Introduction
The major functions of the nervous system are to detect, analyze, and transmit information. Information is gathered by sensory systems, integrated by the brain, and used to generate signals to motor and autonomic pathways for control of movement and of visceral and endocrine functions. These actions are controlled by neurons, which are interconnected to form signaling networks that comprise motor and sensory systems. In addition to neurons, the nervous system contains neuroglial cells that serve a variety of immunologic and support functions and modulate the activity of neurons. Understanding the pathophysiology of nervous system disease requires knowledge of neural and glial cell biology and the anatomy of neural networks. The first part of this chapter reviews several basic aspects of histology, cellular physiology, and anatomy of the nervous system.
Understanding the causes of neurologic diseases requires knowledge of molecular and biochemical mechanisms. Discoveries in the fields of molecular biology and genetics have made available important information about the mechanisms of several disease states. Several neurologic disorders in which some of the molecular mechanisms of pathogenesis are known are discussed later in this chapter including motor neuron disease, Parkinson disease, myasthenia gravis, epilepsy, Alzheimer disease, and stroke. Exciting advances in our understanding and overlap of these diseases are leading to new therapeutic targets and the hope of better treating these devastating diseases.
Normal Structure & Function of the Nervous System
The major function of neurons is to receive, integrate, and transmit information to other cells. Neurons consist of three parts: dendrites, which are elongated processes that receive information from the environment or from other neurons; the cell body, which contains the nucleus; and the axon, which may be up to 1 m long and conducts impulses to muscles, glands, or other neurons (Figure 7–1). Most neurons are multipolar, containing one axon and several dendrites. Bipolar neurons have one dendrite and one axon and are found in the cochlear and vestibular ganglia, retina, and olfactory mucosa. Spinal sensory ganglia contain pseudounipolar neurons that have a single process that emanates from the cell body and divides into two branches, one extending to the spinal cord and the other extending to the periphery. Axons and dendrites usually branch extensively at their ends. Dendritic branching can be very complex, with the result that a single neuron may receive thousands of inputs. Axon branching allows several target cells to simultaneously receive a message from one neuron. Each branch of the axon terminates on the next cell at a synapse, which is a structure specialized for information transfer from the axon to muscle, to glands, or to another neuron. Synapses between neurons most often occur between axons and dendrites but may occur between an axon and a cell body, between two axons, or between two dendrites.
Figure 7–1
Schematic drawing of a Nissl-stained motor neuron. The myelin sheath is produced by oligodendrocytes in the central nervous system and by Schwann cells in the peripheral nervous system. Note the three motor end plates, which transmit the nerve impulse to striated skeletal muscle fibers. (Redrawn, with permission, from Mescher AL. Junqueira’s Basic Histology, 12th ed. McGraw-Hill, 2009.)

Signals are propagated electrically along axons. Like other cells, neurons maintain cell size and osmolarity primarily through the action of Na+-K+ ATPase, which actively pumps Na+ out of cells in exchange for K+. This results in the formation of concentration gradients for Na+ and K+ across the cell membrane. The membrane is practically impermeable to Na+, but the presence of K+ leak channels permits the flow of K+ out of cells. This produces a difference in electrical charge across the membrane that counters transport of K+ from the cell. The flow of ions continues until the opposing electrical force reaches a value that balances the diffusional force and the membrane reaches the equilibrium potential for K+ (EK). EK is calculated by the Nernst equation:
where
R = gas constant (2 kcal mol−1°K−1)
T = absolute temperature (°K)
F = Faraday constant (2.3 × 104 kcal V−1 mol−1)
[K+]o = concentration of K+ outside the cell
[K+]i = concentration of K+ inside the cell
In most neurons, the resting membrane potential (Em) is 50–100 mV and lies close to EK since the leak of K+ is the major determinant of the charge difference across the membrane.
The membrane potential may be altered by increasing the permeability of the membrane to another ion, which drives the resting membrane potential toward the equilibrium potential for that ion. Neurons are highly specialized to use rapid changes in membrane potential to generate electrical signals. This is accomplished by ligand-gated and voltage-gated ion channels that allow the passage of Na+, K+, Ca2+, or Cl− ions in response to electrical or chemical stimuli. These channels are composed of protein complexes embedded in the lipid membrane to form aqueous pores to the inside of the cell. In general, channels are selective for a particular species of ion. An array of charged amino acids within voltage-dependent channels detects changes in voltage and induces a conformational change in the channel to alter ion permeability. Binding sites for neurotransmitters such as glutamate, γ-aminobutyric acid (GABA), glycine, and acetylcholine exist on ligand-gated channels and, when occupied, induce a conformational change to open the channel.
Electrical signals are propagated in neurons because a voltage change across the membrane in one part of a neuron is propagated to other parts. Passive spread of a voltage disturbance weakens with increasing distance from the source unless energy-dependent processes amplify the signal. Passive spread of electrical signals works well over short distances and is a major mechanism of signal propagation in dendrites. However, long-distance communication down axons to nerve terminals requires amplification. This is accomplished through the generation of self-propagating waves of excitation known as action potentials.
An action potential arises primarily from voltage-dependent changes in membrane permeability to Na+ and K+ (Figure 7–2). If a depolarizing stimulus raises the membrane potential to about −45 mV, voltage-gated Na+ channels open, allowing influx of Na+ and further depolarization toward ENa (±50 mV). Nearby areas of membrane are depolarized to the threshold for Na+ channel activation, propagating a wave of depolarization from the initial site. The resting potential is restored quickly by a combination of events. First, Na+ channels close rapidly and remain in an inactive state until the membrane potential returns to negative levels for several milliseconds. Voltage-dependent K+ channels open as the membrane potential peaks, speeding the efflux of K+ from cells and driving the membrane potential back to EK. K+ channels are also inactivated, but more slowly than Na+ channels, and this may transiently hyperpolarize cells. Plasma membrane ion exchangers and ion pumps then counteract the ion fluxes and eventually restore the resting state.
Figure 7–2
Conduction of the nerve impulse through an unmyelinated nerve fiber. In the resting axon, there is a difference of 70 mV between the interior of the axon and the outer surface of its membrane (resting potential). During the impulse passage, more Na+ (thick arrow) passes into the axon interior than the amount of K+ (thin arrow) that migrates in the opposite direction. In consequence, the membrane polarity changes (the membrane becomes relatively positive on its inner surface), and the resting potential is replaced by an action potential (+35 mV here). (Redrawn, with permission, from Junqueira LC et al, eds. Basic Histology, 10th ed. McGraw-Hill, 2003.)

