Introduction
The adrenal gland is actually two endocrine organs, one wrapped around the other. The outer adrenal cortex secretes many different steroid hormones, including glucocorticoids such as cortisol, mineralocorticoids such as aldosterone, and androgens, chiefly dehydroepiandrosterone (DHEA). The glucocorticoids help to regulate carbohydrate, protein, and fat metabolism. The mineralocorticoids help to regulate Na+ and K+ balance and extracellular fluid volume. The glucocorticoids and mineralocorticoids are essential for survival, but no essential role of adrenal androgens has been determined. The inner adrenal medulla, discussed in Chapter 12, secretes catecholamines (epinephrine, norepinephrine, and dopamine).
Mainly because of their potent immunosuppressive and anti-inflammatory effects, glucocorticoids are used commonly in pharmacologic doses to treat diseases such as autoimmune disorders. Interestingly, while the deleterious effects of glucocorticoids in states of hypercortisolism and the beneficial effects of their use in pharmacotherapy are rather well understood, the actual role of endogenous glucocorticoids in metabolic homeostasis during times of minimal stress remains somewhat enigmatic.
The major disorders of the adrenal cortex (Table 21–1) are characterized by excessive or deficient secretion of each type of adrenocortical hormone: hypercortisolism (Cushing syndrome), adrenal insufficiency (Addison disease), hyperaldosteronism (aldosteronism), hypoaldosteronism, and androgen excess.
|
|
|
|
|
|
|
Normal Structure & Function of the Adrenal Cortex
The adrenal glands are paired organs located in the retroperitoneal area near the superior poles of the kidneys (Figure 21–1). They are flattened, crescent-shaped structures, which together normally weigh about 8–10 g. Each is covered by tight fibrous capsules and surrounded by fat. The blood flow to the adrenals is copious.
Figure 21–1
Human adrenal glands. Note location of adrenal at superior pole of each kidney. Adrenocortical tissue is stippled; adrenal medullary tissue is gray. Also shown (turquoise) are extra-adrenal sites at which cortical and medullary tissues are sometimes found. (Redrawn, with permission, from Forsham PH. The adrenal cortex. In: Williams RH, ed. Textbook of Endocrinology, 4th ed. Saunders, 1968.)

Grossly, each gland consists of two concentric layers: The yellow peripheral layer is the adrenal cortex, and the reddish brown central layer is the adrenal medulla. Adrenal cortical tissue is sometimes found at other sites, usually near the kidney or along the path taken by the gonads during their embryonic descent (Figure 21–1).
The adrenal cortex can be subdivided into three concentric layers: zona glomerulosa, zona fasciculata, and zona reticularis (Figure 21–2). The zona glomerulosa is the outermost layer, situated immediately beneath the capsule. Zona glomerulosa cells are columnar or pyramidal in appearance and are arranged in closely packed, rounded, or arched clusters surrounded by capillaries. They secrete mineralocorticoids, primarily aldosterone. The zona fasciculata is the middle layer of the cortex. Zona fasciculata cells are polyhedral in shape and arranged in straight cords or columns, one or two cells thick, running at right angles to the capsule with capillaries between them. The zona reticularis, the innermost layer of the cortex, lies between the zona fasciculata and the adrenal medulla, accounting for only 7% of the mass of the adrenal gland. Zona reticularis cells are smaller than the other two types and are arranged in irregular cords or interlaced in a network. Zona fasciculata and zona reticularis cells secrete both glucocorticoids, primarily cortisol and corticosterone, and androgens such as dehydroepiandrosterone. These steroid hormones are low-molecular-weight lipid-soluble molecules able to diffuse freely across cell membranes.
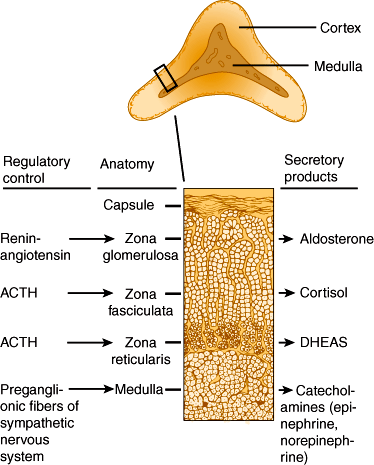
Cortisol and corticosterone are referred to as glucocorticoids because they increase hepatic glucose output by stimulating the catabolism of peripheral fat and protein to provide substrate for hepatic gluconeogenesis. The glucocorticoids help regulate the metabolism of carbohydrates, proteins, and fat. They act on virtually all cells of the body.
The major glucocorticoids secreted by the adrenal cortex are cortisol and corticosterone. Biosynthetic pathways for these hormones are illustrated in Figure 21–3.
Figure 21–3
A: Simplified pathways of steroid synthesis in the different zones of the adrenal cortex. Note the differences in the types of enzyme necessary and the different order of enzymatic reactions in the different zones. B: Enzymes involved in steroid synthesis. Four of the five enzymes involved are cytochrome P450s and the P450s are commonly known by their cytochrome (CYP) numbers, as shown.

