Introduction
The Centers for Disease Control and Prevention estimates that in the United States, more than 10% of people 20 years and older (or more than 20 million individuals) have chronic kidney disease. In addition, many more people suffer from acute kidney injury and other forms of kidney disease annually. Thus, clinicians of all specialties will encounter patients with renal disorders, and it behooves them to be aware of the various risk factors and causes of kidney disease. This is particularly important because with early detection and appropriate management, most forms of kidney disease to prevent or at least slow the rate of progression to kidney failure or other complications.
The kidneys serve a crucial role in filtering blood, and a wide range of diseases of other organ systems and systemic diseases may be manifested in the kidney. For example, renal disease is a prominent presentation of long-standing diabetes mellitus and hypertension and of autoimmune disorders such as systemic lupus erythematosus.
A particular challenge is that patients are typically asymptomatic until relatively advanced kidney failure is present. There are no pain receptors within the substance of the kidney, so pain is not a prominent presenting complaint, except in those renal diseases (eg, nephrolithiasis) in which there is involvement of the ureter or the renal capsule. In early stages of kidney disease, patients may only have abnormalities of urine volume (eg, oliguria) or composition (eg, presence of red blood cells and/or protein). Later, they may manifest systemic symptoms and signs of lost renal function (eg, edema, fluid overload, electrolyte abnormalities, and anemia). Depending on the nature of the renal disease, they may progress to display a wide range of chronic complications resulting from inadequate renal function.
The kidneys play multiple roles in the body, including blood filtration, metabolism and excretion of endogenous and exogenous compounds, and endocrine functions. Perhaps most significantly, the kidneys are the primary regulators of fluid, acid-base, and electrolyte balance in the body, and this remarkable pair of organs maintains homeostasis across a broad array of dietary and environmental changes. An understanding of each of these roles is required to illuminate the pathophysiologic basis behind the many different manifestations of kidney disease.
Checkpoint
1. What are some important causes of renal disease?
2. What are some consequences of renal failure?
Normal Structure & Function of the Kidney
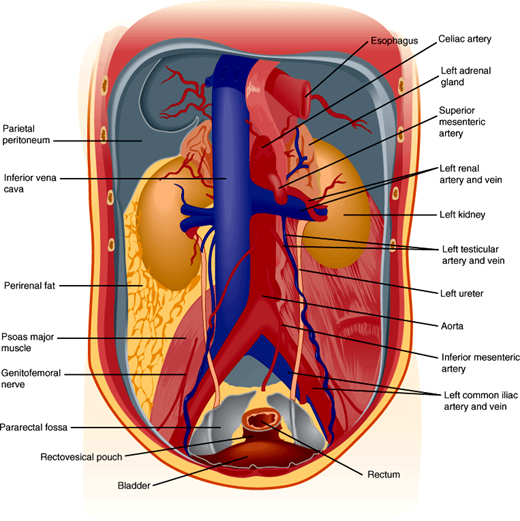
The kidneys maintain homeostasis while functioning under a tremendous range of environmental water and salt availability. For example, the kidneys have the capacity to excrete free water in freshwater fish, varying amounts of water and solute in humans, and an extremely concentrated urine in the kangaroo rat, which can live its entire life without access to water. The kidneys are a pair of encapsulated organs located in the retroperitoneal area (Figure 16–1). A renal artery enters and a renal vein exits from each kidney at the hilum. Approximately 20% of cardiac output goes to the kidneys. Blood is filtered in the kidneys, removing wastes—in particular urea and nitrogen-containing compounds—and regulating extracellular electrolytes and intravascular volume. Because renal blood flow is from cortex to medulla and because the medulla has a relatively low blood flow for a high rate of metabolic activity, the normal oxygen tension in the medulla is lower than in other parts of the kidney. This makes the medulla particularly susceptible to ischemic injury.