Neurons transmit signals chemically to other cells at synapses (Figure 7–3). Presynaptic and postsynaptic cells are electrically isolated from each other and separated by a narrow synaptic cleft. Signaling across the cleft occurs through the release of neurotransmitters from the terminal of the presynaptic neuron. Most neurotransmitters are stored in membrane-bound synaptic vesicles and are released into the synaptic cleft by Ca2+-dependent exocytosis. Depolarization of the nerve terminal opens voltage-gated Ca2+ channels, stimulating Ca2+ influx and neurotransmitter release. Neurotransmitters diffuse across the cleft and bind to receptors on ligand-gated ion channels concentrated at the postsynaptic membrane. This produces local permeability changes, altering the membrane potential of the postsynaptic cell. If the response is depolarizing, an action potential may be generated if there are enough voltage-gated Na+ channels nearby and the membrane potential has been raised to the threshold for their activation. Receptor-gated ion channels are highly selective for a particular neurotransmitter and for the type of ions they pass, which determines whether they generate excitatory or inhibitory responses. In general, excitatory neurotransmitters, such as glutamate, open cation channels that allow influx of Na+ or Ca2+ and generate a depolarizing excitatory postsynaptic potential. Inhibitory neurotransmitters such as GABA and glycine open Cl− channels and generate an inhibitory postsynaptic potential, keeping the postsynaptic membrane near ECl (= −70 mV). Termination of the signal is achieved by removal of the neurotransmitter from the synaptic cleft. Acetylcholine is hydrolyzed by acetylcholinesterase at the postsynaptic membrane. Other neurotransmitters such as glutamate are removed by specific membrane transporters on nerve terminals or glial cells.

Not all neurotransmitter receptors are ion channels. Many receptors are coupled to cellular enzymes that regulate levels of intracellular second messengers to modulate the function of ion channels and many other cell proteins. A major mechanism by which messengers regulate ion channels is by promoting phosphorylation of channel subunits. For example, binding of the neurotransmitter norepinephrine to β-adrenergic receptors activates the enzyme adenylyl cyclase and stimulates the production of cyclic adenosine monophosphate (cAMP). The cAMP, in turn, activates a cAMP-dependent protein kinase that can phosphorylate voltage-gated calcium channels. In many cases, this increases the duration of time the channel remains open once it is activated, resulting in increased Ca2+ influx through the channel. Other neurotransmitter receptors, such as α1-adrenergic, muscarinic cholinergic, or metabotropic glutamate receptors, are coupled to the enzyme phospholipase C, which catalyzes the hydrolysis of the membrane lipid phosphatidylinositol-4,5-bisphosphate. Binding of neurotransmitter to the receptor activates phospholipase C to produce two second messengers: 1,2-diacylglycerol and inositol-1,4,5-trisphosphate. Diacylglycerol activates several enzymes of the protein kinase C family, some of which phosphorylate ion channels and either enhance or suppress their function. Inositol-1,4,5-trisphosphate binds an intracellular receptor that is itself a calcium ionophore, allowing release of calcium from intracellular stores into the cytosol. This calcium signal activates several calcium-dependent enzymes, including phosphatases and kinases that can alter the phosphorylation state and function of several ion channels and other cell proteins.
Astrocytes serve a variety of metabolic, immunologic, structural, and nutritional support functions required for normal function of neurons. They possess numerous processes that radiate from the cell body, surrounding blood vessels and covering the surfaces of the brain and spinal cord (Figure 7–4). Astrocytes express voltage- and ligand-gated ion channels and regulate K+ and Ca2+ concentrations within the interstitial space. Many synapses are invested with astrocytic processes, and this allows astrocytes to modulate neurotransmission by regulating extracellular concentrations of these cations. Astrocytes provide structural and trophic support for neurons through the production of extracellular matrix molecules such as laminin and through release of growth factors such as nerve growth factor, fibroblast growth factors, and brain-derived neurotrophic factor. End-feet of astrocytic processes at blood vessels provide sites for release of cytokines and chemoattractants during central nervous system (CNS) injury. Astrocytes respond to brain injury by increasing in size—and in some cases in number—through a process called reactive astrocytosis. This phenotypic change is characterized by an increase in cells expressing glial-fibrillary acidic protein and by synthesis and release of cytokines that regulate inflammatory responses and entry of hematogenous cells into the CNS. Astrocytes play an important role also in terminating neuronal responses to glutamate, the most abundant excitatory neurotransmitter in the brain. In cell cultures, neurons die in the presence of high levels of glutamate unless astrocytes are present. Glutamate transporters present on astrocyte cell membranes remove glutamate from the synapse. Astrocytes also contain glutamine synthase, which converts glutamate to glutamine, detoxifying the CNS of both glutamate and ammonia.
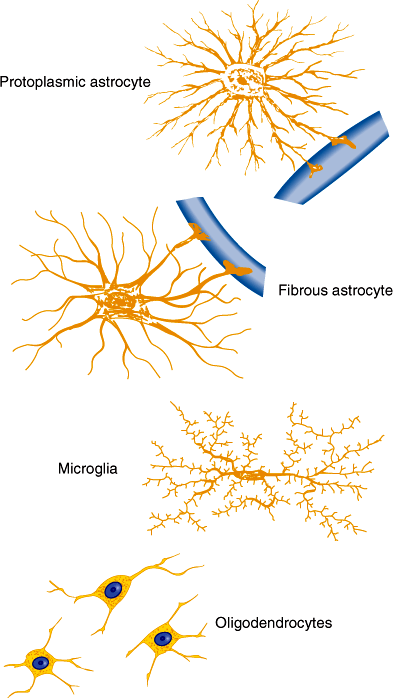
Plasma membranes of oligodendrocytes in the CNS and Schwann cells in the peripheral nervous system envelop axons. For many axons, the membranes of these glial cells are wrapped layer on layer around the axon, forming a myelin sheath (Figure 7–5). Gaps form between myelin sheaths from neighboring glia and produce nodes of Ranvier where a small portion of the axon is exposed to the interstitial space and where voltage-dependent Na+ channels are clustered in the axonal membrane. Between the nodes, myelin insulates the axon from the extracellular space, allowing efficient spread of depolarization from one node to another. This allows action potentials to propagate rapidly by jumping from node to node in a process called saltatory conduction.
Figure 7–5
Myelination of axons. Top left: Unmyelinated axon. Top right: Myelinated axon. Note that the cell membrane of the Schwann cell has wrapped itself around the axon. Bottom: Myelination of several axons in the CNS by an oligodendrogliocyte. (Redrawn, with permission, from Ganong WF. Review of Medical Physiology, 22nd ed. McGraw-Hill, 2005.)

Although peripheral blood lymphocytes and monocytes enter from the circulation and patrol the CNS, microglia, which reside in the CNS, function as the main immune effector cells. They appear to be derived from bone marrow precursors of macrophage-monocyte lineage and invade the CNS during the perinatal period. Microglia cells are activated by brain injury, infection, or neuronal degeneration. Activation is characterized by proliferation, migration into damaged tissue, increased or de novo expression of surface receptors, including CD45 (leukocyte common antigen), MHC class I and class II and immunoglobulin Fc receptors, and secretion of several cytokines, reactive oxygen intermediates, and proteinases. This response functions to remove dead tissue and destroy invading organisms but may contribute to CNS damage, particularly in certain CNS inflammatory and degenerative diseases.