Both cortisol and corticosterone are secreted in an unbound state but circulate bound to plasma proteins. They bind mainly to corticosteroid-binding globulin (CBG) (or transcortin) and to a lesser extent to albumin. Protein binding serves mainly to distribute and deliver the hormones to target tissues, but it also delays their metabolic clearance and prevents marked fluctuations of glucocorticoid levels during episodic secretion by the gland.
CBG (molecular weight ~50,000) is an α-globulin synthesized in the liver. Its production is increased by pregnancy, estrogen or oral contraceptive therapy, hyperthyroidism, diabetes mellitus, certain hematologic disorders, and familial CBG excess. When the CBG level rises, more cortisol is bound, and the free cortisol level falls temporarily. This fall stimulates pituitary adrenocorticotropic hormone (ACTH) secretion and more adrenal cortisol production. Eventually, the free cortisol level and the ACTH secretion return to normal but with an elevated level of protein-bound cortisol. Similarly, when the CBG level falls, the free cortisol level rises. CBG production is decreased in cirrhosis, nephrotic syndrome, hypothyroidism, multiple myeloma, and familial CBG deficiency.
Normally, about 96% of the circulating cortisol is bound to CBG and 4% is free (unbound). The bound hormone is inactive. The free hormone is physiologically active. The normal morning total plasma cortisol level is 5–20 μg/dL (140–550 nmol/L). Because cortisol is protein bound to a greater degree than corticosterone, its half-life in the circulation is longer (~60–90 minutes) than that of corticosterone (~50 minutes).
Glucocorticoids are metabolized in the liver and conjugated to glucuronide or sulfate groups. The inactive conjugated metabolites are excreted in the urine and stool. The metabolism of cortisol is decreased in infancy, old age, pregnancy, chronic liver disease, hypothyroidism, anorexia nervosa, surgery, starvation, and other major physiologic stress. Catabolism of cortisol is increased in thyrotoxicosis. Because of its avid protein binding and extensive metabolism before excretion, less than 1% of secreted cortisol appears in the urine as free cortisol.
Glucocorticoid secretion is regulated primarily by ACTH, a 39-amino-acid polypeptide secreted by the anterior pituitary. Its half-life in the circulation is very short (~10 minutes). The site of its catabolism is unknown. ACTH regulates both basal secretion of glucocorticoids and increased secretion provoked by stress.
ACTH, in turn, is regulated by hypothalamic corticotropin-releasing hormone (CRH), a 41-amino-acid polypeptide secreted into the median eminence of the hypothalamus. CRH secretion by the hypothalamus is regulated by a variety of neurotransmitters (Figure 21–4) in response to physical and emotional stressors. The hypothalamus is subject to regulatory influences from other parts of the brain, including the limbic system. CRH is transported in the portal-hypophysial vessels to the anterior pituitary (see Chapter 19). There, CRH causes a prompt increase in ACTH secretion. This, in turn, leads to a transient increase in cortisol secretion by the adrenal. Arginine-vasopressin (AVP) is an additional hypothalamic peptide that regulates ACTH release.
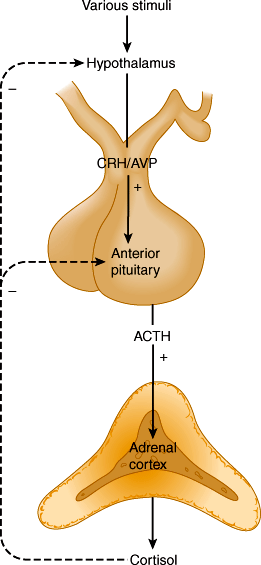
The control of ACTH and CRH/AVP secretion involves three components: episodic secretion and diurnal rhythm of ACTH, stress responses of the hypothalamic-pituitary-adrenal axis, and negative feedback inhibition of ACTH secretion by cortisol.
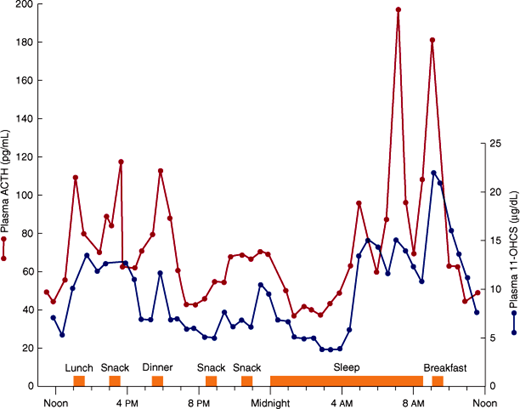
ACTH is secreted in episodic bursts throughout the day, after a diurnal (circadian) rhythm, with bursts most frequent in the early morning and least frequent in the evening (Figure 21–5). The peak level of cortisol in the plasma normally occurs between 6:00 and 8:00 AM (during sleep, just before awakening) and the nadir at around 12:00 AM. The diurnal rhythm of ACTH secretion persists in patients with adrenal insufficiency who are receiving maintenance doses of glucocorticoids but is lost in Cushing syndrome. The diurnal rhythm is altered also by changes in patterns of sleep (eg, shift work), light-dark exposure, or food intake; physical stress such as major illness, surgery, trauma, or starvation; psychologic stress, including severe anxiety, depression, and mania; CNS and pituitary disorders; liver disease and other conditions that affect cortisol metabolism; chronic kidney disease; alcoholism; and antiserotonergic drugs such as cyproheptadine.
