Introduction
Infectious diseases remain one of the leading causes of death in both developed and developing countries. Infections cause significant morbidity and mortality, especially in individuals who are most vulnerable to illness: the very young, the elderly, the immunocompromised, and the disenfranchised.
The pathogenesis of infectious diseases reflects the relationship among the human host, the infectious agent, and the external environment. Figure 4–1 portrays a host-agent-environment paradigm for the study of infectious diseases. The infectious agent can be either exogenous (ie, not normally found on or in the body) or endogenous (ie, one that may be routinely cultured from a specific anatomic site but that does not normally cause disease in the host). Infection results when an exogenous agent is introduced into a host from the environment or when an endogenous agent overcomes innate host immunity to cause disease. Host susceptibility plays an important role in either of these settings.
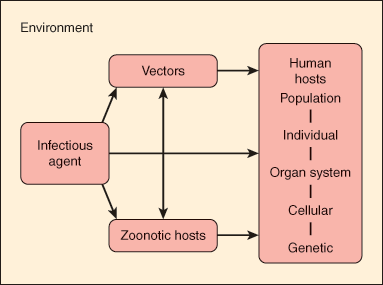
The environment includes vectors (insects and other carriers that transmit infectious agents) and zoonotic hosts or reservoirs (animals that harbor infectious agents and often act to amplify the infectious agent). For example, the white-footed mouse (Peromyscus leucopus) serves as an animal reservoir for Borrelia burgdorferi, the bacterium that causes Lyme disease. The Ixodes tick serves as an insect vector. Infection in the mouse is asymptomatic, and the bacteria can multiply to high levels in this animal. When the tick larva feeds on an infected mouse, it becomes secondarily infected with B burgdorferi, and this infection persists when the tick molts into a nymph. Subsequently, when an infected nymph feeds on a human, the bacterium is transmitted through infected saliva into the host bloodstream, causing disease.
The study of infectious diseases requires understanding of pathogenesis at the level of the population, the individual, the cell, and the gene. For example, at the population level, the spread of tuberculosis in the community is related to the social interactions of an infectious human host. Outbreaks of tuberculosis have occurred in group settings such as homeless shelters, prisons, and nursing homes when an index case comes in close contact with susceptible persons. At the individual level, tuberculosis results from inhalation of respiratory droplets containing airborne tubercle bacilli. At the cellular level, these bacilli activate T cells, which play a critical role in containing the infection. Individuals with an impaired T-cell response (eg, those infected with human immunodeficiency virus [HIV]) are at particularly high risk for developing active tuberculosis at the time of the initial infection or for reactivation of latent tuberculosis as their immunity wanes. Finally, at the genetic level, individuals with specific polymorphisms in a macrophage protein gene may be at significantly higher risk for pulmonary tuberculosis.
Specific microorganisms have a tendency to cause certain types of infections: Streptococcus pneumoniae commonly causes pneumonia, meningitis, and bacteremia but rarely causes endocarditis (infection of the heart valves); Escherichia coli is a common cause of gastrointestinal and urinary tract infections; Plasmodium species infect red blood cells and liver cells to cause malaria; Entamoeba histolytica causes amebic dysentery, liver abscesses, and so on. Table 4–1 presents a clinical approach to taking a patient history that considers features of the host and the environment in identifying the most likely microorganisms associated with specific clinical syndromes.