Checkpoint
1. What are the primary functions of neurons, astrocytes, and microglia?
2. What role does myelin play in axonal conduction?
3. What is responsible for the resting membrane potential and for the generation of action potentials?
4. What are some of the major neurotransmitters in the nervous system, and what effects do they produce when they bind to their receptors?
Functional Neuroanatomy
To understand neuroanatomy, it is useful to study structures as parts of functional systems.
Large alpha motor neurons of the spinal cord ventral horns and brainstem motor nuclei (facial nucleus, trigeminal motor nucleus, nucleus ambiguus, hypoglossal nucleus) extend axons into spinal and cranial nerves to innervate skeletal muscles. Damage to these lower motor neurons results in loss of all voluntary and reflex movement because they comprise the output of the motor system. Neurons in the precentral gyrus and neighboring cortical regions (upper motor neurons) send axons to synapse with lower motor neurons. Axons from these upper motor neurons comprise the corticospinal and corticobulbar tracts. The motor cortex and spinal cord are connected with other deep cerebral and brainstem motor nuclei, including the caudate nucleus, putamen, globus pallidus, red nuclei, subthalamic nuclei, substantia nigra, reticular nuclei, and neurons of the cerebellum. Neurons in these structures are distinct from cortical motor (pyramidal) neurons and are referred to as extrapyramidal neurons. Many parts of the cerebral cortex are connected by fiber tracts to the primary motor cortex. These connections are important for complex patterns of movement and for coordinating motor responses to sensory stimuli.
Each alpha motor neuron axon contacts up to about 200 muscle fibers, and together they constitute the motor unit (Figure 7–6). Axons of the motor neurons intermingle to form spinal ventral roots, plexuses, and peripheral nerves. Muscles are innervated from specific segments of the spinal cord, and each muscle is supplied by at least two roots. Motor fibers are rearranged in the plexuses so that most muscles are supplied by one peripheral nerve. Thus, the distribution of muscle weakness differs in spinal root and peripheral nerve lesions.
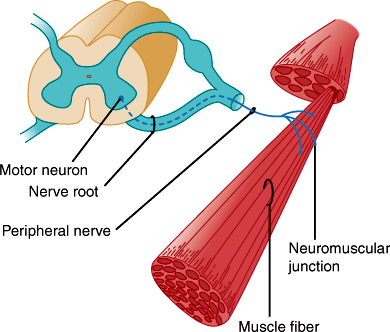
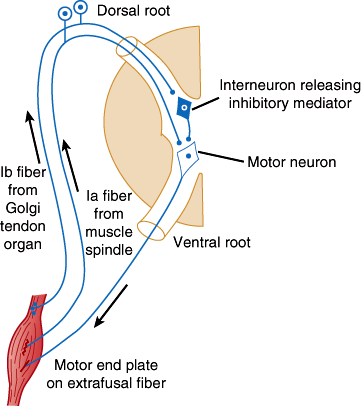
The lower motor neurons are the final common pathway for all voluntary movement. Therefore, damage to lower motor neurons or their axons causes flaccid weakness of innervated muscles. In addition, muscle tone or resistance to passive movement is reduced, and deep tendon reflexes are impaired or lost. Tendon reflexes and muscle tone depend on the activity of alpha motor neurons (Figure 7–7), specialized sensory receptors known as muscle spindles, and smaller gamma motor neurons whose axons innervate the spindles. Some gamma motor neurons are active at rest, making the spindle fibers taut and sensitive to stretch. Tapping on the tendon stretches the spindles, which causes them to send impulses that activate alpha motor neurons. These in turn fire, producing the brief muscle contraction observed during the myotactic stretch reflex. Alpha motor neurons of antagonist muscles are simultaneously inhibited. Both alpha and gamma motor neurons are influenced by descending fiber systems, and their state of activity determines the level of tone and activity of the stretch reflex.
Figure 7–7
Diagram illustrating the pathways responsible for the stretch reflex and the inverse stretch reflex. Stretch stimulates the muscle spindle, and impulses pass up the Ia fiber to excite the motor neuron. It also stimulates the Golgi tendon organ, and impulses passing up the Ib fiber activate the interneuron to release the inhibitory mediator glycine. With strong stretch, the resulting hyperpolarization of the motor neuron is so great that it stops discharging. (Redrawn, with permission, from Barnett KE et al, eds. Ganong’s Review of Medical Physiology, 24th ed. McGraw-Hill, 2012.)
Each point of contact between nerve terminal and skeletal muscle forms a specialized synapse known as a neuromuscular junction composed of the presynaptic motor nerve terminal and a postsynaptic motor end plate (Figure 7–8). Presynaptic terminals store synaptic vesicles that contain the neurotransmitter acetylcholine. The amount of neurotransmitter within a vesicle constitutes a quantum of neurotransmitter. Action potentials depolarize the motor nerve terminal, opening voltage-gated calcium channels and stimulating calcium-dependent release of neurotransmitter from the terminal. Released acetylcholine traverses the synaptic cleft to the postsynaptic (end plate) membrane, where it binds to nicotinic cholinergic receptors. These receptors are ligand-gated cation channels, and, on binding to acetylcholine, they allow entry of extracellular sodium into the motor end plate. This depolarizes the motor end plate, which in turn depolarizes the muscle fiber. After activation, cholinergic receptors are rapidly inactivated, reducing sodium entry. They remain inactive until acetylcholine dissociates from the receptor. This is facilitated by the enzyme acetylcholinesterase, which hydrolyzes acetylcholine and is present in the postsynaptic zone.
Figure 7–8
Sites of involvement in disorders of neuromuscular transmission. Left: Normal transmission involves depolarization-induced influx of calcium (Ca2+) through voltage-gated channels. This stimulates release of acetylcholine (ACh) from synaptic vesicles at the active zone and into the synaptic cleft. ACh binds to ACh receptors and depolarizes the postsynaptic muscle membrane. Right: Disorders of neuromuscular transmission result from blockage of Ca2+ channels (Lambert-Eaton syndrome or aminoglycoside antibiotics), impairment of Ca2+-mediated ACh release (botulinum toxin), or antibody-induced internalization and degradation of ACh receptors (myasthenia gravis). (Redrawn, with permission, from Greenberg DA et al, eds. Clinical Neurology, 8th ed. McGraw-Hill, 2012.)

