Introduction
Cell growth and maturation are normal events in organ development during embryogenesis, growth, and tissue repair and remodeling after injury. Disordered regulation of these processes can result in loss of control over cell growth, differentiation, and spatial confinement. Human neoplasia collectively represents a spectrum of diseases characterized by abnormal growth of cells resulting in distorted tissue architecture. Although cancers are typically classified by their tissues of origin or anatomic location, many features are shared by all types. There is also considerable variation among patients with a given type of cancer in the nature of cellular alterations as well as the clinical presentation and course of disease. The recognition of overt malignancy by physical examination or imaging requires the presence in the body of about 1 billion malignant cells. A preclinical phase may sometimes be recognized. Preclinical signs may consist of, among others, polyps in the colon or dysplastic nevi on the skin—potential precursors of colon carcinoma and malignant melanoma, respectively. Such precursor lesions usually harbor molecular genetic abnormalities and exhibit features of abnormal cell proliferation without the demonstration of invasiveness and may precede the development of an invasive malignancy by months to years, or may not progress to cancer within the individual’s lifetime. More commonly, the preclinical phase goes undetected until invasive cancer, occasionally with regional or distant metastases, is already present. As is the case with other medical disorders, our understanding of the pathophysiology of neoplasia has been based on clinical and pathologic observations of large series of patients. More recently, cellular and molecular features of cancer cells have been described, and their relationships to certain neoplastic entities and clinical situations have extended our knowledge in this field.
Checkpoint
What is the preclinical phase of cancer?
How many malignant cells must be present before overt signs of cancer are evident?
The Molecular & Biochemical Basis of Neoplasia
The process of neoplasia is a result of stepwise alterations in cellular function. These phenotypic changes confer proliferative, invasive, and metastatic potential that are the hallmarks of cancer. It is generally believed—although not conclusively proved—that genetic alterations underlie all cellular and biochemical aberrations responsible for the malignant phenotype. In addition to mutational changes that alter the genetic code, epigenetic changes also underlie cellular and biochemical aberrations that contribute to the malignant phenotype. Epigenetic phenomena influence gene expression and cell behavior, and although once acquired they are transmitted to daughter cells with cell division, they are not changes in the genetic code. An example of this is the silencing of certain genes by hypermethylation of DNA in the promoter region. An increasing number of genetic and cellular changes are being catalogued from the study of cancer cells, both in vivo, from primary tumors of patients, and in vitro, from established cancer cell lines grown in tissue culture. Some alterations are related to particular cellular phenotypes, such as a high proliferative rate or metastatic potential. Some of these changes are specific to a certain tumor type, whereas others are seen across different tumor types. In certain types of tumors, a particular genetic alteration is etiologically linked with, and is pathognomonic of, that cancer type and can play a significant role as a molecular marker of that disease and as a target for drug development. However, most types of cancers do not have unifying molecular characteristics. Although many of the most common types of cancers are categorized by their primary organ site, such as breast or prostate, this classification belies the heterogeneous nature of the cancers that can arise from that organ, and in actuality, what is currently called breast cancer is in fact a compilation of many diseases, previously difficult to categorize. Technological advances in high-throughput analysis of the entire cellular genome and of total cellular gene expression profiles have allowed the characterization of tumors by their molecular signatures. Ongoing studies are attempting to link molecular signatures with important predictive and prognostic clinical parameters. The technologies underlying the molecular analysis of tumors are rapidly evolving both in the depth and scale of the analysis and in their cost and speed. Although molecular profiling of cancers has already led to new classification systems for several types of cancer, these may soon become redundant in favor of a complete individualized molecular tumor characterization for every patient in real time at the time of presentation.
Although the progressive phenotypic characteristics of neoplasia result predominantly through sequential molecular alterations and abnormal function of the proliferating tumor cells, it is now clear that at some level abnormal function of the host stromal cells is fundamentally involved in continued tumor progression. It is not clear whether the abnormal function of stromal cells in tumor progression is due to genetic changes in these host cells or whether it is through cell-to-cell communications established through juxtacrine signaling loops with tumor cells. Stromal cell abnormalities can be nonproliferative, such as secretion of requisite growth factors, or proliferative, such as an expansion of the blood vessel network to support the growth of enlarging tumors or expansion of the extracellular matrix compartment.
Checkpoint
What stepwise phenotype changes are the hallmarks of cancer?