| Component | Feature | Examples of Infection |
|---|---|---|
| History of present illness | Age | West Nile virus neuroinvasive disease much more common in the elderly |
| Pregnancy | Pregnant women at increased risk for serious varicella pneumonia; risks to fetus with various infections or treatments | |
| Site of acquisition (home, skilled nursing facility (SNF), hospital) | Multi-drug–resistant bacteria more commonly cultured from patient in a SNF or hospital | |
| Season | Influenza epidemics limited to fall through early spring | |
| Symptoms (duration, severity, pattern) | Bacterial meningitis excluded if symptoms last >1 week | |
| Past medical history (including medications, allergies, and immunizations) | Immunocompromise (eg, HIV, organ transplant, corticosteroid use, chemotherapy, asplenia) | Pneumocystis jirovecii pneumonia in patients with AIDS |
| Comorbid disease (eg, chronic obstructive lung disease, diabetes mellitus, alcohol abuse) | Limb-threatening soft tissue infections in diabetic patients | |
| Transfusions | Blood-borne infections such as cytomegalovirus or hepatitis C virus | |
| Habits and exposures | Substance use (eg, alcohol, cigarettes, type and route of illicit drugs use) | Endocarditis associated with injection drug use due to seeding of bloodstream with skin bacteria |
| Sexual contacts | Risk for sexually transmitted infections such as syphilis | |
| Outdoor activities | Arthropod-borne infections (eg, Rocky Mountain spotted fever) | |
| Pets | Zoonotic infections (eg, cat scratch disease) | |
| Social history | Occupation | Q-fever in veterinarians |
| Congregated living facility | Transmission of infection from an ill contact (eg, influenza, Shigella, norovirus) | |
| Homelessness | Tuberculosis, scabies | |
| Travel | Internationally acquired infections (eg, malaria) | |
| Family history | Transmittable diseases | Tuberculosis |
| Review of systems | Symptoms by organ system | History of headache raises concern for central nervous system infection; diarrhea raises concern for gastroenteritis |
Host Defenses Against Infection
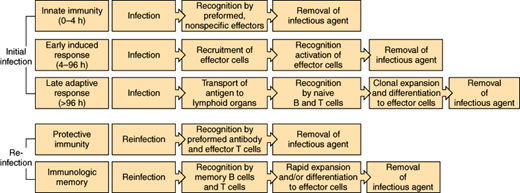
The human body has the ability to control infection through a number of different mechanisms. Physical barriers impede the entry of bacteria from the external environment and from normally colonized sites in the body into sterile anatomic areas. When these physical defenses are breached, the immune system is activated (Figure 4–2). Constitutive or innate immunity, provided by preformed proteins (eg, complement) and immune cells (eg, phagocytes) that are activated by nonspecific foreign proteins, allows an immediate response to foreign material. Induced or adaptive immunity includes both early and late adaptive responses activated by specific antigenic proteins (eg, production of antibodies active against the specific strains of S pneumoniae contained in the pneumococcal vaccine in a previously vaccinated individual). Induction of these specific immune receptor cells may take several days in the immunologically naive host. Protective immunity, which occurs after initial exposure (infection or vaccination) through generation of memory lymphocytes and pathogen-specific antibody, allows a much more rapid response to reinfection. These components of the immune response are discussed in detail later.
Figure 4–2
Phases of the host response to infection. During the earliest stage of initial infection, nonspecific mediators (complement, phagocytes) predominate. Adaptive immunity (production of antibody, stimulation of lymphocytes) requires clonal expansion after recognition of specific antigens. Once immunity toward a specific agent is induced, the immune response remains primed so that the response to reinfection is much more rapid.
The human body normally harbors numerous species of bacteria, viruses, fungi, and protozoa, referred to as the human microbiota. The great majority of these are commensals, defined as organisms that live symbiotically on or within the human host but rarely cause disease (Figure 4–3). Anatomic sites where bacteria are normally found include the skin (staphylococci and diphtheroids), oropharynx (streptococci, anaerobes), large intestine (enterococci, enteric bacilli), and vagina (lactobacilli).
Figure 4–3
Commensal bacteria secrete toll-like receptor (TLR) ligands, which bind to TLR on the surface of normal intestinal tissue. This interaction stimulates basal signaling, which protects against cellular injury. Disruption of TLR signaling or antibiotic associated eradication of commensal bacteria result in compromised ability of the intestinal epithelium to withstand injury and repair cell damage. (Redrawn, with permission, from Madara J. Building an intestine—Architectural contributions of commensal bacteria. N Engl J Med. 2004;351:1686.)

Determining when an isolate is a component of the normal flora rather than an invasive pathogen may be difficult. For example, culture of staphylococci from a blood sample may represent skin contamination at the time of phlebotomy or may indicate a potentially life-threatening bloodstream infection. Helpful clues include burden of organism (eg, number of positive blood cultures), symptoms and signs of infection (eg, cough, fever), and the presence of inflammatory cells (eg, polymorphonuclear cells in the sputum and an increased proportion of immature neutrophils in the blood). Isolation of an obligate pathogen such as Mycobacterium tuberculosis from any anatomic site is diagnostic of infection. Fortunately, few microorganisms are absolute pathogens. For example, Neisseria meningitidis, a major bacterial cause of meningitis, can be cultured from the oropharynx of as many as 10% of asymptomatic individuals, in which case it represents transient normal flora. Even if asymptomatic, the host can serve as a carrier, transferring bacteria to susceptible individuals. Infections resulting from commensals that rarely cause disease (eg, Candida albicans) or organisms ubiquitous in the environment that are generally not considered human pathogens (eg, Aspergillus) are termed opportunistic infections. These infections occur almost exclusively in immunocompromised hosts such as HIV-infected patients or transplant recipients. The agents are opportunists in that they take advantage of impaired host immunity to cause infection but rarely cause disease in a healthy host.