Neuromuscular transmission may be disturbed in several ways (Figure 7–8). In the Lambert-Eaton myasthenic syndrome, antibodies to calcium channels inhibit calcium entry into the nerve terminal and reduce neurotransmitter release. In these cases, repetitive nerve stimulation facilitates accumulation of calcium in the nerve terminal and increases acetylcholine release. Clinically, limb muscles are weak, but if contraction is maintained, power increases. Electrophysiologically, there is an increase in the amplitude of the muscle response to repetitive nerve stimulation. Aminoglycoside antibiotics also impair calcium channel function and cause a similar syndrome. Proteolytic toxins produced by Clostridium botulinum cleave specific presynaptic proteins, preventing neurotransmitter release at both neuromuscular and parasympathetic cholinergic synapses. As a result, patients with botulism develop weakness, blurred vision, diplopia, ptosis, and large unreactive pupils. In myasthenia gravis, autoantibodies to the nicotinic acetylcholine receptor (AChR) block neurotransmission by inhibiting receptor function and activating complement-mediated lysis of the postsynaptic membrane. Myasthenia gravis is discussed in greater detail later in this chapter.
Motor nerves exert trophic influences on the muscles they innervate. Denervated muscles undergo marked atrophy, losing more than half of their original bulk in 2–3 months. Nerve fibers are also required for organization of the muscle end plate and for the clustering of cholinergic receptors to that region. Receptors in denervated fibers fail to cluster and become spread across the muscle membrane. Muscle fibers within a denervated motor unit may then discharge spontaneously, giving rise to a visible twitch (fasciculation) within a portion of a muscle. Individual fibers may also contract spontaneously, giving rise to fibrillations, which are not visible to the examiner but can be detected by electromyography. Fibrillations usually appear 7–21 days after damage to lower motor neurons or their axons.
Checkpoint
5. From where do lower motor neurons emanate, and to where do they send axons?
6. Describe four mechanisms that can disturb the function of the neuromuscular junction.
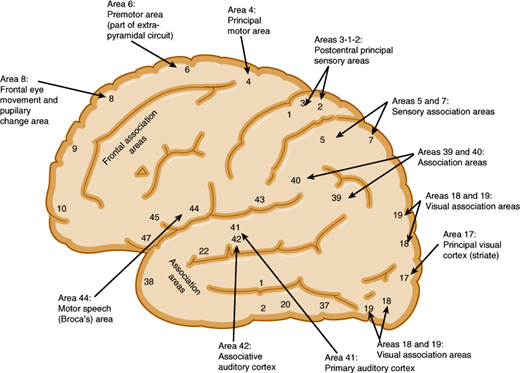
The motor cortex is the region from which movements can be elicited by electrical stimuli (Figure 7–9). This includes the primary motor area (Brodmann area 4), premotor cortex (area 6), supplementary motor cortex (medial portions of 6), and primary sensory cortex (areas 3, 1, and 2). In the motor cortex, groups of neurons are organized in vertical columns, and discrete groups control contraction of individual muscles. Planned movements and those guided by sensory, visual, or auditory stimuli are preceded by discharges from prefrontal, somatosensory, visual, or auditory cortices, which are then followed by motor cortex pyramidal cell discharges that occur several milliseconds before the onset of movement.
Cortical motor neurons contribute axons that converge in the corona radiata and descend in the posterior limb of the internal capsule, cerebral peduncles, ventral pons, and medulla. These fibers constitute the corticospinal and corticobulbar tracts and together are known as upper motor neuron fibers (Figure 7–10). As they descend through the diencephalon and brainstem, fibers separate to innervate extrapyramidal and cranial nerve motor nuclei. The lower brainstem motor neurons receive input from crossed and uncrossed corticobulbar fibers, although neurons that innervate lower facial muscles receive primarily crossed fibers.

In the ventral medulla, the remaining corticospinal fibers course in a tract that is pyramidal in shape in cross section—thus, the name pyramidal tract. At the lower end of the medulla, most fibers decussate, although the proportion of crossed and uncrossed fibers varies somewhat between individuals. The bulk of these fibers descend as the lateral corticospinal tract of the spinal cord.
Different groups of neurons in the cortex control muscle groups of the contralateral face, arm, and leg. Neurons near the ventral end of the central sulcus control muscles of the face, whereas neurons on the medial surface of the hemisphere control leg muscles (Figure 7–10). Because the movements of the face, tongue, and hand are complex in humans, a large share of the motor cortex is devoted to their control. A somatotopic organization is also apparent in the lateral corticospinal tract of the cervical cord, where fibers to motor neurons that control leg muscles lie laterally and fibers to cervical motor neurons lie medially.
Upper motor neurons are the final common pathway between cortical and subcortical structures, such as the basal ganglia, in the planning, initiation, sequencing, and modulation of all voluntary movement. Much has been learned about the normal function of upper motor neurons through the study of animals and humans with focal brain lesions. Upper motor neuron pathways can be interrupted in the cortex, subcortical white matter, internal capsule, brainstem, or spinal cord. Unilateral upper motor neuron lesions spare muscles innervated by lower motor neurons that receive bilateral cortical input, such as muscles of the eyes, jaw, upper face, pharynx, larynx, neck, thorax, and abdomen. Unlike paralysis resulting from lower motor neuron lesions, paralysis from upper motor neuron lesions is rarely complete for a prolonged period of time. Acute lesions, particularly of the spinal cord, often cause flaccid paralysis and absence of spinal reflexes at all segments below the lesion. With spinal cord lesions, this state is known as spinal shock. After a few days to weeks, a state known as spasticity appears, which is characterized by increased tone and hyperactive stretch reflexes. A similar but less striking sequence of events can occur with acute cerebral lesions.
Upper motor neuron lesions cause a characteristic pattern of limb weakness and change in tone. Antigravity muscles of the limbs become more active relative to other muscles. The arms tend to assume a flexed, pronated posture, and the legs become extended. In contrast, muscles that move the limbs out of this posture (extensors of the arms and flexors of the legs) are preferentially weakened. Tone is increased in antigravity muscles (flexors of the arms and extensors of the legs), and if these muscles are stretched rapidly, they respond with an abrupt catch, followed by a rapid increase and then a decline in resistance as passive movement continues. This sequence constitutes the “clasp knife” phenomenon. Clonus—a series of involuntary muscle contractions in response to passive stretch—may be present, especially with spinal cord lesions.
Pure pyramidal tract lesions in animals cause temporary weakness without spasticity. In humans, lesions of the cerebral peduncles also cause mild paralysis without spasticity. It appears that control of tone is mediated by other tracts, particularly corticorubrospinal and corticoreticulospinal pathways. This may explain why the degrees of weakness and spasticity often do not correspond in patients with upper motor neuron lesions.
The distribution of paralysis resulting from upper motor neuron lesions varies with the location of the lesion. Lesions above the pons impair movements of the contralateral lower face, arm, and leg. Lesions below the pons spare the face. Lesions of the internal capsule often impair movements of the contralateral face, arm, and leg equally, because motor fibers are packed closely together in this region. In contrast, lesions of the cortex or subcortical white matter tend to differentially affect the limbs and face because the motor fibers are spread over a larger area of brain. Bilateral cerebral lesions cause weakness and spasticity of cranial, trunk, and limb muscles, which leads to dysarthria, dysphonia, dysphagia, bifacial paresis, and sometimes reflexive crying and laughing (pseudobulbar palsy).