Figure 21–5
Fluctuations in plasma ACTH and glucocorticoids (11-OHCS) throughout the day. Note the greater ACTH and glucocorticoid rises in the morning before awakening. (Redrawn, with permission, from Krieger DT et al. Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab. 1971;32:266.)
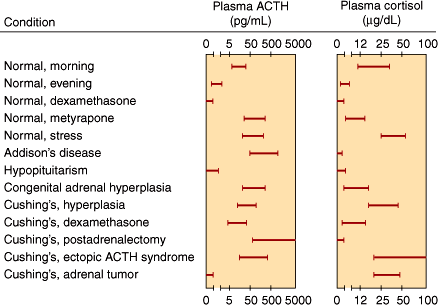
Normally, the morning plasma ACTH concentration is about 25 pg/mL (5.5 pmol/L). Plasma ACTH and cortisol values in various normal and abnormal states are shown in Figure 21–6.
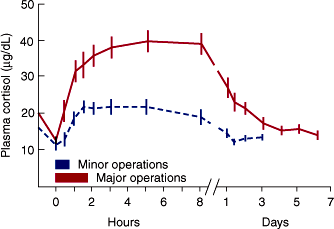
Plasma ACTH and cortisol secretion are also triggered by various forms of stress. Emotional stress (such as fear and anxiety) and bodily injury (such as surgery or hypoglycemia) release CRH from the hypothalamus. Similarly, vasopressin is released in response to volume depletion. ACTH secretion induced by these hormones, in turn, stimulates a transient increase in cortisol secretion (Figure 21–7). If the stress is prolonged, it may abolish the normal diurnal rhythm of ACTH and cortisol secretion.
A rising level of plasma cortisol inhibits release of ACTH from the pituitary by both inhibiting CRH release from the hypothalamus and interfering with the stimulatory action of CRH on the pituitary (Figure 21–4). The fall in plasma ACTH leads to a decline in adrenal secretion of cortisol. Conversely, the loss of negative feedback resulting from a drop in plasma cortisol induces a net increase in ACTH secretion. In untreated chronic adrenal insufficiency, there is a marked increase in the rate of ACTH synthesis and secretion.
ACTH and CRH secretion are also inhibited by chronic pharmacologic treatment with exogenous corticosteroids in proportion to their glucocorticoid potency. When prolonged corticosteroid treatment is stopped, the adrenal is atrophic and unresponsive and the patient is at risk for acute adrenal insufficiency. Chronic suppression of the HPA axis by exogenous glucocorticoids also impacts on hypothalamic CRH and pituitary ACTH secretion and it may take some time to recover after cessation of glucocorticoid treatment. Such adrenal insufficiency after abrupt glucocorticoid withdrawal can be life threatening. The time to recovery to full physiological function of the HPA axis is dependent on duration and dose of glucocorticoid treatment. Moreover, there are significant interindividual differences in these parameters. While there are no useful predictors to facilitate determining which patients are at risk for prolonged adrenal insufficiency, there is some evidence that alternate-day glucocorticoid treatment tends to preserve some adrenal function. Another well-accepted method of preventing long-term suppression of the HPA axis following glucocorticoid therapy is to slowly taper the dosage of exogenous glucocorticoids. Tapering exogenous glucocorticoid has a dual function. A short-term taper (days to a few weeks) of pharmacologic doses of glucocorticoids prevents a rebound flare of the underlying treated disease (eg, autoimmune disorder). A slow taper of exogenous glucocorticoid from physiologic replacement doses to complete discontinuation serves the purpose of allowing the endogenous HPA axis to recover. Such tapering only supports the recovery of HPA-axis function if it is done slowly (weeks to months) with doses below the daily physiologic glucocorticoid equivalent (eg, 5.0–7.5 mg of prednisone).
Circulating ACTH binds to high-affinity receptors (ACTH receptor or melanocortin MC2 receptor) on adrenocortical cell membranes, activating adenylyl cyclase, increasing intracellular cyclic adenosine monophosphate (cAMP). There is a dual response to ACTH stimulation: a) immediate production and release of cortisol, and b) induction of steroidogenic enzyme synthesis.
Prolonged hypersecretion or administration of ACTH causes initial hypertrophy followed by hyperplasia of the zona fasciculata and zona reticularis. Growth factors such as additional POMC peptides and insulin-like growth factors play important roles in this process. Conversely, prolonged ACTH deficiency results in adrenocortical atrophy.
The physiologic effects of glucocorticoids in various tissues are the result of their binding to the ubiquitous cytosolic glucocorticoid receptors (GRs) (Figure 21–8). The hormone-GR complexes then enter the nucleus and can act by two main mechanisms: a) transactivation, in which the GRs bind to nuclear DNA and promote the transcription of DNA, production of mRNAs, and hence synthesis of proteins; or b) transrepression, in which gene transcription is inhibited through interference with other transcription factors.