The site from which an organism is cultured is important in differentiating colonization from infection. Growth of any microorganism from a normally sterile site such as blood, cerebrospinal fluid, synovial (joint) fluid, or deep tissues of the body is diagnostic of infection. For example, Bacteroides, the predominant genus of bacteria in the colon, may cause intra-abdominal abscesses and sepsis when the integrity of the colonic mucosa is breached. Staphylococcus epidermidis, a common skin commensal, can cause bacteremia after intravascular catheter placement. Knowledge of the common endogenous flora may be useful in determining the cause of an infection and may aid in the choice of empiric antibiotic therapy.
When the delicate symbiosis between the commensal and the host is disturbed, the normal flora may be overgrown by either endogenous or exogenous organisms. This phenomenon, which may be transient or persistent, is called colonization. For example, broad-spectrum antibiotics will destroy normal vaginal flora, such as lactobacilli, and allow overgrowth of Candida (yeast) species. When replacement of the normal flora occurs in the hospital environment, the organisms are said to be nosocomially acquired. The distinction between hospital-acquired and community-acquired infections has blurred in recent years, because of an increase in medical care in the home or skilled nursing facility among patients who previously would have required long-term hospitalization. For this reason, the broader term “healthcare-associated infections” is used to encompass both hospitalized patients and patients with frequent medical interactions (eg, residence in nursing home, outpatient hemodialysis, home intravenous antibiotics). Healthcare-associated infections are significant because the organisms are often resistant to multiple antibiotics. Not uncommonly, colonization will progress to symptomatic infection. For example, individuals hospitalized for extended periods often become colonized with gram-negative bacteria such as Pseudomonas aeruginosa. These individuals are then at increased risk for life-threatening infections such as pseudomonas pneumonia.
Host defense mechanisms that serve to inhibit colonization by pathogenic bacteria include: (1) mechanical clearance, (2) phagocytic killing, and (3) depriving organisms of necessary nutrients. Successful colonizers have adapted to evade or overcome these defenses. For example, gonococci, the bacteria that cause gonorrhea, avoid excretion in the urine by adhering to the mucosal epithelium of the urogenital tract with pili. Pneumococci resist phagocytosis by encapsulation within a slime layer that impairs uptake by neutrophils. Some staphylococci elaborate enzymes known as hemolysins that destroy host red blood cells, thus giving them access to a needed source of iron.
Colonization of sites that are normally sterile or have very few microbes is generally easier because there is no competition for nutrients from endogenous flora. However, host defenses at these sites are often vigorous. For instance, the stomach is normally sterile because few microbes can survive at the normal gastric pH of 4.0. However, if antacids are used to decrease gastric acidity, colonization of the stomach and trachea with gram-negative bacteria rapidly occurs.
The normal flora prevents colonization through numerous mechanisms. These organisms often have a selective advantage over colonizers in that they are already established in an anatomic niche. This means that they are bound to receptors on the host cell and are able to metabolize local nutrients. Many species of the normal flora are able to produce bacteriocins, proteins that are toxic to other bacterial strains or species. Finally, the normal flora promotes production of antibodies that may cross-react with colonizing organisms. For instance, an antibody produced against E coli, a gram-negative bacterium normally found in the large intestine, cross-reacts with the polysaccharide capsule of a meningitis-producing strain of N meningitidis. When the normal flora is altered (eg, by the administration of broad-spectrum antibiotics), one bacterial species may predominate or exogenous bacteria may gain a selective advantage, permitting colonization and predisposing the host to infection.
Constitutive defenses of the human body are nonspecific barriers against infectious diseases that do not require prior contact with the microorganism. These defenses consist of simple physical (eg, skin) and chemical (eg, acidic gastric secretions) barriers that prevent easy entry of microorganisms into the body. Some infectious agents use a vector (such as an insect) to bypass structural barriers and gain direct access to the blood or subcutaneous tissues of the body. Once an agent has entered the body, the major constitutive defenses are the acute inflammatory response and the complement system. These defenses can neutralize the agent, recruit phagocytic cells, and induce a more specific response through humoral and cell-mediated immunity. The constitutive defenses of the body are important from an evolutionary perspective in enabling humans to encounter and adapt to a variety of new and changing environments.
The squamous epithelium of the skin is the first line of defense against microorganisms encountered in the outside world. As keratinized epithelial surface cells desquamate, the skin maintains its protective barrier by generating new epithelial cells beneath the surface. The skin is also bathed with oils and moisture from the sebaceous and sweat glands. These secretions contain fatty acids that inhibit bacterial growth. Poor vascular supply to the skin may result in skin breakdown and increased susceptibility to infection. For example, chronically debilitated or bedridden patients may suffer from decubitus ulcers as a result of constant pressure on dependent body parts, predisposing to severe infections by otherwise harmless skin flora.