Checkpoint
7. Define the motor cortex and describe its organization.
8. Fibers from which nuclei and in which tracts constitute upper motor neurons? What is their path?
9. Describe the somatotopic organization of motor neurons in the cortex.
10. What are the characteristics of weakness and tone in upper motor neuron lesions?
11. How is the distribution of paralysis and spasticity affected by the location of an upper motor neuron lesion?
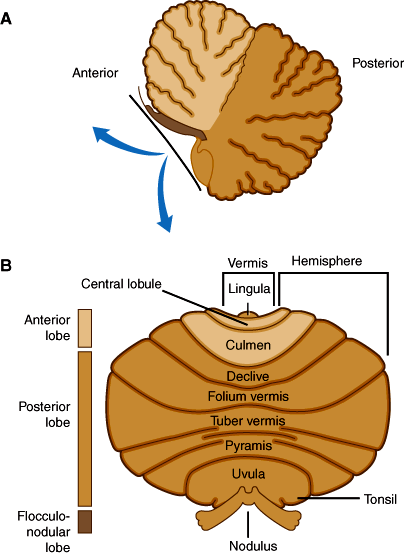
The cerebellar cortex can be divided into three anatomic regions (Figure 7–11B). The flocculonodular lobe, composed of the flocculus and the nodulus of the vermis, has connections to vestibular nuclei and is important for the control of posture and eye movement. The anterior lobe (Figure 7–11A) lies rostral to the primary fissure and includes the remainder of the vermis. It receives proprioceptive input from muscles and tendons via the dorsal and ventral spinocerebellar tracts and influences posture, muscle tone, and gait. The posterior lobe, which comprises the remainder of the cerebellar hemispheres, receives major input from the cerebral cortex via the pontine nuclei and middle cerebellar peduncles and is important for the coordination and planning of voluntary skilled movements initiated from the cerebral cortex.
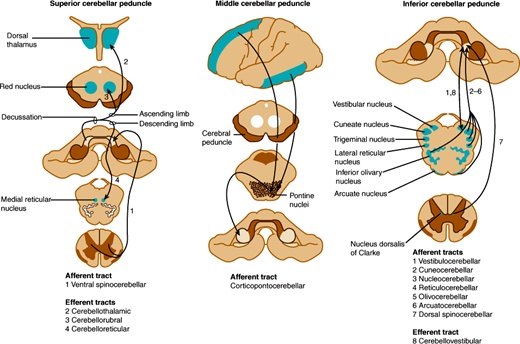
Efferent fibers from these lobes project to deep cerebellar nuclei, which in turn project to the cerebrum and brainstem through two main pathways (Figure 7–12). The fastigial nucleus receives input from the vermis and sends fibers to bilateral vestibular nuclei and reticular nuclei of the pons and medulla via the inferior cerebellar peduncles. Other regions of the cerebellar cortex send fibers to the dentate, emboliform, and globose nuclei, whose efferents form the superior cerebellar peduncles, enter the upper pons, decussate completely in the lower midbrain, and travel to the contralateral red nucleus. At the red nucleus, some fibers terminate, whereas others ascend to the ventrolateral nucleus of the thalamus, whence thalamic neurons send ascending efferent fibers to the motor cortex of the same side. A smaller group of fibers descend after decussation in the midbrain and terminate in reticular nuclei of the lower brainstem. Thus, the cerebellum controls movement through connections with cerebral motor cortex and brainstem nuclei.
Figure 7–12
Cerebellar connections in the superior, middle, and inferior cerebellar peduncles. The peduncles are indicated by gray shading and the areas to and from which they project by blue shading. (Redrawn, with permission, from Greenberg DA et al, eds. Clinical Neurology, 8th ed. McGraw-Hill, 2012.)
The cerebellum is responsible for the coordination of muscle groups, control of stance and gait, and regulation of muscle tone. Rather than causing paralysis, damage to the cerebellum interferes with the performance of motor tasks. The major manifestation of cerebellar disease is ataxia, in which simple movements are delayed in onset and their rates of acceleration and deceleration are decreased, resulting in intention tremor and dysmetria (“overshooting”). Lesions of the cerebellar hemispheres affect the limbs, producing limb ataxia, whereas midline lesions affect axial muscles, causing truncal and gait ataxia and disorders of eye movement. Cerebellar lesions are often associated with hypotonia as a result of depression of activity of alpha and gamma motor neurons. If a lesion of the cerebellum or cerebellar peduncles is unilateral, the signs of limb ataxia appear on the same side as the lesion. However, if the lesion lies beyond the decussation of efferent cerebellar fibers in the midbrain, the clinical signs are on the side opposite the lesion.
Checkpoint
12. What is the overall role of the cerebellum?
13. What are the anatomic regions of the cerebellum, what do they control, and through which other regions of the brain do they make connections?
14. What are the consequences of damage to the cerebellum, and what symptoms and signs are seen in patients with cerebellar lesions?
15. Below what point do unilateral cerebellar lesions manifest on the opposite side?
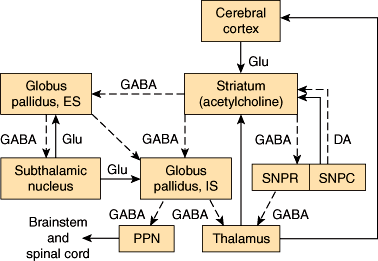
Several subcortical, thalamic, and brainstem nuclei are critical for regulating voluntary movement and maintaining posture. These include the basal ganglia (ie, the caudate nucleus and putamen [corpus striatum]), globus pallidus, substantia nigra, and subthalamic nuclei. They also include the red nuclei and the mesencephalic reticular nuclei. The major pathways that involve the basal ganglia form three neuronal circuits (Figure 7–13). The first is the cortical-basal ganglionic-thalamic-cortical loop. Inputs mainly from premotor, primary motor, and primary sensory cortices (areas 1, 2, 3, 4, and 6) project to the corpus striatum, which sends fibers to the medial and lateral portions of the globus pallidus. Fibers from the globus pallidus form the ansa and fasciculus lenticularis, which sweep through the internal capsule and project onto ventral and intralaminar thalamic nuclei. Axons from these nuclei project to the premotor and primary motor cortices (areas 4 and 6), completing the loop. In the second loop, the substantia nigra sends dopaminergic fibers to the corpus striatum, which has reciprocal connections with the substantia nigra. The substantia nigra also projects to the ventromedial thalamus. The third loop is composed of reciprocal connections between the globus pallidus and the subthalamic nucleus. The subthalamic nucleus also sends efferents to the substantia nigra and corpus striatum.