Maintaining genomic integrity is a fundamental cellular task. A complex cellular apparatus serves to recognize DNA damage or errors in replication, activate checkpoints to halt further cell replication, and implement corrective measures or signal suicidal cell death. One of the earliest phenomena observed in the course of tumor initiation is the development of defects in the genes involved in the machinery that guards the genome. This malfunction creates a degree of instability inherent in the genome that greatly increases the spontaneous rate at which genomic mutations or structural alterations occur and subsequently enables tumors to potentially acquire defects in an unlimited number of additional genes that may confer to them a growth advantage. Exposure to ionizing radiation and chemical carcinogens are environmental factors that can accelerate the accumulation of deleterious mutations. The cataloguing of these mutated genes has been a fundamental task of molecular oncology because it identifies genes whose functions are relevant to tumor cells. Genes that confer an advantage to tumor cells through a loss-of-function alteration are named tumor suppressor genes. Genes that confer an advantage through a gain-of-function event are named proto-oncogenes, and their activated counterparts are named oncogenes. Tumor suppressor genes are much more common because genes can be inactivated through a variety of types of mutations. These include nonsense mutations, frame-shift mutations, or deletion mutations, resulting in loss of the protein product of the gene. Alternatively, the gene can be silenced by promoter methylation or can be entirely or partially lost by deletion. Proto-oncogenes can be activated through mutation, gene amplification and overexpression, chromosomal translocation, and possibly other mechanisms. Examples of oncogenes and tumor suppressor genes are listed in Tables 5–1 and 5–2. In general, during the gain-of-function alteration of a proto-oncogene, only one allele is mutated. In contrast, during the loss-of-function alteration of a tumor suppressor gene both alleles need to be inactivated. In certain cases, loss of one allele can result in reduction of gene expression. For some genes, this gene-dosage reduction is sufficient to permit tumorigenic growth.
| Oncogene | Cellular Function | Tumor Types Activated | Mechanism of Activation |
|---|---|---|---|
| EGFR/HER1 | Growth factor receptor | Glioblastoma, lung and breast cancer | Mutation, amplification |
| HER2/Neu | Growth factor receptor | Breast, ovarian, gastric cancer | Amplification |
| PRAD1/Cyclin D1 | Cell cycle regulator | Breast and esophageal cancer, lymphoma, parathyroid adenoma | Amplification, translocation |
| K-Ras, N-Ras, H-Ras | G protein, signal transduction | Multiple tumor types | Mutation |
| B-Raf | Signal transduction | Multiple tumor types, melanomas | Mutation |
| Src | Adhesion and cytoskeletal signaling, other functions | Colon, breast, lung cancer, sarcoma, melanoma | Unknown, rarely mutated |
| Myc | Transcription factor | Multiple tumor types | Amplification, mutation |
| Myb | Transcription factor | Leukemia | Amplification, overexpression |
| Fos | Transcription factor | Multiple tumor types | Overexpression |
| Int2/FGF3 | Growth factor | Esophageal, gastric, head and neck cancers | Amplification |
| Fes/Fps | Signal transduction | Leukemia | Unknown |
| menin | Transcription factor | Pituitary, pancreas, parathyroid tumors | Mutation |
| Ret | Growth factor receptor | Parathyroid, medullary thyroid carcinoma, pheochromocytoma | Mutation |
| Tumor Suppressor Gene | Cellular Function | Tumor Types Inactivated | Mechanism of Inactivation | Hereditary Syndromes with a Germline Inactivated Allele |
|---|---|---|---|---|
| p53 | Cell cycle regulator | Multiple tumor types | Mutation | Li-Fraumeni |
| Rb | Cell cycle regulator | Retinoblastoma, small-cell lung cancer, sarcoma | Deletion, mutation | Familial retinoblastoma |
| APC | Cell adhesion | Colon cancer | Deletion, mutation | Familial adenomatous polyposis |
| PTEN | Signal transduction, adhesion signaling | Glioblastomas, prostate cancer, breast cancer | Deletion, mutation | Cowden |
| hMSH2 | DNA mismatch repair | Colon cancer, endometrial cancer, melanoma | Mutation | Hereditary nonpolyposis colon cancer (Lynch syndrome) |
| hMLH1 | DNA mismatch repair | Colon cancer, melanoma | Mutation | Hereditary nonpolyposis colon cancer (Lynch syndrome) |
| BRCA1 | DNA ds-break repair | Breast and ovarian cancers | Mutation | Familial breast/ovarian |
| BRCA2 | DNA ds-break repair | Breast and ovarian cancers | Mutation | Familial breast/ovarian |
| WT1 | Transcription factor | Wilms tumor | Deletion, mutation | Childhood Wilms tumor |
| NF1 | GTPase activator | Sarcoma, glioma | Deletion, mutation | Neurofibromatosis |
| NF2 | Cytoskeletal protein | Schwannoma | Mutation | Neurofibromatosis |
| VHL | Ubiquitin ligase | Kidney cancer, multiple tumor types | Mutation | Von Hippel-Lindau disease |
| p16/CDKN2 | Cell cycle regulator | Melanoma, pancreatic and esophageal cancers | Mutation, deletion, methylation | Familial melanoma |
In addition to being generated through the mutation of cellular proto-oncogenes, oncogenes can also be acquired through the introduction of foreign genomic material, typically transmitted by viruses. Although virally induced tumors are common in animals, only a few human tumors are directly caused by viral infection. Causative viruses and their associated malignancies are listed in Table 5–3. One such virus, human T-cell leukemia virus, is closely related to HIV and can cause a type of T-cell leukemia as a result of proteins encoded by the viral genome that are able to activate latent human genes. Human papillomavirus has long been linked epidemiologically to cervical cancer, and the serotypes most often linked have been found to encode proteins that can bind and inactivate host tumor suppressor gene products. The ability of viruses to modulate the host cellular machinery—and in some cases retain altered mammalian genes that are oncogenic—is likely to have developed over the course of mammalian evolution, because an actively proliferating cell provides the optimal conditions for replication of virions and propagation of viral infections.