The mucous membranes also provide a physical barrier to microbial invasion. The mucous membranes of the mouth, pharynx, esophagus, and lower urinary tract are composed of several layers of epithelial cells, whereas those of the lower respiratory tract, the GI tract, and the upper urinary tract are delicate single layers of epithelial cells. These membranes are covered by a protective layer of mucus, which traps foreign particles and prevents them from reaching the lining epithelial cells. Because the mucus is hydrophilic, many substances produced by the body easily diffuse to the surface, including enzymes with antimicrobial activity such as lysozyme and peroxidase.
When a microorganism crosses the epidermis or the epithelial surface of the mucous membranes, it encounters other components of the host constitutive defenses. These responses are constitutive because they are nonspecific and do not require prior contact with the organism to be effective. Clinically, signs of inflammation (heat, erythema, pain, and swelling) are the characteristic features of localized infection, secondary tissue injury, and the body’s response to this injury. Blood supply to the affected area increases in response to vasodilation, and the capillaries become more permeable, allowing antibodies, complement, and white blood cells to cross the endothelium and reach the site of injury. An important consequence of inflammation is that the pH of the inflamed tissues is lowered, creating an inhospitable environment for the microbe. The increased blood flow to the area allows continued recruitment of inflammatory cells as well as the necessary components for tissue repair and recovery.
When a microorganism enters host tissue, it activates the complement system and components of the coagulation cascade and induces the release of chemical mediators of the inflammatory response. These mediators result in the increased vascular permeability and vasodilation characteristic of inflammation. For example, the anaphylatoxins C3a, C4a, and C5a, produced by the activation of complement, stimulate the release of histamine from mast cells. Histamine dilates the blood vessels and further increases their permeability. Bradykinin is also released, increasing vascular permeability.
Proinflammatory cytokines include interleukin-1 (IL-1), IL-6, tumor necrosis factor, and interferon-γ. These factors, singly or in combination, promote fever, produce local inflammatory signs, and trigger catabolic responses. During severe infection, a change in hepatic synthesis of proteins occurs, resulting in an increase in some proteins and a decrease in others. Most notable is the increase in “acute-phase reactants” that include rheumatoid factor, C-reactive protein, ferritin, and various proteinase inhibitors. The erythrocyte sedimentation rate, a nonspecific marker of inflammation, also rises, whereas the serum levels of various elements such as zinc and iron decrease. A catabolic state is further augmented by simultaneous increases in levels of circulating cortisol, glucagon, catecholamines, and other hormones.
Mild-to-moderate inflammatory responses serve important host defense functions. For example, elevated body temperature may inhibit viral replication. Inflammatory hyperemia and systemic neutrophilia optimize phagocyte delivery to sites of infection. The decreased availability of iron inhibits the growth of microbes such as Yersinia that require this element as a nutrient. However, when the inflammatory responses become extreme, extensive tissue damage can result, as in the case of sepsis.
The complement system is composed of a series of plasma protein and cell membrane receptors that are important mediators of host defenses and inflammation (Figure 4–4). Most of the biologically significant effects of the complement system are mediated by the third component (C3) and the terminal components (C5–9). To carry out their host defense and inflammatory functions, C3 and C5–9 must first be activated. Two pathways of complement activation have been recognized and have been termed the classic and alternative pathways. The classic pathway is activated by antigen–antibody complexes or antibody-coated particles, and the alternative pathway is activated by mechanisms independent of antibodies, usually by interaction with bacterial surface components. Both pathways form C3 convertase, which cleaves the C3 component of complement, a key protein common to both pathways. The two pathways then proceed in identical fashion to bind late-acting components to form a membrane attack complex (C5–9), which results in target cell lysis.