Figure 7–13
Diagrammatic representation of the principal connections of the basal ganglia. Solid lines indicate excitatory pathways; dashed lines indicate inhibitory pathways. The transmitters are indicated in the pathways, where they are known. Glu, glutamate; DA, dopamine. Acetylcholine is the transmitter produced by interneurons in the striatum (ie, the putamen and the caudate nucleus, which have similar connections). SNPR, substantia nigra, pars reticulata; SNPC, substantia nigra, pars compacta; ES, external segment; IS, internal segment. The subthalamic nucleus also projects to the pars compacta of the substantia nigra; this pathway has been omitted for clarity. (Redrawn, with permission, from Barnett KE et al, eds. Ganong’s Review of Medical Physiology, 24th ed. McGraw-Hill, 2012.)
Basal ganglia circuits regulate the initiation, amplitude, and speed of movements. Diseases of the basal ganglia cause abnormalities of movement and are collectively known as movement disorders. They are characterized by motor deficits (bradykinesia, akinesia, loss of postural reflexes) or abnormal activation of the motor system, resulting in rigidity, tremor, and involuntary movements (chorea, athetosis, ballismus, and dystonia).
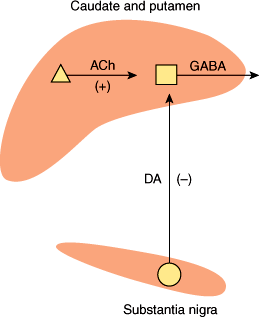
Several neurotransmitters are found within the basal ganglia, but their role in disease states is only partly understood. Acetylcholine is present in high concentrations within the corpus striatum, where it is synthesized and released by large Golgi type 2 neurons (Figure 7–14). Acetylcholine acts as an excitatory transmitter at medium-sized spiny striatal neurons that synthesize and release the inhibitory neurotransmitter GABA and project to the globus pallidus. Dopamine is synthesized by neurons of the substantia nigra, whose axons form the nigrostriatal pathway that terminates in the corpus striatum. Dopamine released by these fibers inhibits striatal GABAergic neurons. In Parkinson disease, degeneration of nigral neurons leads to loss of dopaminergic inhibition and a relative excess of cholinergic activity. This increases GABAergic output from the striatum and contributes to the paucity of movement that is a cardinal manifestation of the disease. Anticholinergics and dopamine agonists tend to restore the normal balance of striatal cholinergic and dopaminergic inputs and are effective in treatment. The pathogenesis of Parkinson disease is discussed later in this chapter.
Figure 7–14
Simplified neurochemical anatomy of the basal ganglia. Dopamine (DA) neurons exert a net inhibitory effect and acetylcholine (ACh) neurons a net excitatory effect on the GABAergic output from the striatum. In Parkinson disease, DA neurons degenerate. The net effect is to increase GABAergic output from the striatum. (Redrawn, with permission, from Greenberg DA et al, eds. Clinical Neurology, 5th ed. McGraw-Hill, 2002.)
Huntington disease is inherited as an autosomal dominant disorder. When disease onset occurs later in life, patients develop involuntary, rapid, jerky movements (chorea) and slow writhing movements of the proximal limbs and trunk (athetosis). When disease onset occurs earlier in life, patients develop signs of parkinsonism with tremor (cogwheeling) and stiffness. The spiny GABAergic neurons of the striatum preferentially degenerate, resulting in a net decrease in GABAergic output from the striatum. This contributes to the development of chorea and athetosis. Dopamine antagonists, which block inhibition of remaining striatal neurons by dopaminergic striatal fibers, reduce the involuntary movements. Neurons in deep layers of the cerebral cortex also degenerate early in the disease, and later this extends to other brain regions, including the hippocampus and hypothalamus. Thus, the disease is characterized by cognitive defects and psychiatric disturbances in addition to the movement disorder.
The gene for the disease is located on chromosome 4p and encodes for a 3144-amino acid protein, huntingtin, which is widely expressed and interacts with several proteins involved in intracellular trafficking and endocytosis, gene transcription, and intracellular signaling. The protein contains a trinucleotide (CAG) repeat of 11–34 copies that encodes a polyglutamine domain and is expanded in patients with the disease. Deletion of the gene in mice causes embryonic death, whereas heterozygous animals are healthy. Transgenic mice with an expanded repeat develop a neurodegenerative disorder, suggesting that the disease results from the toxic effect of a gain of function mutation.
The mechanisms by which mutant huntingtin causes disease are not certain. The mutant protein is degraded, and the resulting fragments that contain the glutamine repeats form aggregates, which are deposited in nuclear and cytoplasmic inclusions. These fragments may bind abnormally to other proteins and interfere with normal protein processing or disrupt mitochondrial function. Nuclear fragments may interfere with nuclear functions such as gene expression. For example, in the cerebral cortex, mutant huntingtin reduces the production of brain-derived neurotrophic factor by suppressing its transcription. In addition, normal huntingtin is protective for cortical and striatal neurons and blocks the processing of procaspase 9, thereby reducing apoptosis (programmed cell death). Therefore, both loss of neurotrophic support and enhanced caspase activity could promote striatal cell loss in Huntington disease.
Checkpoint
16. Which are the component nuclei of the basal ganglia, and what is their functional role?
17. What are the clinical consequences of lesions in the basal ganglia?
18. What are some of the neurotransmitters within the basal ganglia, and what is their role in disorders of basal ganglia function?
Somatosensory pathways confer information about touch, pressure, temperature, pain, vibration, and the position and movement of body parts. This information is relayed to thalamic nuclei and integrated in the sensory cortex of the parietal lobes to provide conscious awareness of sensation. Information is also relayed to cortical motor neurons to adjust fine movements and maintain posture. Some ascending sensory fibers, particularly pain fibers, enter the midbrain and project to the amygdala and limbic cortex, where they contribute to emotional responses to pain. In the spinal cord, painful stimuli activate local pathways that induce the firing of lower motor neurons and cause a reflex withdrawal. Thus, somatosensory pathways provide tactile information, guide movement, and serve protective functions.
A variety of specialized end organs and free nerve endings transduce sensory stimuli into neural signals and initiate the firing of sensory nerve fibers. Fibers that mediate cutaneous sensation from the trunk and limbs travel in sensory or mixed sensorimotor nerves to the spinal cord (Figure 7–15). Cutaneous sensory nerves contain small myelinated Aδ fibers that transmit information about pain and temperature, larger myelinated fibers that mediate touch and pressure sensation, and more numerous unmyelinated pain and autonomic C fibers. Myelinated proprioceptive fibers and afferent and efferent muscle spindle fibers are carried in the larger sensorimotor nerves. The cell bodies of the sensory neurons are in the dorsal root ganglia, and their central projections enter the spinal cord via the dorsal spinal roots. Innervation of the skin, muscles, and surrounding connective tissue is segmental, and each root innervates a region of skin known as a dermatome (Figure 7–16). Cell bodies of the sensory neurons that innervate the face reside in the trigeminal ganglion and send their central projections in the trigeminal nerve to the brainstem. The trigeminal innervation of the face is subdivided into three regions, each innervated by one of the three divisions of the trigeminal nerve.