| Virus Type | Virus Family | Associated Cancer Type |
|---|---|---|
| HTLV-I | Retrovirus (RNA virus) | T-cell leukemia/lymphoma |
| Hepatitis B | Hepadnavirus (hepatotropic DNA virus) | Hepatocellular carcinoma |
| Hepatitis C | Hepadnavirus | Hepatocellular carcinoma |
| Epstein-Barr | Herpesvirus (DNA virus) | Nasopharyngeal carcinoma |
| Burkitt lymphoma | ||
| Immunoblastic lymphoma | ||
| Hodgkin disease | ||
| HHV-8 (KSHV) | Herpesvirus | Kaposi sarcoma |
| Body cavity lymphoma | ||
| HPV serotypes 16, 18, 33, 39 | Papillomavirus (DNA virus) | Cervical carcinoma |
| Anal carcinoma | ||
| HPV serotypes 5, 8, 17 | Papillomavirus | Skin cancer |
The diploid human genome naturally contains defective alleles of many genes, and although defective alleles are for the most part biologically silent, in the case of tumor suppressor genes, a defective allele can confer significant cancer risk to an individual and all family members harboring such an allele. The loss of function of a gene in adult tissues is statistically much more probable when only one functional allele exists in all cells from the beginning of life, and inherited susceptibility to cancer is almost always a result of germline passage of a defective tumor suppressor gene allele. Many of the identified tumor suppressor genes that are frequently inactivated in sporadic human tumors have also been linked to specific hereditary cancer syndromes. In families with these syndromes, a defective allele of the responsible tumor suppressor gene is passed in the germline, and members who harbor this heterozygous genotype inherit a high risk for tumors in which the second allele has also been lost. An inherited mutation in one allele of the p53 gene can cause the rare Li-Fraumeni syndrome, characterized by the early development of bone, breast, brain, and soft tissue tumors (sarcomas) along with other organ-specific tumors (such as adrenal cancer). Inherited mutations in single alleles of the BRCA1 or BRCA2 genes confer a high risk for breast or ovarian cancers. The hereditary cancer syndromes linked with many tumor suppressor genes are listed in Table 5–2. The list includes predominantly high-penetrance genes, which confers a very high risk of disease when inherited, and families carrying these alleles are noticeable by their high incidences of cancers associated with these genes. In fact, cancer susceptibility syndromes have been defined clinically to describe such families, often well before the discovery of the genes underlying their pathogenesis. However, it is now well recognized that many human cancers, possibly a majority, are etiologically linked with moderate- or low-penetrance genes or possibly a combination of low-penetrance genes. Cancers arising from such genes do not cluster as tightly within families and therefore are not among the clinically defined susceptibility syndromes. Rather, they are being increasingly recognized by modern-day large-scale gene germline sequencing efforts.
In contrast to single alleles of defective tumor suppressor genes, single alleles of mutationally activated oncogenes are not biologically silent and, if present in the germline, can have profound clinical manifestations, even embryonic demise. Because of this fact, inherited syndromes related to germline transmission of activated oncogenes are much more uncommon. A rare example, however, is the familial syndrome of multiple endocrine neoplasia type II, in which heterozygotes carrying an activated RET oncogene on chromosome 10 are at increased risk of developing two rare neural crest tumors: pheochromocytoma and medullary carcinoma of the thyroid, together with parathyroid tumors.
Proteins encoded by proto-oncogenes and tumor suppressor genes perform diverse cellular functions. Not surprisingly, these include proteins that recognize and repair DNA damage, proteins that regulate the cell cycle, proteins that mediate growth factor signal transduction pathways and that regulate programmed cell death, and proteins involved in cell adhesion, proteolytic proteins, and transcription factors. The function of many proto-oncogenes and tumor suppressor genes remains unknown. Mutations that confer selective advantage to tumors are those that result in increased genomic instability, elimination of cell cycle checkpoints, inactivation of programmed cell death (apoptotic) pathways, increased growth factor signaling, decreased cell adhesion, and increased extracellular proteolysis. The expression and functions of many genes can be simultaneously affected through deregulation of transcription factors. With rapid advances in sequencing technologies and high-throughput capabilities to study normal and tumor genomes, aggressive efforts are in progress to identify all the tumor suppressor genes and proto-oncogenes in the human genome.