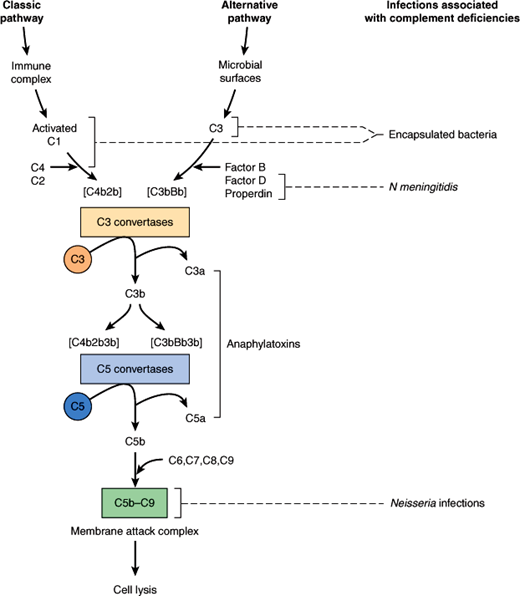
Once activated, complement functions to enhance the antimicrobial defenses in several ways. Complement facilitates phagocytosis through proteins called opsonins, which coat invading microorganisms, making them susceptible to engulfment and destruction by neutrophils and macrophages. The complement-derived membrane attack complex inserts itself into the membrane of a target organism, leading to increased permeability and subsequent lysis of the cell. Complement also acts indirectly through production of substances that are chemotactic for white blood cells and through promotion of the inflammatory response.
Inherited disorders of complement are associated with an increased risk of bacterial infection. The specific infections seen in complement-deficient patients relate to the biologic functions of the missing component (Figure 4–4). Patients with a deficiency of C3 or of a component in either of the two pathways necessary for the activation of C3 typically have increased susceptibility to infections with encapsulated bacteria such as S pneumoniae and Haemophilus influenzae. In contrast, patients with deficiencies of C5–9 have normal resistance to encapsulated bacteria because C3b-mediated opsonization is intact. These patients, however, are unusually susceptible to life-threatening infections with N meningitidis and N gonorrhoeae because they are unable to form a membrane attack complex and, therefore, cannot lyse the Neisseria cell membrane.
After the natural barriers of the skin or mucous membranes have been penetrated, the phagocytic cells—neutrophils, monocytes, and macrophages—constitute the next line of host defense. The process of internalizing organisms by these cells (phagocytosis) involves attachment of the organism to the cell surface. This triggers extension of a pseudopod to enclose the bacterium in an endocytic vesicle, or phagosome. The circulating polymorphonuclear neutrophil (PMN) is an important component of the host immune response that in the absence of infection circulates in a quiescent state. When chemotactic factors, arachidonic acid metabolites, or complement cleavage fragments interact with specific PMN membrane receptors, the neutrophil rapidly becomes activated and moves toward the chemoattractants. After phagocytosis, the mechanisms by which the phagolysosome destroys the microorganism can be divided into oxygen-independent and oxygen-dependent processes. Functional defects in circulating neutrophils or decreases in absolute number of neutrophils are important risk factors for infection.
Neutropenia, defined as an absolute neutrophil count of less than 1000 cells/μL, is a common predisposing factor for life-threatening bacterial and fungal infections. The risk of infection is inversely proportionate to the number of neutrophils, rising significantly with neutrophil counts less than 500 cells/μL. The longer the duration of profound neutropenia, the greater is the risk of infection. At the first sign of infection (eg, fever), these patients should be given broad-spectrum antibacterial agents to cover gram-negative bacterial pathogens. In addition to impaired immunity, neutropenic hosts often have additional risk factors for infection such as the need for long-term indwelling central venous catheters (predisposing infection with skin bacteria) and the frequent use of parenteral nutrition (predisposing to fungal infection).
Several inherited disorders of neutrophil function have been described. Chédiak-Higashi syndrome is a rare autosomal recessive hereditary disorder in which the neutrophils have a profound defect in the formation of intracellular granules. Opsonized bacteria such as Staphylococcus aureus are ingested normally, but viable bacteria persist intracellularly, presumably because of the inability of the neutrophil’s intracellular granules to fuse with phagosomes to form phagolysosomes. Patients with Chédiak-Higashi syndrome experience recurrent bacterial infections, most frequently involving the skin and soft tissues and the upper and lower respiratory tracts.
Myeloperoxidase deficiency is the most common neutrophil disorder, with a prevalence of one case per 2000 individuals. In this disorder, phagocytosis, chemotaxis, and degranulation are normal, but microbicidal activity for bacteria is delayed. In general, these patients do not suffer from recurrent infections. In contrast, chronic granulomatous disease is a genetically heterogeneous group of inherited disorders characterized by the failure of phagocytic cells to produce superoxides. The defect involves neutrophils, monocytes, eosinophils, and some macrophages. Oxygen-dependent intracellular killing is impaired, and these patients are susceptible to recurrent, often life-threatening infections. Patients with chronic granulomatous disease also tend to form granulomas in tissues, particularly in the lungs, liver, and spleen, and are particularly susceptible to infection with S aureus and Aspergillus species.