Figure 7–15
Schematic illustration of a spinal cord segment with its dorsal root, ganglion cells, and sensory organs. Sensory organs shown (from top to bottom) are the pacinian corpuscle, muscle spindle, tendon organ, encapsulated ending, and free nerve endings. The somatotopic arrangement of fibers in the dorsal columns, spinothalamic tract, and corticospinal tract is also shown. (Redrawn, with permission, from Waxman SG. Clinical Neuroanatomy, 26th ed. McGraw-Hill, 2010.)
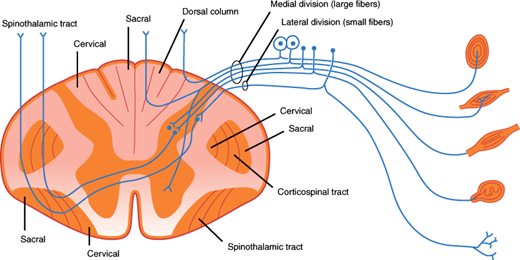

The dorsal roots enter the dorsal horn of the spinal cord (Figure 7–15). Large myelinated fibers divide into ascending and descending branches and either synapse with dorsal gray neurons within a few cord segments or travel in the dorsal columns, terminating in the gracile or cuneate nuclei of the lower medulla on the same side. Secondary neurons of the dorsal horn also send axons up the dorsal columns. Fibers in the dorsal columns are displaced medially as new fibers are added, so that in the cervical cord, leg fibers are located medially and arm fibers laterally (Figure 7–15). The gracile and cuneate nuclei send fibers that cross the midline in the medulla and ascend to the thalamus as the medial lemniscus (Figure 7–17). The dorsal column-lemniscal system carries information about pressure, limb position, vibration, direction of movement, recognition of texture and shape, and two-point discrimination.
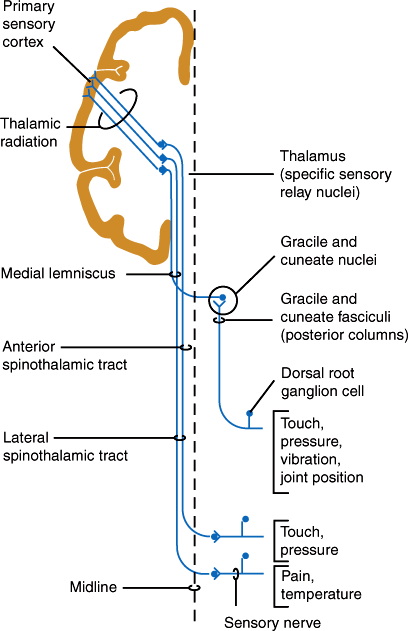
Thinly myelinated and unmyelinated fibers enter the lateral portion of the dorsal horn and synapse with dorsal spinal neurons within one or two segments. The majority of secondary fibers from these cells cross in the anterior spinal commissure and ascend in the anterolateral spinal cord as the lateral spinothalamic tracts. Crossing fibers are added to the inner side of the tract, so that in the cervical cord the leg fibers are located superficially and arm fibers are deeper. These fibers carry information about pain, temperature, and touch sensation.
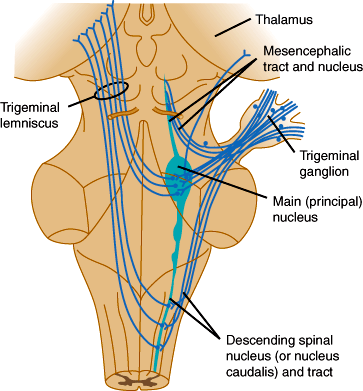
Sensation from the face is carried by trigeminal sensory fibers that enter the pons and descend to the medulla and upper cervical cord (Figure 7–18). Fibers carrying information about pain and temperature sensation terminate in the nucleus of the spinal tract of cranial nerve V, which is continuous with the dorsal horn of the cervical cord. Touch, pressure, and postural information is conveyed by fibers that terminate in the main sensory and mesencephalic nuclei of the trigeminal nerve. Axons arising from trigeminal nuclei cross the midline and ascend as the trigeminal lemniscus just medial to the spinothalamic tract. Fibers from the spinothalamic tract, medial lemniscus, and trigeminal lemniscus merge in the midbrain and terminate along with sensory fibers ascending from the spinal cord in the posterior thalamic nuclei, mainly in the nucleus ventralis posterolateralis. These thalamic nuclei project to the primary somatosensory cortex (Brodmann areas 3, 1, and 2) and to a second somatosensory area on the upper bank of the sylvian fissure (lateral cerebral sulcus). The primary somatosensory region is organized somatotopically like the primary motor cortex.
Free nerve endings of unmyelinated C fibers and small-diameter myelinated Aδ fibers in the skin convey sensory information in response to chemical, thermal, and mechanical stimuli. Intense stimulation of these nerve endings evokes the sensation of pain. In contrast to skin, most deep tissues are relatively insensitive to chemical or noxious stimuli. However, inflammatory conditions can sensitize sensory afferents from deep tissues to evoke pain on mechanical stimulation. This sensitization appears to be mediated by bradykinin, prostaglandins, and leukotrienes released during the inflammatory response. Information from primary afferent fibers is relayed via sensory ganglia to the dorsal horn of the spinal cord and then to the contralateral spinothalamic tract, which connects to thalamic neurons that project to the somatosensory cortex.
Damage to these pathways produces a deficit in pain and temperature discrimination and may also produce abnormal painful sensations (dysesthesias) usually in the area of sensory loss. Such pain is termed neuropathic pain and often has a strange burning, tingling, or electric shocklike quality. It may arise from several mechanisms. Damaged peripheral nerve fibers become highly mechanosensitive and may fire spontaneously without known stimulation. They also develop sensitivity to norepinephrine released from sympathetic postganglionic neurons. Electrical impulses may spread abnormally from one fiber to another (ephaptic conduction), enhancing the spontaneous firing of multiple fibers. Neuropeptides released by injured nerves may recruit an inflammatory reaction that stimulates pain. In the dorsal horn, denervated spinal neurons may become spontaneously active. In the brain and spinal cord, synaptic reorganization occurs in response to injury and may lower the threshold for pain. In addition, inhibition of pathways that modulate transmission of sensory information in the spinal cord and brainstem may promote neuropathic pain.
Pain-modulating circuits exert a major influence on the perceived intensity of pain. One such pathway (Figure 7–19) is composed of cells in the periaqueductal gray matter of the midbrain that receive afferents from frontal cortex and hypothalamus and project to rostroventral medullary neurons. These in turn project in the dorsolateral white matter of the spinal cord and terminate on dorsal horn neurons. Additional descending pathways arise from other brainstem nuclei (locus ceruleus, dorsal raphe nucleus, and nucleus reticularis gigantocellularis). Major neurotransmitters utilized by these systems include endorphins, serotonin, and norepinephrine, providing the rationale for the use of opioids, serotonin agonists, and serotonin and norepinephrine reuptake inhibitors in the treatment of pain.