Figure 21–8
Mechanism of glucocorticoid action. Glucocorticoid (GC) hormone binds to the cytosolic intracellular glucocorticoid receptor (GR), which dimerizes and then translocates to the nucleus and increases transcription of glucocorticoid-responsive target genes (eg, PEPCK, transactivation) or inhibits gene transcription of genes (eg, collagenase, interleukin-2, transrepression) by interference with other transcription factors (eg, nuclear factor kappa-B [NFκB] or activator protein 1 [AP1]). (Arrows depict gene transcription, crossed-out arrows depict inhibited gene transcription; RE, response element.)

The effects of glucocorticoids on target tissues are summarized in Table 21–2. Under physiologic circumstances, the effects of glucocorticoid are not very well understood but appear to be mainly permissive. The effects of glucocorticoids secreted at supraphysiologic levels, however, are well described. In most tissues, glucocorticoids have a catabolic effect, promoting degradation of protein and fat to provide substrate for intermediary metabolism. In the liver, however, glucocorticoids have a synthetic effect, promoting the uptake and use of carbohydrates (in synthesis of glucose and glycogen), amino acids (in synthesis of RNA and protein enzymes), and fatty acids (as an energy source).
| Target Tissue | Effect | Mechanism |
|---|---|---|
| Muscle |
|
|
| Fat |
|
|
| Liver |
|
|
| Immune system |
|
|
|
| |
|
| |
| ||
| Cardiovascular |
| |
| Renal |
|
During fasting, glucocorticoids help to maintain plasma glucose levels by several mechanisms (Table 21–2). In peripheral tissues, glucocorticoids antagonize the effects of insulin. Glucocorticoids inhibit glucose uptake in muscle and adipose tissue. The brain and heart are spared from this antagonism, and the extra supply of glucose helps these vital organs to cope with stress. In diabetics, the insulin antagonism may worsen control of blood sugar levels, raise plasma lipid levels, and increase the formation of ketone bodies. However, in nondiabetics, the rise in blood glucose levels stimulates a compensatory increase in insulin secretion that prevents these sequelae.
Small amounts of glucocorticoids must be present for other metabolic processes to occur (permissive action). For example, glucocorticoids must be present for catecholamines to produce their calorigenic, lipolytic, pressor, and bronchodilator effects and for glucagon to increase hepatic gluconeogenesis.
Glucocorticoids are also required to resist various stresses. Indeed, the increased secretion of pituitary ACTH and consequent increase in circulating glucocorticoids after injury are essential to survival. Hypophysectomized or adrenalectomized individuals treated with only maintenance doses of glucocorticoids may die when exposed to such stress. This underscores the crucial role of glucocorticoids as stress hormones.
Checkpoint
What are the histologic layers of the adrenal cortex, and what steroids does each secrete?
What three roles are proposed for steroid-binding proteins?
In what conditions is corticosteroid-binding globulin increased? Decreased?
In what conditions is cortisol metabolism increased? Decreased?
Describe the diurnal rhythm of ACTH secretion, and name the conditions in which it is altered.
What stress responses trigger ACTH secretion?
Describe the negative feedback control of the hypothalamic-pituitary-adrenal axis.
Describe the major physiologic effects of glucocorticoids.
The primary function of mineralocorticoids is to regulate Na+ excretion and maintain a normal intra-vascular volume. However, other factors affect Na+ excretion besides the mineralocorticoids, such as the glomerular filtration rate, atrial natriuretic peptide, presence of an osmotic diuretic, and changes in tubular reabsorption of Na+ that are not regulated by mineralocorticoid.
Aldosterone is the principal mineralocorticoid secreted by the adrenal. Deoxycorticosterone also has minor mineralocorticoid activity, as does corticosterone.
Aldosterone is bound to plasma proteins (albumin and corticosteroid-binding globulin) to a lesser extent than glucocorticoids. The amount of aldosterone secreted under normal circumstances is small (~0.15 mg/24 h). The normal average plasma concentration of (free and bound) aldosterone is 0.006 μg/dL (0.17 nmol/L). Free (unbound) aldosterone comprises 30–40% of the total.
The half-life of aldosterone is short (~20– 30 minutes). Aldosterone is catabolized principally in the liver, and its metabolites are excreted in the urine. Less than 1% of secreted aldosterone is excreted in urine in the free form.
Aldosterone secretion is regulated primarily by the renin-angiotensin system but also by pituitary ACTH and by the plasma electrolytes, K+ and, to a lesser extent, Na+.
The renin-angiotensin system regulates aldosterone secretion in a feedback fashion (Figure 21–9). Renin is a proteolytic enzyme produced from a larger protein, prorenin. Renin is excreted by the juxtaglomerular cells of the kidney in response to decreases in renal perfusion pressure and reflex increases in renal nerve discharge. Once in the circulation, renin acts on angiotensinogen, to form angiotensin I, a decapeptide. In the lung and elsewhere, angiotensin I is converted by angiotensin-converting enzyme (ACE) to angiotensin II, an octapeptide. Angiotensin II binds to zona glomerulosa cell membrane receptors and stimulates synthesis and secretion of aldosterone. Aldosterone promotes Na+ and water retention, causing plasma volume expansion, which then shuts off renin secretion. In the supine state, there is a diurnal rhythm of aldosterone and renin secretion; the highest values are in the early morning before awakening.