Although constitutive host defenses against infectious agents are generally nonspecific and do not require prior exposure to the invading agent, induced defenses are highly specific and are qualitatively and quantitatively altered by prior antigenic exposure. Details of the pathophysiology of the host immune system are covered in Chapter 3. Recurrent infections or infections with unusual organisms may be clues to an underlying defect in the induced immune response (Table 4–2).
| Host Defect | Examples of Related Immunodeficiency States | Commonly Associated Infections |
|---|---|---|
| T-lymphocyte deficiency or dysfunction | AIDS | Viral: reactivation of herpes group viruses (HSV, VZV, CMV) |
| Solid organ transplant | ||
| Corticosteroid use | Bacterial: Listeria monocytogenes, Mycobacterium tuberculosis | |
| Idiopathic CD4+ leukopenia | Fungal: Candida esophagitis, Aspergillus, cryptococcal meningitis | |
| Parasitic: Toxoplasma gondii | ||
| B-cell deficiency or dysfunction | Common variable immunodeficiency | Viral: enteroviruses |
| Agammaglobulinemia | Bacteria: Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis, Mycoplasma pneumonia | |
| Chronic lymphocytic leukemia (secondary hypogammaglobulinemia) | ||
| Parasites: Giardia lamblia | ||
| Mixed T- and B-cell deficiency or dysfunction | Ataxia-telangiectasia | Recurrent sinopulmonary infections |
| Severe combined immunodeficiency | Chronic diarrhea | |
| Mucocutaneous candidiasis | ||
| Viral: respiratory viruses, herpes group viruses |
An infectious disease occurs when a pathogenic organism causes inflammation or organ dysfunction. This may be caused directly by the infection itself, as when the etiologic agent multiplies in the host, or indirectly as a result of the host’s inflammatory response. Many infections are subclinical, not producing any obvious manifestations of disease. To cause overt infection, all microorganisms must go through the following stages (Table 4–3): The microorganism must (1) encounter the host, (2) gain entry into the host, (3) multiply and spread from the site of entry, and (4) cause host tissue injury, either directly (eg, cytotoxins) or indirectly (host inflammatory response). The severity of infection ranges from asymptomatic to life-threatening, and the course may be characterized as acute, subacute, or chronic. Whether infection is subclinical or overt, the outcome is either (1) resolution (eg, eradication of the infecting pathogen), (2) chronic active infection (eg, HIV or hepatitis), (3) prolonged asymptomatic excretion of the agent (eg, carrier state with Salmonella typhi), (4) latency of the agent within host tissues (eg, latent tuberculosis or varicella zoster virus), or (5) host death from infection.
| Stage of Infection | Factors Influencing Stage of Infection |
|---|---|
| Encounter |
|
| Entry |
|
| Multiplication and spread |
|
| Injury |
|
| Course of infection |
|
| Outcome of infection |
|
Except for congenital infections (acquired in utero) humans first encounter microorganisms at birth. During parturition, the newborn comes into contact with microorganisms present in the mother’s vaginal canal and on her skin. Most of the bacteria the newborn encounters do not cause harm, and for those that might cause infection, the newborn usually has passive immunity through antibodies acquired from the mother in utero. For example, neonates are protected against infection with H influenzae by maternal antibodies for the first 6 months of life until passive immunity wanes and the risk of infection with this bacterium increases. On the other hand, newborns whose mothers are vaginally colonized with group B streptococci are at increased risk in the perinatal period for serious infections such as sepsis or meningitis with this organism. For this reason, it is recommended that (1) vaginal cultures be done to screen for group B streptococci for all pregnant women and (2) intrapartum antibiotic prophylaxis be administered to those with positive detection of group B streptococci.
Direct entry of microorganisms into the host (ie, bypassing the usual chemical and physical barriers) may occur when (1) an insect vector directly inoculates the infectious agent into the host (mosquitoes transmitting malaria), (2) bacteria gain direct access to host tissues through loss of integrity of the skin or mucous membranes (trauma or surgical wounds), or (3) microbes gain access via instruments or catheters that allow communication between usually sterile sites and the outside world (indwelling venous catheters). Ingression occurs when an infectious agent enters the host via an orifice contiguous with the external environment. This primarily involves inhalation of infectious aerosolized droplets (M tuberculosis) or ingestion of contaminated foods (salmonella, hepatitis A virus).
Other infectious agents directly infect mucous membranes or cross the epithelial surface to cause infection. This commonly occurs in sexually transmitted diseases. For example, HIV can cross vaginal mucous membranes by penetration of virus-laden macrophages from semen.