Tumor suppressor genes include proteins involved in DNA damage control, cell cycle control, programmed cell death, and cell adhesion. Examples include both the retinoblastoma protein and the p16 cell cycle inhibitor, which function in regulation of the G1 checkpoint of the cell cycle. Loss of these genes can result in unchecked progression through the G1/S checkpoint. The p53 tumor suppressor gene is a critical guardian of genomic integrity and serves to recognize DNA damage and consequently inhibit cell cycle progression and induce programmed cell death. Loss of p53 can result in continued cell replication despite DNA damage and failure to activate programmed cell death. The fundamental importance of p53 function and of genomic stability in the oncogenic process is underscored by the fact that p53 mutations are the most common mutations in human cancers and are seen in more than half of all human tumors. The PTEN tumor suppressor gene is a phosphatase involved in the regulation of an important survival signaling pathway. Loss of PTEN function can result in unopposed survival signaling and failure to activate programmed cell death. Cadherins are proteins involved in cell–cell adhesion. Loss of cadherins can result in reduced cell adhesion, cell detachment, and metastasis. Table 5–2 presents a small list of examples of tumor suppressor genes. When fully identified, the entire list of human tumor suppressor genes will be much larger.
Proto-oncogenes include proteins involved in various steps of the extracellular growth factor signaling pathway from the membrane receptors to the membrane intermediates to the proteins mediating the cytoplasmic signaling cascades. The epidermal growth factor receptor (EGFR) binds a number of extracellular ligands and, in cooperation with its homolog, HER2, signals proliferative and apoptotic pathways. Overactivity of EGFR or HER2 can lead to unregulated control of growth and apoptotic signaling. The gene for EGFR or HER1 is mutated or amplified in nearly half of all glioblastomas, is amplified in a fraction of breast cancers and other epithelial cancers, and is mutationally activated in a fraction of lung cancers. The HER2 gene is amplified in 20% of breast cancers and confers an aggressive phenotype. Ras is a membrane-bound signaling switch that functions immediately downstream of membrane receptors at a key branch point of cytoplasmic signaling. Mutational activation of Ras causes overactive cytoplasmic signaling and deregulation of proliferative and apoptotic pathways. Ras appears to be critically important in tumorigenesis because nearly one-third of all human tumors harbor mutationally activated Ras. Raf is a serine-threonine kinase that functions downstream of Ras. Mutational activation of Raf similarly can lead to overactive signaling and deregulation of proliferative and apoptotic pathways and is commonly seen in many tumors. Table 5–1 presents a partial list of oncogenes identified in human malignancies, along with the tumor types in which they are commonly observed and the cellular function encoded by their proto-oncogene counterparts.
Another pathway frequently activated in many human cancers is the PI3 kinase signaling pathway. This pathway controls many cellular processes required for malignant transformation, particularly because it functions to allow the cell to deal with and respond to stress. Activation of this pathway allows cells to adapt to and survive in conditions of low oxygen, low nutrients, and other environmental stresses and signals processes leading to increased protein synthesis, increased energy production, use of alternative metabolic pathways, cell survival, and cell proliferation. This pathway can be activated by upstream signals or can be activated within the pathway by the mutational activation of PI3K or its downstream signal Akt or by mutational inactivation of its negative regulator PTEN.
It is now clear that the inactivation of a single tumor suppressor gene or the activation of a single oncogene is insufficient for the development of most types of human tumors. In fact, the process entails the sequential acquisition of a number of hits over a period of time leading to sequential cellular phenotypic changes from atypia to dysplasia to hyperplasia to in situ cancer to invasive and subsequently metastatic cancer. The largest body of evidence to support this theory has been generated from the molecular study of colon cancer and identifiable preneoplastic lesions, including adenomas and colonic polyps. In this model, the progressive development of neoplasia from premalignant to malignant to invasive lesions is associated with an increasing number of genetic abnormalities, including both oncogene activation and tumor suppressor gene inactivation. This theory is further supported by the identification of inherited abnormalities of several tumor suppressor genes, all associated with a strong familial tendency to develop colon cancer at a young age.
Some forms of human cancer appear to be more simplistic in evolution. A translocation of the long arm of chromosome 9 to the long arm of chromosome 22 leads to fusion of the BCR gene with the c-Abl gene and results in expression of the BCR-Abl oncoprotein seen in chronic myelogenous leukemia (CML). The expression of this oncogene in hematopoietic cells in animal models reproduces the disease. This oncogenic event is seen in virtually 100% of cases of this disease, and a treatment that inhibits the kinase activity of this oncoprotein produces remissions in nearly 100% of the patients. Thus, in contrast to the multistep process involved in most types of carcinogenesis, the steps necessary for the development of CML may be much simpler.
The identification of tumor suppressor genes and oncogenes as the fundamental enablers of tumorigenesis has led to the hypothesis that cancer can be successfully treated by treatments that counteract the biochemical sequelae of these molecular abnormalities. This has fueled attempts to develop therapeutic agents that can inhibit the function of activated oncoproteins or that can restore the function of inactivated tumor suppressor proteins.