Receptors in the muscles, tendons, and joints provide information about deep pressure and the position and movement of body parts. This allows one to determine an object’s size, weight, shape, and texture. Information is relayed to the spinal cord via large Aα and Aβ myelinated fibers and to the thalamus by the dorsal column-lemniscal system. Detecting vibration requires sensing touch and rapid changes in deep pressure. This depends on multiple cutaneous and deep sensory fibers and is impaired by lesions of multiple peripheral nerves, the dorsal columns, medial lemniscus, or thalamus but rarely by lesions of single nerves. Vibratory sense is often impaired together with proprioception.
Primary sensory cortex provides awareness of somatosensory information and the ability to make sensory discriminations. Touch, pain, temperature, and vibration sense are considered the primary modalities of sensation and are relatively preserved in patients with damage to sensory cortex or its projections from the thalamus. In contrast, complex tasks that require integration of multiple somatosensory stimuli and of somatosensory stimuli with auditory or visual information are impaired. These include the ability to distinguish two points from one when touched on the skin (two-point discrimination), localize tactile stimuli, perceive the position of body parts in space, recognize letters or numbers drawn on the skin (graphesthesia), and identify objects by their shape, size, and texture (stereognosis).
The patterns of sensory loss often indicate the level of nervous system involvement. Symmetric distal sensory loss in the limbs, affecting the legs more than the arms, usually signifies a generalized disorder of multiple peripheral nerves (polyneuropathy). Sensory symptoms and deficits may be restricted to the distribution of a single peripheral nerve (mononeuropathy) or two or more peripheral nerves (mononeuropathy multiplex). Symptoms limited to a dermatome indicate a spinal root lesion (radiculopathy).
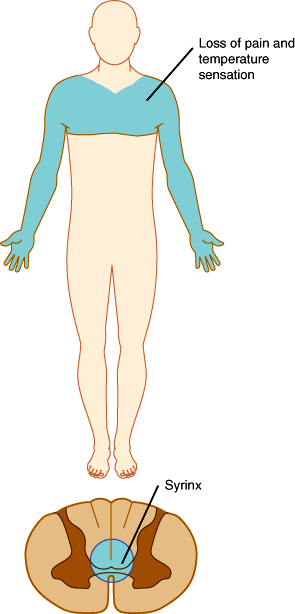
In the spinal cord, segregation of fiber tracts and the somatotopic arrangement of fibers give rise to distinct patterns of sensory loss. Loss of pain and temperature sensation on one side of the body and of proprioception on the opposite side occurs with lesions that involve one half of the cord on the side of the proprioceptive deficit (Brown-Séquard syndrome; Figure 7–20). Compression of the upper spinal cord causes loss of pain, temperature, and touch sensation first in the legs, because the leg spinothalamic fibers are most superficial. More severe cord compression compromises fibers from the trunk. In patients with spinal cord compression, the lesion is often above the highest dermatome involved in the deficit. Thus, radiographic studies should be tailored to visualize the cord at and above the level of the sensory deficit detected on examination. Intrinsic cord lesions that involve the central portions of the cord often impair pain and temperature sensation at the level of the lesion because the fibers crossing the anterior commissure and entering the spinothalamic tracts are most centrally situated. Thus, enlargement of the central cervical canal in syringomyelia typically causes loss of pain and temperature sensation across the shoulders and upper arms (Figure 7–21).
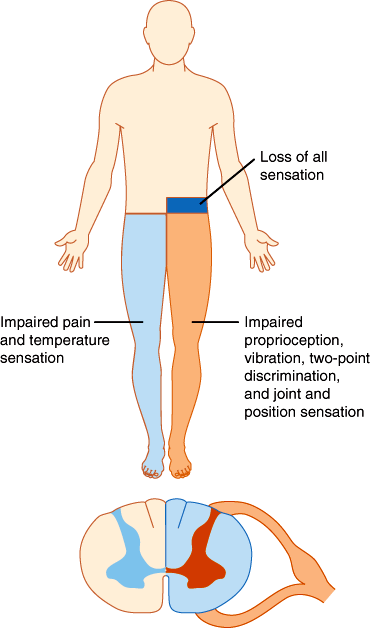
Figure 7–21
Syringomyelia (the presence of a cavity in the spinal cord resulting from breakdown of gliomatous new formations, presenting clinically with pain and paresthesias followed by muscular atrophy of the hands) involving the cervicothoracic portion of the cord. (Redrawn, with permission, from Waxman SG. Clinical Neuroanatomy, 26th ed. McGraw-Hill, 2010.)
Brainstem lesions involving the spinothalamic tract cause loss of pain and temperature sensation on the opposite side of the body. In the medulla, such lesions can involve the neighboring spinal trigeminal nucleus, resulting in a “crossed” sensory deficit involving the ipsilateral face and contralateral limbs. Above the medulla, the spinothalamic and trigeminothalamic tracts lie close together, and lesions there cause contralateral sensory loss of the face and limbs. In the midbrain and thalamus, medial lemniscal fibers run together with pain and temperature fibers, and lesions are more likely to impair all primary sensation contralateral to the lesion. Because sensory fibers converge at the thalamus, lesions there tend to cause fairly equal loss of pain, temperature, and proprioceptive sensation on the contralateral half of the face and body. Lesions of the sensory cortex in the parietal lobe impair discriminative sensation on the opposite side of the body, whereas detection of the primary modalities of sensation may remain relatively intact.
Checkpoint
19. What fibers carry pain, and how are they segregated from fibers that carry proprioception information in the spinal cord?
20. What are the differences in characteristics of sensory loss at different levels of the nervous system?
21. What is the function of the sensory cortex in the parietal lobe, and what are the clinical features of damage to this region?
The visual system provides our most important source of sensory information about the environment. The visual system and pathways for the control of eye movements are among the best characterized pathways in the nervous system. Familiarity with these neuroanatomic features is often extremely valuable in localization of neurologic disease.
The cornea and lens of the eye refract and focus images on the photosensitive posterior portion of the retina. The posterior retina contains two classes of specialized photoreceptor cells, rods and cones, which transduce photons into electrical signals. At the retina, the image is reversed in the horizontal and vertical planes so that the inferior visual field falls on the superior portions of the retina and the lateral field is detected by the nasal half of the retina.
Fibers from the nasal half of the retina traverse the medial portion of the optic nerve and cross to the other side at the optic chiasm (Figure 7–22). Each optic tract contains fibers from the same half of the visual field of both eyes. The optic tracts terminate in the lateral geniculate nuclei of the thalamus. Lateral geniculate neurons send fibers to the primary visual cortex in the occipital lobe (area 17, calcarine cortex; see Figure 7–9). These fibers form the optic radiations, which extend through the white matter of the temporal lobes and the inferior portion of the parietal lobes.