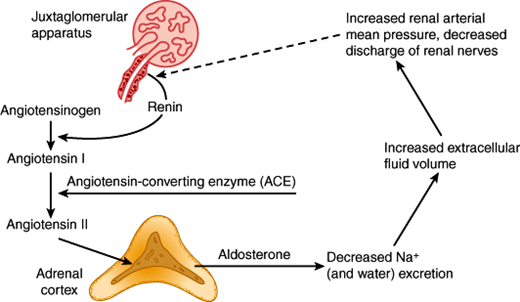
The physiologic stimuli for the renin-angiotensin system to increase aldosterone secretion include factors that reduce renal perfusion such as extracellular fluid volume depletion, dietary Na+ restriction, and decreases in intra-arterial vascular pressure (eg, resulting from hemorrhage or upright posture). Other disease states that cause reduced renal perfusion include renal artery stenosis, salt-losing disorders, heart failure, and hypoproteinemic states (cirrhosis of the liver, or nephrotic syndrome). These disorders increase renin secretion, producing secondary hyperaldosteronism.
ACTH also stimulates mineralocorticoid output. More ACTH is needed to stimulate mineralocorticoid than glucocorticoid secretion, but the amount required is still within the range of normal ACTH secretion. The effect of ACTH on aldosterone secretion is transient, however. Even if ACTH secretion remains elevated, aldosterone production declines to normal within 48 hours, perhaps because renin secretion decreases in response to hypervolemia.
An increase in plasma K+ concentration—or a fall in plasma Na+—stimulates aldosterone release. Although minor changes of plasma K+ (≤1 mEq/L) have an effect, major changes in plasma Na+ (drops of about 20 mEq/L) are needed to stimulate aldosterone secretion. Na+ depletion increases the affinity and number of angiotensin II receptors on adrenocortical cells.
Aldosterone, like other steroid hormones, acts by binding to a mineralocorticoid receptor (MR) in the cytosol. The expression of the MR is restricted to a small number of tissues, such as the kidney. Interestingly, glucocorticoids also have a high affinity to the MR, but usually do not exert mineralocorticoid effects because mineralocorticoid-sensitive tissues express the enzyme 11-hydroxysteroid dehydrogenase type 2, which metabolizes and inactivates glucocorticoids before it can bind to the MR. The aldosterone-MR complex moves into the nucleus of the target cell and increases transcription of DNA, induction of mRNA, and stimulation of protein synthesis by ribosomes. The aldosterone-stimulated proteins have two effects: a rapid effect to increase the activity of epithelial sodium channels (ENaCs) by increasing the insertion of ENaCs into the cell membrane from a cytosolic pool, and a slower effect to increase the synthesis of ENaCs. One of the genes activated by aldosterone is the gene for serum- and glucocorticoid-regulated kinase (sgk), a serine-threonine protein kinase. The sgk gene product increases ENaC activity (Figure 21–10). Aldosterone also increases the mRNAs for the three subunits that comprise the ENaCs.
Figure 21–10
Mechanism of action of aldosterone in an epithelial cell of the renal tubule’s collecting duct. In the kidney, aldosterone acts primarily on the principal cell of the collecting ducts. Under the influence of aldosterone, increased amounts of Na+ are exchanged for K+ and H+ in the renal tubules, producing a K+ diuresis and an increase in urine acidity. Na+ enters via the epithelial sodium channels (ENaCs) in the apical membrane and is pumped into the interstitial fluid by Na+-K+ ATPases in the basolateral membrane. Aldosterone activates the genome to produce sgk and other proteins, and the number of active ENaCs is increased. (Redrawn and modified, with permission, from Ganong WF. Review of Medical Physiology, 22nd ed. McGraw-Hill, 2005.)
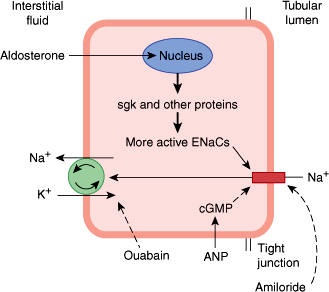
The fact that the principal effect of aldosterone on Na+ transport takes 10–30 minutes to develop and even longer to peak indicates that it depends on the synthesis of new proteins by the genomic mechanism. However, aldosterone also binds directly to distinct membrane receptors with a high affinity for aldosterone, and, by a rapid nongenomic action, increases the activity of membrane Na+-K+ exchangers to increase intracellular Na+.