Although structurally altered genes, classified as oncogenes or tumor suppressor genes, are key mediators of neoplasia, the role of unaltered genes is not to be dismissed and is likely equally important in carcinogenesis. Signaling proteins of all kinds may drive the oncogenic process through abnormal signaling: abnormal in time, duration, or intensity; abnormal tissue expression; or abnormal subcellular compartment localization. The regulation of growth in complex organisms requires specialized proteins for the normal growth, maturation, development, and function of cells and specialized tissue. The complexity of the human organism requires that these proteins be expressed at precisely coordinated points in space and time. An essential component of this regulation is the system of hormones, growth factors, and growth inhibitors. On binding to specific receptor proteins on the cell surface or in the cytoplasm, these factors lead to a complex set of signals that can result in a variety of cellular effects, including mitogenesis, growth inhibition, changes in cell cycle regulation, apoptosis, differentiation, and induction of a secondary set of genes. The actual end effects are dependent not only on the particular type of interacting factor and receptor but also on the cell type and milieu in which factor–receptor coupling occurs. This system allows for cell-to-cell interactions, whereby a factor secreted by one cell or tissue can enter the bloodstream and influence another set of distant cells (endocrine action) or act on adjacent cells (paracrine action). An autocrine action is also possible when a cell produces a factor that binds to a receptor on or in the same cell. Altered concentration of these growth factors as well as overexpression or mutations of the receptors can change the signaling behavior, contributing to a malignant phenotype. Only a subset of growth factor receptors are proto-oncogenes. However, many additional growth factors and growth factor receptors appear to be important in tumor growth and progression, although not classified as proto-oncogenes, because they serve tumorigenic causes without incurring mutations or without overexpression.
An important class of growth factor signaling molecules are the growth factor receptor tyrosine kinases (RTKs). A number of tyrosine kinase receptor families exist, and in experimental models most are capable of transforming cells if activated or overexpressed. Although all of these abnormalities are not necessarily seen in naturally occurring human tumors, these experimental data highlight the potential inherent in these proteins and the important role they may be playing in tumor cells despite lacking the oncogene label. Members of the HER family of RTKs are commonly mutated or amplified in human tumors and exemplify the important role of RTKs in human neoplasia. In many other tumors, they likely play an important role despite having a normal sequence and expression level. For example, HER1 (also called EGFR) is not mutated or overexpressed in colon cancers, but it is sometimes activated by autocrine signaling in the cancer cells, and EGFR-targeted therapies are used to treat this type of cancer. The platelet-derived growth factor (PDGF) receptors, fibroblast growth factor receptors, vascular endothelial growth factor receptors, and insulin-like growth factor receptor are all families of RTKs that function similar to HER family RTKs. These receptors are, in general, not reported to be mutated or amplified in human tumors. However, there is increased expression in many tumors or aberrant expression in tumors from tissue types that ordinarily would not be expected to express that receptor. Alternatively, excessive production of receptor ligands is due to a variety of mechanisms (ie, loss of epigenetic silencing of the gene coding for the ligand or excessive gene transcription of the same gene). In experimental systems, each of these RTK systems has oncogenic potential, building a circumstantial case that they may be important players in human tumors.
Some growth factor signaling pathways function to inhibit cell growth and provide negative regulation in response to extracellular stimuli. Desensitization of cells to such growth inhibitors is common in tumors. An example of this is the transforming growth factor-β (TGF-β). TGF-β has diverse biological effects. It potently inhibits cell proliferation but also stimulates the production and deposition of extracellular matrix (ECM) and adhesion factors. These functions are important in tissue remodeling during embryogenesis and wound repair. In some tumor types, the antiproliferative response to TGF-β is lost early on because of mutations in its downstream signaling components. However, continued secretion, and often oversecretion, of TGF-β by the tumor and stromal tissues leads to an increase in the production of ECM and adhesion factors and promotes the invasive and metastatic property of tumors.