The anatomic unit of kidney function is the nephron, a structure consisting of a tuft of capillaries termed the glomerulus, the site at which blood is filtered, and a renal tubule from which water and salts in the filtrate are reclaimed (Figure 16–2). Each human kidney has approximately 1 million nephrons.
Figure 16–2
Structures of the kidney. A: Landmarks of the normal kidney. B: Glomerulus and glomerular capillary. C: Detailed structure of the glomerulus and the glomerular filtration membrane composed of endothelial cell, basement membrane, and podocyte. Note that for clarity the distal tubule is separated from the glomerulus in A; however, its true anatomic relationship, which is essential for physiologic function, is illustrated in B. (Redrawn, with permission, from Chandrasoma P et al, eds. Concise Pathology, 3rd ed. Originally published by Appleton & Lange. Copyright © 1998 by The McGraw-Hill Companies, Inc.)
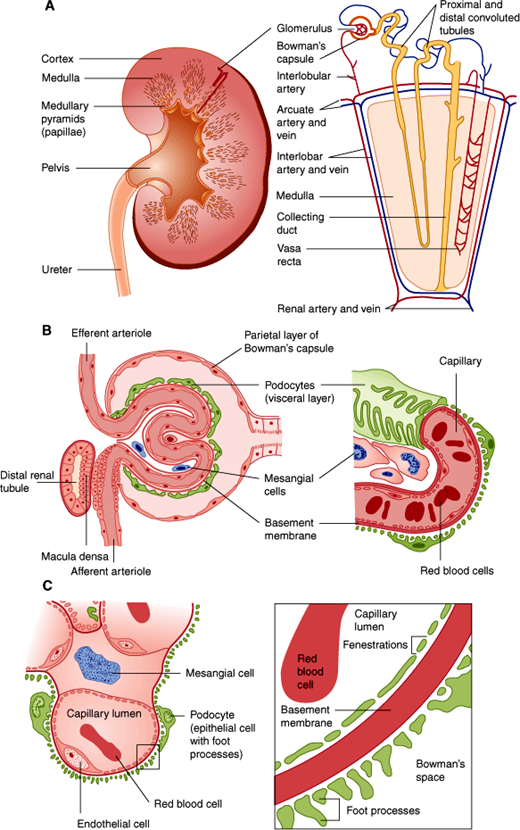
A glomerulus consists of an afferent and an efferent arteriole and an intervening tuft of capillaries lined by endothelial cells and covered by epithelial cells that form a continuous layer with those of the Bowman capsule and the renal tubule. The space between capillaries in the glomerulus is called the mesangium. Material comprising a basement membrane is located between the capillary endothelial cells and the epithelial cells (Figure 16–2).
Closer examination of glomerular histology and cell biology reveals unique features not found in most peripheral capillaries (Figure 16–2). First, the glomerular capillary endothelium is fenestrated. However, because the endothelial cells have a coat of negatively charged glycoproteins and glycosaminoglycans, they normally exclude plasma proteins such as albumin. On the other side of the glomerular basement membrane are the epithelial cells. Termed “podocytes” because of their numerous extensions or foot processes, these cells are connected to one another by modified desmosomes.
The mesangium is an extension of the glomerular basement membrane but is less dense and contains two distinct cell types: intrinsic glomerular cells and tissue macrophages. Both cell types contribute to the development of immune-mediated glomerular disease by their production of, and response to, cytokines such as transforming growth factor-β (TGF-β).
Understanding the complex organization of the glomerulus is crucial for understanding both normal renal function and also the characteristics of different glomerular diseases. Thus, in some conditions immune complexes may accumulate under the epithelial cells, whereas in others they accumulate under the endothelial cells. Likewise, because immune cells are not able to cross the glomerular basement membrane, immune complex deposition under the epithelial cells is generally not accompanied by a cellular inflammatory reaction (see later discussion).