After the initial encounter with the host, the infectious agent must successfully multiply at the site of entry. The process whereby the newly introduced microorganism successfully competes with normal flora and is able to multiply is termed colonization (eg, pneumococci colonizing the upper respiratory tract). When the microorganism multiplies at a normally sterile site, it is termed infection (eg, pneumococci multiplying in the alveoli, causing pneumonia). Factors that facilitate the multiplication and spread of infection include inoculum size (the quantity of infectious organisms introduced), host anatomic factors (eg, impaired ciliary function in children with cystic fibrosis), availability of nutrients for the microbe, physicochemical factors (eg, gastric pH), microbial virulence factors, and microbial sanctuary (eg, abscesses). An abscess is a special case in which the host has contained the infection but is unable to eradicate it, and these localized infections generally require surgical drainage. Once introduced, infections can spread along the epidermis (impetigo), along the dermis (erysipelas), along subcutaneous tissues (cellulitis), along fascial planes (necrotizing fasciitis), into muscle tissue (myositis), along veins (suppurative thrombophlebitis), into the blood (bacteremia, fungemia, viremia, etc), along lymphatics (lymphangitis), and into organs (eg, pneumonia, brain abscesses, hepatitis).
Infections cause direct injury to the host through a variety of mechanisms. If organisms are present in sufficient numbers and are of sufficient size, mechanical obstruction can occur (eg, children with roundworm gastrointestinal infections may present with bowel obstruction). More commonly, pathogens cause an intense secondary inflammatory response, which may result in life-threatening complications (eg, children with H influenzae epiglottitis may present with mechanical airway obstruction secondary to intense soft tissue swelling of the epiglottis). Some bacteria produce neurotoxins that affect host cell metabolism rather than directly causing cell damage (eg, tetanus toxin antagonizes inhibitory neurons, causing unopposed motor neuron stimulation, manifested clinically as sustained muscle rigidity). Host cell death can occur by a variety of mechanisms. Shigella produces a cytotoxin that causes death of large intestine enterocytes, resulting in the clinical syndrome of dysentery. Poliovirus-induced cell lysis of the anterior horn cells of the spinal cord causes flaccid paralysis. Gram-negative bacterial endotoxin can initiate a cascade of cytokine release, resulting in sepsis syndrome and septic shock.
The time course of an infection can be characterized as acute, subacute, or chronic, and its severity may vary from asymptomatic to life-threatening. Many infections that begin as mild and easily treatable conditions readily progress without prompt treatment. Small, seemingly insignificant skin abrasions superinfected with toxic shock syndrome toxin (TSST-1)–producing S aureus can result in fulminant infection and death. Even indolent infections, such as infective endocarditis resulting from Streptococcus viridans, can be fatal unless they are recognized and appropriately treated.
There are three potential outcomes of infection: recovery, chronic infection, and death. Most infections resolve, either spontaneously (eg, rhinovirus, the leading cause of the common cold) or with medical therapy (eg, after treatment of streptococcal pharyngitis with penicillin). Chronic infections may be either saprophytic, in which case the organism does not adversely affect the health of the host; or parasitic, causing tissue damage to the host. An example of the former is Salmonella typhi, which may be harbored asymptomatically in the gallbladder of about 2% of individuals after acute infection. Chronic infection with the hepatitis B virus may be either saprophytic, in which case the human host is infectious for the virus but has no clinical evidence of liver damage, or parasitic, with progressive liver damage and cirrhosis. A final form of chronic infection is tissue latency. Varicella-zoster virus, the agent causing chickenpox, survives in the dorsal root ganglia, with reactivation causing a dermatomal eruption with vesicles or shallow ulcerations, commonly known as shingles or zoster. When the ability of the immune system to control either the acute or the chronic infection is exceeded, the infection may result in host death.