Another important class of receptors is the large superfamily of nuclear hormone receptors. These include the cellular receptors for a variety of hormones, among them estrogen and progesterone, androgens, glucocorticoids, thyroid hormone, and retinoids. The actions of estrogen are fundamentally important in the development of breast cancer. In women, oophorectomy early in life offers substantial protection against the development of breast cancer, and in animal models mammary carcinogenesis is significantly retarded in the absence of estrogen. Approximately half of all breast cancers are dependent on estrogen for proliferation. Although these data clearly implicate the estrogen signaling pathway in breast carcinogenesis, specific abnormalities of the estrogen receptor (ER) are not seen in breast cancers; therefore, the ER does not qualify as a tumor suppressor protein or an oncoprotein. It is possible that, although the loss of certain tumor suppressor genes or activation of certain oncogenes leads to the development of breast cancer, continued ER function is essential throughout this process and without ER function it cannot proceed. Alternatively, it is possible that abnormal ER signaling, perhaps as a result of altered cofactors, cross-talk, or phosphorylation status, drives breast carcinogenesis. Although the mechanism by which estrogen and its receptor drive breast cancers has not yet been determined, its fundamental role in this disease is well established. Furthermore, treatments that work through inhibiting the production of the active ligand or that inhibit the function of the ER are the most effective therapies for breast cancer yet developed and are highly active in the prevention and treatment of breast cancer. The androgen receptor (AR), similarly, plays a critical role in the development of prostate cancer, although occasional activating mutations and amplification of the AR have been reported in prostate cancers. On the contrary, the ability of retinoids (ligands for retinoic acid receptors) that are well known to participate in the differentiation of a variety of tissues during development to cause the differentiation of certain tumors in tissue culture models has been exploited as a treatment approach for acute promyelocytic leukemia (APL). APL is characterized by a t(15;17) chromosomal translocation resulting in the fusion of the PML gene with the retinoic acid receptor-α (RAR-α) gene. The resulting fusion protein blocks the differentiation of hematopoietic progenitor cells and eventually leads to the development of APL. This fusion protein is not by itself transforming in experimental models and cannot be categorized as a classic oncogene or tumor suppressor gene, but it is etiologically involved in the pathogenesis of APL. Because the fusion protein contains the ligand-binding domain of RAR-α, it remains sensitive to ligand and treatment of patients with the ligand all-trans retinoic acid results in differentiation of tumor cells and complete remission in most patients with this disease.
Other functional membrane proteins not related to growth can also be present on tumors cells. The MDR-1 gene product belongs to a class of ATP-dependent channel transporter proteins and is present on some normal epithelial cells. Its physiologic role may be to pump toxic molecules out of the cell, but in some tumor cells, its overexpression causes efflux of certain chemotherapeutic agents, leading to drug resistance. In some situations, its expression can be induced by long-term exposure to chemotherapy.
The preservation of tissue structure in multicellular organisms involves the orderly arrangement of cells within an architectural framework. This higher-level order is required to maintain tissue structure and organ function, and mechanisms are in place to enable remodeling during embryogenesis or during wound repair. A number of protein families serve to constitute the ECM, to embed cells within the ECM, to attach cells to each other, and to dissolve and reestablish the ECM when necessary. Abnormalities of these proteins frequently occur in later stages of tumorigenesis, account for the loss of architecture, and mediate the invasive and metastatic phenotype of tumor cells. Integrins are a large family of membrane proteins that bind ECM ligands, anchor cells to the ECM, and activate intracellular signaling pathways in response to ECM signals. Cells have the ability to express any of a large repertoire of integrin combinations and the specificity of integrin expression is not well understood. However, tumor cells can reshuffle their integrin expression profiles in favor of an invasive or metastatic phenotype. Cadherins are a family of membrane proteins that function in epithelial cell-to-cell adhesion. Loss of E-cadherin expression is seen in some human epithelial tumors leading to a more invasive phenotype. The expression and activity of many secreted and membrane-anchored proteases are increased in tumor cells. This includes the matrix metalloprotease family and the serine protease family of proteins. Increased protease activity leads to ECM degradation, triggering of the plasminogen activation cascade, and activation of transmembrane receptors through cleavage and shedding of their extracellular domains. Through abnormalities in deposition of ECM, of expression of cell adhesion proteins, and in activity of membrane and secreted proteases, cancer cells develop an invasive and ultimately a metastatic phenotype.
In addition to abnormalities in cell proliferation and survival, signal transduction, adhesion, and migration, tumor cells have changes in metabolic pathways in order to meet their increased metabolic requirements. Oxygen pressure is reduced in tumor tissues, and tumor hypoxia signals changes in gene expression for adaptation to the hypoxic environment. Tumor cells secrete angiogenic growth factors, which signal the proliferation of vascular structures into tumor tissue for nutrition and oxygenation. The identification of tumor factors that signal pathologic neovascularization has been of particular interest because such factors could be targets for therapeutic drug development and produce treatments that inhibit tumor angiogenesis. The best studied proangiogenic factor is the vascular endothelial growth factor (VEGF), a mitogen to endothelial cells that is often secreted by tumor cells and activates the VEGF receptors in endothelial cells, leading to de novo vascularization. Although most cells may not ordinarily express VEGF, malignant transformation often results in the induction of VEGF expression by tumor cells, either directly through the effects of oncogenes or the loss of tumor suppressor genes or indirectly as a result of hypoxia and the induction of hypoxia-induced gene transcription. Other growth factors also have proangiogenic effects, including epidermal growth factor, fibroblast growth factor, PDGF, transforming growth factor-α, and others.