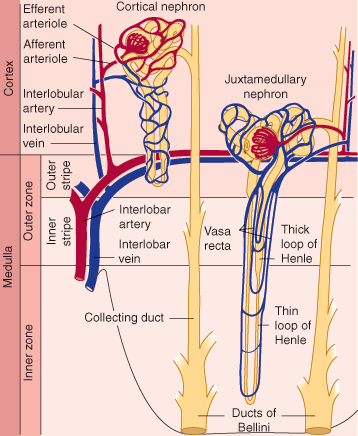
The renal tubule itself has a number of different structural regions: the proximal convoluted tubule, from which most of the electrolytes and water are reclaimed; the loop of Henle; and a distal convoluted tubule and collecting duct (Figure 16–3), where the urine is concentrated and additional electrolyte and water changes are made in response to hormonal control.
Approximately 100–120 mL/min of glomerular filtrate is generated in a normal adult with two fully functional kidneys. The approximate mass cutoff of substances for filtration is 70 kDa. However, substances smaller than this are often retained, sometimes because of charge effects or because they are tightly bound to other proteins to give them a larger effective size.
After filtration at the glomerulus, there is extensive reabsorption of filtered substances along the renal tubular network. The degree of reabsorption varies by substance and anatomic location in the tubules, thus allowing for differential regulation of constituent components. Most (60–70%) of the filtered Na+—and, under normal conditions, almost all of the K+ and glucose—is actively resorbed from the tubular fluid via co-transporter mechanisms in the proximal tubule. Water is resorbed passively and along osmotic gradients established by the reabsorption of Na+. In addition to absorption, a number of substances are secreted into the tubular fluid through the action of transporters along the renal tubule. Examples of substances that are secreted include organic anions and cations such as creatinine, histamine, and many drugs and toxins.
Normally, about 30 mL/min of isotonic filtrate is delivered to the loops of Henle, where a countercurrent multiplier mechanism achieves concentration of the urine. The loop of Henle passes down into the medulla of the kidney, where secretion of Na+ from the cells in the thick ascending limb establishes a hypertonic concentration gradient to reabsorb water from the tubular fluid across the cells of the descending limb.
Under normal circumstances, no more than 5–10 mL/min of glomerular filtrate is delivered to the collecting ducts. Water absorption in the collecting ducts occurs directly through water channels controlled by vasopressin (also known as antidiuretic hormone [ADH]). Under the control of aldosterone, Na+ resorption from tubular fluid and K+ and H+ transport into tubular fluid occur in different types of cells in the renal collecting ducts. Even though it deals with less than one tenth of the total glomerular filtrate, the collecting duct is the site of regulation of urine volume and the site at which water, Na+, acid-base, and K+ balance are achieved. The crucial role of the collecting duct in regulation of kidney function depends on two features: First, the collecting duct is under hormonal control, in contrast to the proximal tubule, whose actions are generally a simple function of volume and composition of tubular fluid and constitutively active transporters. Second, the collecting duct is the last region of the renal tubule traversed before the remaining 1–2 mL/min of the original glomerular filtrate exits into the ureters as urine.
The kidney plays an important role in blood pressure regulation by virtue of its effect on Na+ and water balance, major determinants of blood pressure. First, the Na+ concentration in the proximal tubular fluid is sensed at the macula densa (Figure 16–2), part of the juxtaglomerular apparatus. The juxtaglomerular apparatus also assesses the perfusion pressure of the blood, an important indicator of intravascular volume status under normal circumstances. Through the action of these two sensors, either low Na+ or low perfusion pressure acts as a stimulus to renin release. Renin, a protease made in the juxtaglomerular cells, cleaves angiotensinogen in the blood to generate angiotensin I, which is then cleaved to angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II raises blood pressure by triggering vasoconstriction directly and by stimulating aldosterone production and secretion in the adrenal cortex, resulting in Na+ and water retention by the collecting duct (see Chapter 21). All of these effects expand the extracellular fluid (ECF) and hence renal perfusion pressure, completing a homeostatic negative feedback loop that alleviates the initial stimulus for renin release. However, these mechanisms can also be maladaptive and contribute to the pathophysiology of various disease states.