| Organ System | History | Physical Examination | Laboratory or Radiographic Data |
|---|---|---|---|
| General | Fever | Fever | Positive blood cultures |
| Chills | Tachycardia | ↑ White blood cell count | |
| Fatigue | Diaphoresis | ↑ Rheumatoid factor | |
| Malaise | Rigors | ||
| HEENT | Blurred vision | Subconjunctival hemorrhage | |
| Roth spots (funduscopic examination) | |||
| Endophthalmitis | |||
| Respiratory | Shortness of breath | Diminished breath sounds | Pleural-based cavitary lesions (septic pulmonary emboli) |
| Pleuritic chest pain | Crackles | Pulmonary edema (heart failure) | |
| Cardiac | Shortness of breath | Murmur (systolic or diastolic) | Vegetation on echocardiogram |
| ↑ Jugular venous pressure | Prolonged PR interval on electrocardiogram (heart block with myocardial ring abscess) | ||
| Lower extremity edema | |||
| Gastrointestinal | Abdominal pain | Splenomegaly | Splenic infarct or abscess on CT scan |
| Genitourinary | Flank pain | Costovertebral angle tenderness | ↑ Blood urea nitrogen |
| Blood in urine | ↑ Serum creatinine | ||
| Hematuria | |||
| ↓ Serum complement levels (C3, C4, CH50) due to immune-complex glomerulonephritis | |||
| Musculoskeletal | Joint pain | Joint effusion, erythema, warmth | Arthrocentesis (↑ white blood cell count; bacteria on Gram stain; positive cultures) |
| Back pain | Spinal tenderness to palpation | MRI of spine (discitis, osteomyelitis, epidural abscess) | |
| Cutaneous | Rash | Splinter hemorrhages (nail beds) | |
| Janeway lesions (painless hemorrhagic macules on palms and soles) | |||
| Petechiae | |||
| Osler nodes (painful nodules on fingers and toes) | |||
| Neurologic | Headache | Altered consciousness | MRI of brain (septic emboli, mycotic aneurysm) |
| Confusion | Focal weakness | ||
| Seizure |
All infectious agents, regardless of specific mechanisms, must successfully reproduce and evade host defense mechanisms. This knowledge helps the physician to prevent infections (eg, vaccinate against influenza virus); to treat and cure infection (eg, antibiotics for E coli urinary tract infection); and when infection cannot be cured, to prevent further transmission, recurrence, or reactivation (eg, barrier protection to reduce the sexual spread of genital herpes simplex infection).
Checkpoint
By what three general mechanisms do hosts resist colonization by pathogenic bacteria?
What are three ways in which the normal flora contributes to the balance between health and disease?
Which specific host defenses against infection do not require prior contact with the infecting organism?
What are the categories of outcomes from an infection?
Pathophysiology of Selected Infectious Disease Syndromes
Infective endocarditis refers to a bacterial or, rarely, a fungal infection of the cardiac valves. Infection of extracardiac endothelium is termed “endarteritis” and can cause disease that is clinically similar to endocarditis. The most common predisposing factor for infective endocarditis is the presence of structurally abnormal cardiac valves. Consequently, patients with a history of rheumatic or congenital heart disease, a prosthetic heart valve, or a history of prior endocarditis are at increased risk for infective endocarditis. Infection involves the left side of the heart (mitral and aortic valves) almost exclusively, except in patients who are injection drug users or, less commonly, in patients with valve injury from a pulmonary artery (Swan-Ganz) catheter, in whom infection of the right side of the heart (tricuspid or pulmonary valve) may occur.
The most common infectious agents causing native valve infective endocarditis are gram-positive bacteria, including viridians group streptococci, S aureus, and enterococci. The specific bacterial species causing endocarditis can often be anticipated on the basis of host factors. Injection drug users commonly introduce skin bacteria such as S aureus into the blood when nonsterile needles are used or the skin is not adequately cleaned before needle insertion. Patients with recent dental work are at risk for transient bacteremia with normal oral flora, particularly viridians group streptococci, with subsequent endocarditis. Genitourinary tract infections with enterococci may lead to bacteremia and subsequent seeding of damaged heart valves. Patients with prosthetic heart valves are also at increased risk for infective endocarditis resulting from skin flora such as S epidermidis or S aureus. Before the availability of antibiotics, infective endocarditis was a uniformly fatal disease. Even with antibiotics, the case fatality rate for endocarditis approaches 25%, and definitive cure often requires both prolonged intravenous antibiotic administration and urgent surgery to replace infected cardiac valves.
Several hemodynamic factors predispose patients to endocarditis: (1) a high-velocity jet stream causing turbulent blood flow, (2) flow from a high-pressure to a low-pressure chamber, and (3) a comparatively narrow orifice separating the two chambers that creates a pressure gradient. The lesions of infective endocarditis tend to form on the surface of the valve in the cardiac chamber with the lower pressure (eg, on the ventricular surface of an abnormal aortic valve and on the atrial surface of an abnormal mitral valve). Endothelium damaged by turbulent blood flow results in exposure of extracellular matrix proteins, promoting the deposition of fibrin and platelets, which form sterile vegetations (nonbacterial thrombotic endocarditis or marantic endocarditis). Infective endocarditis occurs when microorganisms are deposited onto these sterile vegetations during the course of bacteremia (Figure 4–5). Not all bacteria adhere equally well to these sites. For example, E coli, a frequent cause of urosepsis, is rarely implicated as a cause of endocarditis. Conversely, virulent organisms such as S aureus can invade intact endothelium, causing endocarditis in the absence of preexisting valvular abnormalities.