The molecular changes of neoplastic cells and their phenotypic behavior is a constantly evolving process. Every cell division can result in additional genomic abnormalities and a variety of phenotypic consequences. Certain genotypes result in proliferative, survival, or other biological attributes that favor its clonal expansion. Such neoplastic clones eventually overtake the tumor cell population and change its clinical behavior. This remodeling process occurs repeatedly with repeated cell division, recreating a process akin to evolution, albeit in a much faster timeframe. Attributes that are acquired early during the evolution of cancer include enhanced proliferation and survival. Changes that are acquired midpoint include the ability to overcome spatial limitations by invading surrounding tissues, the ability to survive under conditions of low oxygen and nutrients, and the ability to evade host immune defenses. Changes acquired later in the progression of neoplasia are the ability to travel to distant organs and the ability to resist anticancer treatments.
The changing nature of cancer with repeated cell proliferation cycles along constantly expanding cell lineages creates heterogeneity in the whole tumor cell population. Tumor cell heterogeneity is a common characteristic of many types of cancer. Although many or most of the cells that arise from cancer cell division themselves proceed to multiply, the changes with repeated cycles of cell division often lead to the loss of some of the more fundamental properties of the ancestral cancer cells. For example, many of the cells in a tumor are unable to give rise to a new tumor if isolated. In fact, only a small proportion of cancer cells appear to be capable of starting new colonies of cancer cells if isolated or if metastasized to a new site in the body. Such cells, named cancer stem cells, typically do not proliferate as fast but are capable of self-renewal and of generating daughter cells that can proliferate much faster and produce new tumors.
Current efforts explore the hypotheses that 1) defects in normal tissue stem cells within an organ give rise to cancer stem cells (see the Hematologic Malignancy section for a more detailed discussion) and 2) terminally differentiated cells (non-stem cells) usurp the machinery used by normal stem cells in the process of becoming cancer stem cells.
Checkpoint
What is an oncogene?
What is a tumor suppressor gene?
What are the genetic mechanisms by which oncogenes can be activated or tumor suppressor genes inactivated?
Which is the more common mechanism of oncogene inactivation in humans: viral infection or somatic alteration?
What is a cancer stem cell?
What is the molecular basis for most inherited susceptibilities to certain cancers?
Name some factors that support or inhibit tumor growth but are not directly implicated in tumorigenesis.
What is the role of proteolytic enzymes in metastasis?
Give some examples of early, middle, and late changes in the progression of neoplasia.
Classification of Neoplasia
Neoplasia describes a large number of human diseases with extremely diverse characteristics. Therefore, the classification of neoplastic diseases into categories and subcategories is of great value in understanding them, diagnosing them, studying them, and developing treatments for them. Malignant transformation, by definition, results in abnormal cellular behavior. Tumor cells that have retained many of their specialized tissue functions and that are very similar appearing to their normal cellular counterparts are identified as well differentiated. Conversely, tumor cells that have lost much of their functions and that bear little similarity to their normal counterparts are identified as poorly differentiated. Poorly differentiated tumors are sometimes so abnormal that their cell or organ of origin cannot be recognized. However, although poorly differentiated tumor cells may have lost much of their specialized functions, their cellular ancestry can often still be recognized by more primitive characteristics.
The broadest classification of tumors relies on the most fundamental characterization of cell types based on their primitive embryologic origins. During early embryonic development, three cell lineages are established: ectoderm, endoderm, and mesoderm. All subsequent cells, including adult tumors, can be traced to one of these three cellular origins. As such, tumors are broadly classified into the categories of carcinoma if they originate from ectodermal or endodermal tissues or as sarcomas if they originate from mesodermal tissues. Even if completely unrecognizable by morphologic analysis, fundamental differences in the expression of certain proteins, especially intermediate filaments such as keratins and vimentin, will identify the lineage of origin.
Carcinomas are the most common cancer type and include all the common epithelial tissue cancers such as lung, colon, breast, and prostate cancers. Sarcomas arise from mesenchymal cell types, which are predominantly the connective tissues. Malignancies of blood cells, including leukemias and lymphomas, are technically a subtype of sarcomas because they are of mesenchymal origin. However, because of the highly specialized nature of hematologic cell types, they are generally grouped together and considered to be the entity of hematologic neoplasms, which includes leukemias and lymphomas. Further classification of carcinomas and sarcomas is based on the organ of origin. In the growing infant and child, mesenchymal tissues are very active in growth and remodeling, and mesenchymal tumors are common, including tumors of the muscle, cartilage, bone, and blood. In adults, the mesenchymal tissues are not very active, and epithelial tumors are by far the most common, including tumors of the lung, breast, prostate, and colon. Developments in gene expression profiling of tumors have enabled classification of tumors based on characteristic molecular portraits, and further work in this area may result in an entirely new classification of human tumors based on their gene expression profiles.