Notably, the trigger for activation of the renin-angiotensin-aldosterone system is the physiologic signal of low effective circulating volume, which may not be synonymous with low total body volume. Edematous states (eg, heart failure, nephrotic syndrome, and cirrhosis) develop due to a pathophysiologic factor favoring fluid movement out of the intravascular space and into the interstitium or third spaces. In the case of heart failure, this is an elevated hydrostatic component related to cardiac congestion. In nephrotic syndrome, it is a fall in oncotic pressure due to loss of protein in the urine. In cirrhosis, there can be a combination of lower oncotic pressures from decreased protein production and increased hydrostatic pressure from hepatic congestion leading to third-spacing of fluid. In all of these conditions, the resultant decreased effective circulating volume signals the kidneys to progressively retain Na+ and water until a new equilibrium is established between the vascular space and interstitial space.
Another trigger for activation of the renin-angiotensin-aldosterone system is renovascular disease, an important cause of secondary hypertension. In renovascular disease, a fixed vascular abnormality in the renal arterial circulation (most commonly, atherosclerosis) results in impaired blood flow, generating the signal of low effective circulating volume despite a normal circulating volume. The resultant activation of the renin-angiotensin-aldosterone system leads to hypertension because of the direct vascular effects of angiotensin and because of the increased circulating volume due to an aldosterone-mediated increase in Na+ reabsorption.
Intravascular volume depletion also triggers vasopressin release. Receptors in the carotid body and elsewhere sense a fall in blood pressure and activate autonomic neural pathways, including fibers that go to the hypothalamus, where vasopressin release is controlled. Vasopressin is released and travels via the bloodstream throughout the body. At the collecting duct, vasopressin facilitates insertion of water channels, thereby increasing the number of water channels. This results in reabsorption of free water. Further discussions of water balance and the role of vasopressin are presented in Chapter 19.
Along with the pulmonary system, the kidneys play a primary role in acid-base homeostasis. In normal conditions, the arterial blood pH is maintained within the range of 7.35–7.45 through a buffering system in which bicarbonate plays a key role:
H+ + HCO3− ↔ H2CO3 ↔ H2O + CO2
For example, a drop in pH (increase in H+ concentration) leads to an increase in CO2, which can be exhaled from the lungs. This immediate buffering effect depletes the body’s stores of bicarbonate. The kidneys subsequently excrete additional H+ and thus serve to replete bicarbonate stores. In this system, the pulmonary response to acid-base imbalance is rapid (seconds to minutes), whereas the kidney’s response is delayed (hours to days). However, the lungs can only excrete volatile acids, and removal of nonvolatile (or “fixed”) acids relies on the kidneys.
During the ingestion of a normal daily diet, humans generate an obligate acid load from the metabolism of protein. To maintain homeostasis, this acid load is excreted in the kidneys. The primary site of acid excretion is the distal collecting duct, where H+ is secreted into the tubular lumen where it combines primarily with ammonia (NH3) to form ammonium (NH4+), which is subsequently excreted in the urine.
In addition to acid excretion, the kidneys regulate acid-base through the reabsorption and regeneration of bicarbonate, primarily in the proximal tubule. Insight into the functional roles of the proximal and distal renal tubules in maintaining acid-base balance can be seen in the clinical features of the various forms of renal tubular acidosis (Table 16–1).