The target organs for the mineralocorticoids include the kidney, colon, duodenum, salivary glands, and sweat glands. In the distal renal tubules and collecting ducts, aldosterone acts to promote the exchange of Na+ for K+ and H+, causing Na+ retention, K+ diuresis, and increased urine acidity. Elsewhere, it acts to increase the reabsorption of Na+ from the colonic fluid, saliva, and sweat. The mineralocorticoids may also increase K+ and decrease Na+ concentrations in muscle and brain cells. Aldosterone action on epithelial cells of the choroid plexus alters the composition of cerebrospinal fluid in a fashion thought to contribute to blood-pressure regulation. In the heart, aldosterone has been shown to induce heart remodeling and interstitial and perivascular fibrosis of the myocardium.
Checkpoint
How is aldosterone secretion regulated?
How does the effect of ACTH on aldosterone secretion differ from the effect on glucocorticoid secretion?
What are the overall effects of aldosterone?
Pathophysiology of Selected Adrenocortical Disorders
Characteristic syndromes are produced by excessive or deficient secretion of each type of adrenal hormone. Excessive glucocorticoid secretion (Cushing syndrome) results in a moon-faced, plethoric appearance, with truncal obesity, purple abdominal striae, hypertension, osteoporosis, mental aberrations, protein depletion, and glucose intolerance or frank diabetes mellitus.
Excessive mineralocorticoid secretion in hyperaldosteronism leads to Na+ retention, usually without edema, and K+ depletion, resulting in hypertension, muscle weakness, polyuria, hypokalemia, metabolic alkalosis, and sometimes hypocalcemia and tetany.
Excessive androgen secretion causes virilization or hirsutism and precocious pseudopuberty or a disorder of sexual development (46,XX DSD [disorder of sexual development], formerly known as female pseudohermaphroditism).
Deficient glucocorticoid secretion resulting from autoimmune or other destruction of the adrenal glands (Addison disease) causes symptoms of weakness, fatigue, malaise, anorexia, nausea and vomiting, weight loss, hypotension, hypoglycemia, and marked intolerance of physiologic stress (eg, infection). Elevation of plasma ACTH may produce hyperpigmentation.
Associated mineralocorticoid deficiency leads to renal Na+ wasting and K+ retention and can produce manifestations of severe dehydration, hypotension, decreased cardiac size, hyponatremia, hyperkalemia, and metabolic acidosis. Deficient mineralocorticoid secretion also occurs in patients with renal disease and low circulating renin levels (hyporeninemic hypoaldosteronism).
Cushing syndrome is the clinical condition resulting from chronic exposure to excessive circulating levels of glucocorticoids (Figure 21–11). It is also called hypercortisolism. The most common cause of the syndrome is excess secretion of ACTH from the anterior pituitary gland (Cushing disease).
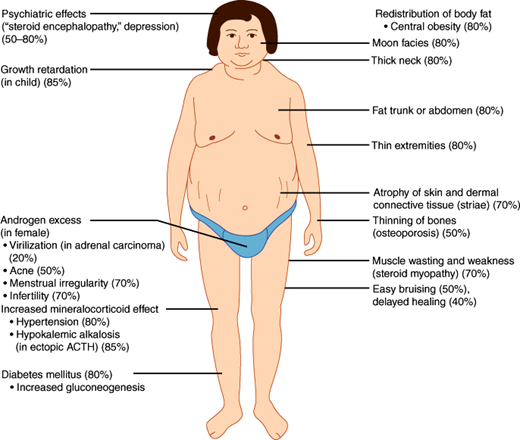
Cushing syndrome may occur either spontaneously or as the result of chronic glucocorticoid administration (iatrogenic Cushing syndrome). The overall incidence of spontaneous Cushing syndrome is approximately two to four cases per million population. It is nine times more common in women than in men. The major causes of Cushing syndrome are summarized in Table 21–3.
| NONIATROGENIC |
| ACTH dependent |
| 1. Cushing disease (ACTH-secreting pituitary adenoma): |
|
| 2. Ectopic ACTH syndrome: |
|
| ACTH independent |
| 3. Functioning adrenocortical tumor: |
|
| IATROGENIC |
| 4. Exogenous glucocorticoid administration: |
|
Uncommonly, patients with Cushing syndrome have diffuse hyperplasia of pituitary corticotroph cells responsible for ACTH hypersecretion. The hyperplasia is probably due to hypersecretion of CRH by the hypothalamus or nonhypothalamic tumors that secret ectopic CRH. Chronic CRH hypersecretion does not cause pituitary adenomas.
Cushing disease is the most common cause of noniatrogenic hypercortisolism. It is four to six times more prevalent in women than in men. Patients with Cushing disease have a pituitary adenoma causing excessive secretion of ACTH (Figure 21–12). Such adenomas are located in the anterior pituitary, are usually less than 10 mm in diameter (microadenomas), and are composed of basophilic corticotroph cells containing ACTH in secretory granules. Macroadenomas are less common and carcinomas extremely rare. Pituitary adenomas are common, found in 10–25% of unselected autopsy series, and in about 10% of asymptomatic individuals subjected to magnetic resonance imaging (MRI). Use of molecular biology techniques to determine the clonal origin of corticotroph tumors has shown that ACTH-secreting pituitary adenomas are monoclonal, arising from a single progenitor cell. Presumably, somatic mutations are required for tumorigenesis.
