mutations in the SOD1 gene. This mutation is seen in about 20% of patients with familial ALS. However, this mutation is quite complex. Researchers understand how the gene functions, but the mutation is an acquisition of a toxic property, meaning that the gene gains a function, rather than losing its normal function. Researchers are perplexed about what that gain of function is, so genetic studies continue.
The first and most important breakthrough so far in ALS genetic research was the discovery of mutations in the SOD1 gene. This mutation is seen in about 20% of patients with familial ALS. However, this mutation is quite complex. Researchers understand how the gene functions, but the mutation is an acquisition of a toxic property, meaning that the gene gains a function, rather than losing its normal function. Researchers are perplexed about what that gain of function is, so genetic studies continue.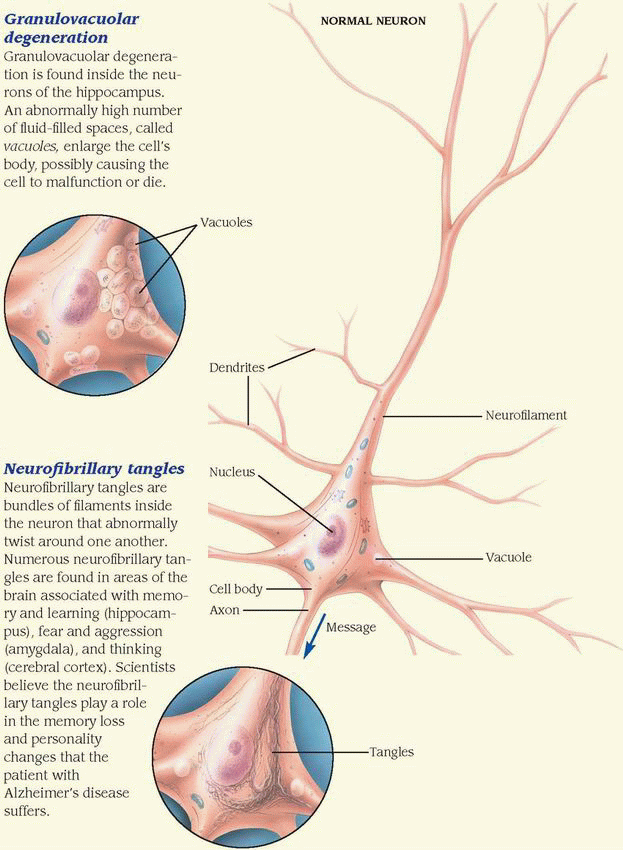
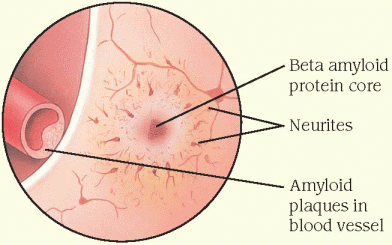
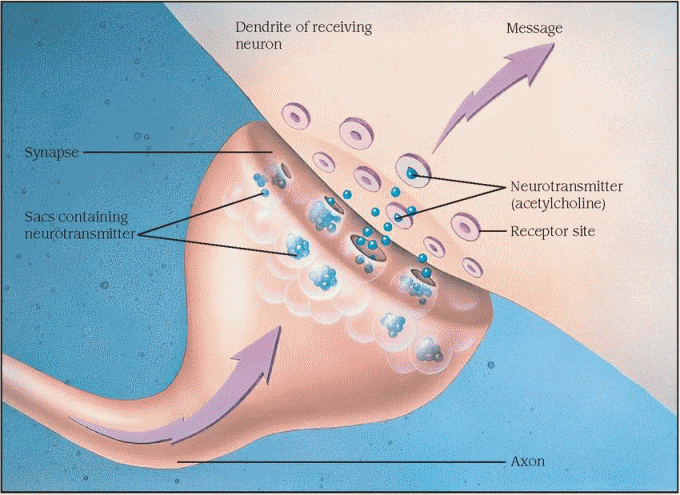
 Suspect CP whenever an infant exhibits an alteration in neurologic function during clinical observation. This may include difficulty in sucking or moving voluntarily. Infants particularly at risk include those with a low birth weight, low Apgar score at 5 minutes, seizures, and metabolic disturbances. However, all infants should have a screening test for CP as a regular part of their 6-month checkup.
Suspect CP whenever an infant exhibits an alteration in neurologic function during clinical observation. This may include difficulty in sucking or moving voluntarily. Infants particularly at risk include those with a low birth weight, low Apgar score at 5 minutes, seizures, and metabolic disturbances. However, all infants should have a screening test for CP as a regular part of their 6-month checkup.