Epithelial cells are in constant turnover, arising from a basal layer that continually generates new cells. The mature and functional layer of cells performs specialized tissue or organ functions, and with senescence it is eventually sloughed off. Proliferating epithelial cells normally observe anatomic boundaries such as the basement membrane that underlies the basal layer of cells in the epithelium. The potential to divide, migrate, and differentiate is tightly controlled. The stimulus to divide may be autonomous or exogenous as a response to factors from adjacent or distant cells. Inhibitory signals and factors may also be present and serve to function as negative regulators to check uncontrolled growth. The neoplastic phenotype of epithelial cells can be seen as a spectrum from hyperplastic to preinvasive to frankly invasive and metastatic neoplasia, as illustrated in Figure 5–1. Because of their embryonic origins, malignancies of epithelial origin are termed carcinomas. Hyperplasia can be a normal physiologic response in some situations, such as that which occurs in the lining of the uterus in response to estrogens before the ovulatory phase of the menstrual cycle. It may also be a pathologic finding and associated with a predisposition to progress to invasive carcinoma. In such instances of hyperplasia, there are usually accompanying disorders of maturation that may be recognizable by microscopic examination. These changes are termed dysplasia, atypical hyperplasia, or metaplasia depending on the type of epithelium in which they are observed. More aggressive proliferation without the ability to invade through the basement membrane is termed preinvasive carcinoma, or carcinoma in situ. Technically, these cells do not have the capacity to invade the basement membrane and metastasize, although they may over time progress to invasive carcinoma. The term “invasive carcinoma” implies that tissue boundaries, especially the basement membrane, have been breached. Metastatic carcinoma occurs via the lymphatic system to regional lymph nodes and via the bloodstream to distant organs and other tissues. This pattern of metastasis, however, is not unique to epithelial malignancies. Epithelial neoplasms in general have a variable propensity to spread to regional nodes and distant sites. It is assumed that the natural history of most tumors is to follow this pattern of spread over time. The specific genotypic and phenotypic changes necessary to accomplish this spread are not well understood; they may, in some cases, be shared across tumor types, and in other cases they are unique to a given neoplasia. Certain molecular characteristics have been linked to clinical characteristics, although the exact mode of action is not fully understood.
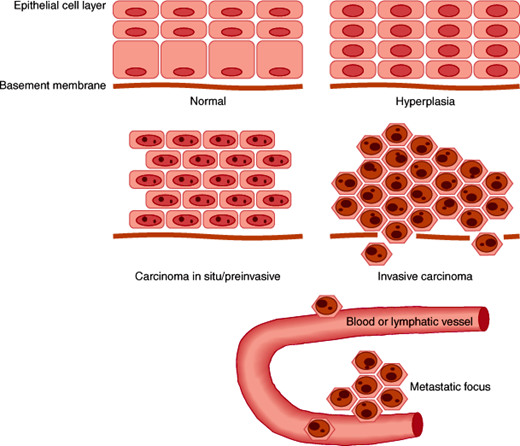
From a pathophysiologic standpoint, certain structural and functional characteristics must be acquired by malignant cells, as outlined in Table 5–4. An increase in growth rate through several mechanisms has been described for different tumor types. It is known that the proliferative fraction (the percentage of cells in S phase, or actively synthesizing DNA) is elevated, and more so in histologically and clinically aggressive tumors. Changes in the tightly regulated cell cycle machinery have been observed, including abnormal levels of cyclins and other proteins that regulate cyclin-dependent kinases responsible for entry of the cell into S phase. Likewise, alterations of intermediate signaling proteins have been noted that couple external growth factor and hormonal stimuli to proliferation. The ability of cells to migrate and pass through cellular and ECM barriers can be enhanced in tumor cells. This can occur through the activation of proteolytic enzyme cascades from within the tumor cell or by the action of stromal cells that are directed to do so as a result of factors produced by nearby tumor cells. Through similar mechanisms, malignant cells can induce the formation of a microvasculature that is essential to support the continued growth of a tumor colony. Other functions necessary to breach the immune defenses and survive destruction by antitumor drugs can be mediated by the genetic program already possessed in latent form by tumor cells. Examples include modulation of antigens and alterations in drug metabolism or metabolic pathways that are targeted by certain drugs.
|
As described earlier, there is evidence that discrete phenotypic changes that arise from specific genetic alterations account for the progression from hyperplasia to metastatic neoplasia. Moreover, there is an interplay between these genetic changes and the inherent program of gene expression of a given epithelial type. Other highly regulated functions of epithelial cells include active or passive transport of ions or molecules as well as synthesis and secretion of specific proteins. These functions may also be lost, altered, or even enhanced for specific tumor types and likewise can create specific pathophysiologic and clinical entities. Two epithelial neoplasms are discussed in further detail. Colon cancer is an example of an epithelial neoplasm for which precursor lesions have been well studied because we can seek out and biopsy such lesions by colonoscopy. Breast epithelial tissue is responsive to steroid hormones and growth factors that may play a role in the development and behavior of breast cancer.
Checkpoint
What factors determine the malignant potential of epithelial versus mesenchymal tumors?
What is the term applied to malignancies of epithelial origin?
What is the spectrum of characteristics of the neoplastic phenotype in epithelial cells?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