Figure 21–12
Hypothalamic, pituitary, and adrenal cortical relationships. Solid arrows indicate stimulation; dashed arrows, inhibition. Normal: Corticotropin-releasing hormone (CRH) elaborated by the median eminence of the hypothalamus stimulates secretion of adrenocorticotropic hormone (ACTH) by the anterior pituitary (AP). ACTH triggers the synthesis and release of cortisol, the principal glucocorticoid of the adrenal cortex. A rising level of cortisol inhibits the stimulatory action of CRH on ACTH release (or cortisol may inhibit CRH release), completing a negative feedback loop. Addison disease: In primary destructive disease of the adrenal cortex, the level of plasma cortisol is very low, and the effect of CRH on the anterior pituitary proceeds without inhibition, causing a marked increase in the secretion of ACTH. High levels of ACTH produce characteristic skin pigmentary changes. Cushing disease: The primary lesion may be at the level of the pituitary or hypothalamus. In either case, production of ACTH and cortisol is excessive. The former causes bilateral adrenal hyperplasia and the latter causes clinical manifestations of hypercortisolism. Cells of the anterior pituitary are relatively resistant to the high levels of circulating cortisol. Ectopic ACTH: In this syndrome, ACTH or an ACTH-like peptide is elaborated by a tumor such as carcinoma of the lung. The adrenals are stimulated, circulating cortisol is increased, and pituitary ACTH secretion is inhibited. Ectopic CRH: In this rare syndrome, CRH is elaborated by a tumor such as a bronchial carcinoid. The pituitary is stimulated, and there is elaboration of excess ACTH. The adrenals are stimulated, and circulating cortisol is increased. The hypercortisolism causes diminished hypothalamic CRH production; however, the negative feedback on the pituitary production of ACTH is overcome by the ectopic CRH. Adrenal adenoma or carcinoma: An adenoma or carcinoma of the adrenal cortex may produce cortisol autonomously. When the rate of production exceeds physiologic quantities, Cushing syndrome results; the effect of CRH on the anterior pituitary is inhibited by the high levels of circulating cortisol, with resultant diminished ACTH secretion and atrophy of normal adrenal tissue. Iatrogenic Cushing syndrome: Exogenous corticosteroid administration in excess of physiologic quantities of cortisol leads directly to peripheral manifestations of hypercortisolism and inhibits the effect of CRH on the anterior pituitary, with resultant diminished ACTH secretion, diminished cortisol production, and atrophy of normal adrenal tissue. (Redrawn and modified, with permission, from Burns TW, Carlson HE. Endocrinology. In: Sodeman WA et al, eds. Pathologic Physiology: Mechanisms of Disease. Saunders, 1985.)

In Cushing disease, the chronic ACTH hypersecretion causes bilateral hyperplasia of the adrenal cortex. Combined adrenal weights (normal: 8–10 g) range from 12 g to 24 g. The adrenal hyperplasia is most typically micronodular, but in some patients, particularly those with long-standing Cushing disease, macronodular hyperplasia develops.
In the ectopic ACTH syndrome, a nonpituitary tumor synthesizes and hypersecretes biologically active ACTH or an ACTH-like peptide (Figure 21–12). The neoplasms most frequently responsible are small cell carcinomas of the lung and bronchial carcinoid tumors. Ectopic ACTH hypersecretion is more common in men, largely owing to the more frequent occurrence of these lung tumors in men. Other associated tumors are listed in Table 21–3. Chronic ACTH hypersecretion causes marked bilateral adrenocortical hyperplasia, with combined adrenal weights ranging from 24–50 g or more. The ACTH secreted by the nonpituitary tumor causes adrenal hyperfunction, and the high circulating cortisol levels suppress hypothalamic secretion of CRH and the pituitary secretion of ACTH. Pituitary corticotroph cells have a decreased ACTH content.
The ectopic CRH syndrome is a rare cause of Cushing syndrome (see Figure 21–12). Most cases have been associated with bronchial carcinoid tumors.
Both adrenocortical adenomas and carcinomas may cause Cushing syndrome by elaborating cortisol autonomously (Figure 21–12). Adenomas are usually 3–6 cm in diameter, weigh 10–70 g, are encapsulated, and consist predominantly of zona fasciculata cells. They are relatively inefficient in cortisol synthesis. Adrenocortical carcinomas are usually large, weighing 100 g to several kilograms, and are often palpable as an abdominal mass by the time Cushing syndrome becomes clinically manifest. Grossly, they are highly vascular, with areas of necrosis, hemorrhage, cystic degeneration, and calcification. They are highly malignant lesions, tending to invade the adrenal capsule, neighboring organs and blood vessels and metastasize to the liver and lungs.
ACTH-independent adrenal micronodular hyperplasia is a rare cause of Cushing syndrome (also termed primary pigmented nodular adrenocortical disease). Pathologically, it is characterized by multiple small, pigmented, usually bilateral cortisol-secreting nodules. About half of cases occur sporadically in children and young adults. The remainder occur as an autosomal dominant disorder in association with blue nevi; pigmented lentigines (freckles) of the skin and mucosal surfaces of the head and face; cutaneous, mammary, and atrial myxomas; pituitary somatotroph adenomas; and tumors of peripheral nerves, testes, and other endocrine glands (Carney complex).
Another rare cause of Cushing syndrome is bilateral adrenal macronodular hyperplasia. In this condition, both glands are markedly enlarged, with bulging nodules found at cut section. Microscopically, the nodules reveal a variegated histologic pattern characterized by trabecular, adenoid, and zona glomerulosa–like structures. Occasionally, the hyperplasia may be unilateral. Some patients with macronodular hyperplasia do not show typical cushingoid features. In these cases, the macronodular hyperplasia is most often discovered incidentally on ultrasound or computed tomography (CT) examination of the abdomen and can be considered benign.
The various causes of Cushing syndrome can be divided into two categories: ACTH dependent and ACTH independent. The causes of ACTH-dependent Cushing syndrome include Cushing disease (95% of ACTH-dependent cases), ectopic ACTH hypersecretion (5%), and ectopic CRH secretion (rare), all of which are characterized by chronic ACTH hypersecretion and increased secretion of cortisol. Causes of ACTH-independent Cushing syndrome include glucocorticoid-secreting adrenocortical adenomas and carcinomas and adrenal micronodular and macronodular hyperplasia, all of which are characterized by autonomous secretion of cortisol and suppression of pituitary ACTH (Figures 21–12 and 21–13).
Figure 21–13
Basal plasma ACTH concentrations in patients with various types of noniatrogenic Cushing syndrome. The colored zone represents the normal range. (Redrawn, with permission, from Scott AP et al. Pituitary adrenocorticotropin and the melanocyte stimulating hormones. In: Parsons JA, ed. Peptide Hormones. University Park Press, 1979.)
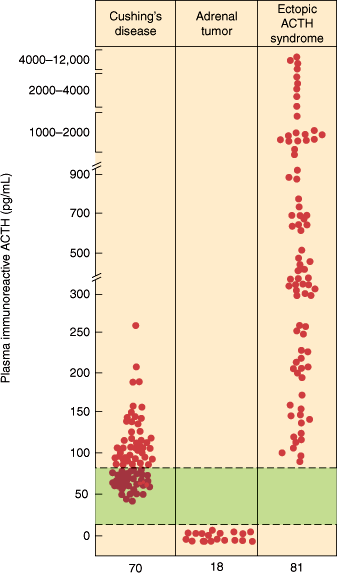
In Cushing disease, there is a persistent overproduction of ACTH by the pituitary adenoma. The ACTH hypersecretion is disorderly, episodic, and random; the normal diurnal rhythm of ACTH and cortisol secretion is usually absent, and midnight values of cortisol are elevated and can be used in diagnostic procedures. Plasma levels of ACTH and cortisol vary and may at times be within the normal range (Figure 21–13). However, a 24-hour urine free cortisol measurement confirms hypercortisolism. The excessive cortisol does not suppress ACTH secretion by the pituitary adenoma.
Most (90%) patients with Cushing disease have exaggerated plasma ACTH and cortisol responses to CRH stimulation and incompletely suppressed secretion of ACTH and cortisol by exogenous glucocorticoids (eg, 1-mg dexamethasone suppression test). Although these findings suggest that the pituitary adenoma cells are unusually sensitive to CRH and relatively resistant to glucocorticoids, the findings may simply be due to the increased number of ACTH-secreting cells. About 10% of patients with pituitary microadenomas do not exhibit major increases in plasma ACTH in response to CRH. Presumably, the clonal cells of such patients have a receptor or postreceptor defect.
Despite ACTH hypersecretion, the pituitary and adrenals fail to respond normally to stress. Stimuli such as hypoglycemia or surgery fail to increase ACTH and cortisol secretion, probably because chronic hypercortisolism has suppressed CRH secretion by the hypothalamus. Hypercortisolism also inhibits other normal pituitary and hypothalamic functions, affecting thyrotropin, growth hormone, and gonadotropin release. Surgical removal of the ACTH-producing pituitary adenoma reverses these abnormalities.
In the ectopic ACTH syndrome, hypersecretion of ACTH and cortisol is random and episodic and quantitatively greater than in patients with Cushing disease (Figure 21–13). Indeed, plasma levels and urinary excretion of cortisol, adrenal androgens, and other steroids are often markedly elevated. Ectopic ACTH secretion by tumors is usually not suppressible by exogenous glucocorticoids such as dexamethasone (Figure 21–14).
Figure 21–14
Diagnostic evaluation for suspected Cushing syndrome. Initial tests (1-mg overnight dexamethasone suppression test, or 24-hour urine cortisol, or midnight salivary cortisol level) will confirm or exclude hypercortisolism. Then, the plasma ACTH level will differentiate adrenal (ACTH-independent) from ACTH-dependent causes. In the case of elevated or normal ACTH levels, localization by inferior petrosal sinus sampling will identify or exclude a pituitary origin. Boxes enclose clinical diagnoses, and ovals indicate diagnostic tests.