| Type 1 (Distal) | Type 2 (Proximal) | Type 4 | |
|---|---|---|---|
| Basic defect | Decreased distal acidification | Diminished proximal HCO3– reabsorption | Aldosterone deficiency or resistance |
| Urine pH during acidemia | >5.3 | Variable: >5.3 if above reabsorptive threshold: <5.3 if below | Usually <5.3 |
| Plasma [HCO3–], untreated | Often extremely low ~10 mEq/L | Usually 14–20 mEq/L | Usually >15 mEq/L |
| Fractional excretion of HCO3– at normal plasma [HCO3–] | <3% in adults; may reach 5–10% in young children | >15–20% | <3% |
| Diagnosis | Response to NaHCO3 or NH4CI | Response to NaHCO3 | Measure plasma aldosterone concentration |
| Plasma [K+] | Usually reduced or normal; elevated with voltage defect | Normal or reduced | Elevated |
| Dose of HCO3– to normalize plasma [HCO3–], mEq/kg per day | 1–2 in adults; 4–14 in children | 10–15 | 1–3; may require no alkali if hyperkalemia corrected |
| Nonelectrolyte complications | Nephrocalcinosis and nephrolithiasis | Rickets or osteomalacia | None |
Metabolic acidosis is a common and potentially severe condition that warrants careful evaluation in the clinical setting. Several mechanisms can lead to the development of metabolic acidosis. First, the excess production of endogenous acids can exceed the ability of the kidneys to excrete H+. This can occur in advanced kidney failure where the kidneys’ ability to generate ammonium is diminished. Conversely, metabolic acidosis can develop from excess production of endogenous acids even in the face of intact renal function (eg, in lactic acidosis from tissue ischemia or in diabetic ketoacidosis). Second, metabolic acidosis can result from the ingestion of exogenous acids (eg, in intoxication with methanol or ethylene glycol, which are metabolized to formic acid and oxalic acid, respectively). Third, metabolic acidosis can develop through the loss of bicarbonate, which can occur from failure to reabsorb bicarbonate in the kidney (ie, proximal renal tubular acidosis) or from gastrointestinal (GI) loss of bicarbonate-rich fluids (eg, severe diarrhea, pancreatic fistula). Fourth, the administration of large amounts of bicarbonate-depleted solution to patients can lead to a dilutional acidosis.
Potassium balance is primarily regulated in the distal collecting duct, where it is secreted into the lumen in response to aldosterone-mediated Na+ reabsorption. Therefore, aldosterone is the primary hormonal regulator of K+. Indeed, in addition to the angiotensin-mediated stimulus discussed above, hyperkalemia is a signal for aldosterone release, while hypokalemia provides negative feedback for such release (see Chapter 21). The kidneys’ ability to regulate K+ balance is such that K+ excretion can be upregulated to exceed even the amount filtered in the glomerulus.
Hypokalemia can develop as a result of intracellular K+ shifting (eg, alkalosis, use of β-agonist therapy), extrarenal losses (eg, diarrhea), or renal losses. In general, increased delivery of Na+ to the distal tubules/collecting duct will result in increased K+ secretion, with the most common causes of renal K+ wasting being diuretic use and osmotic kaliuresis. Hyperaldosteronism, either primary aldosteronism from an adrenal tumor (ie, Conn syndrome) or secondary (eg, hyperreninemic) hyperaldosteronism, frequently presents with hypokalemia due to unregulated Na+ reabsorption with resultant secretion of both K+ and H+. Therefore, the clinical presentation of hypokalemia with hypertension and metabolic alkalosis should prompt an evaluation for a state of aldosterone excess. Further discussion of the role of the renin-angiotensin-aldosterone system in the regulation of potassium and intravascular volume is presented in Chapter 21.
Hyperkalemia can occur from extracellular shifting of potassium (eg, acidosis), cellular release of potassium (eg, hemolysis), increased ingestion of potassium, or decreased renal excretion of potassium (eg, renal insufficiency or kidney failure). Numerous drugs can also interfere with renal K+ excretion.
The kidney plays a number of important roles in Ca2+ and phosphate homeostasis. First, the kidney is the site of 1α-hydroxylation or 24-hydroxylation of 25-hydroxycholecalciferol, the hepatic metabolite of vitamin D3. This produces calcitriol (or 1,25-dihydroxy vitamin D), the biologically active form of vitamin D that increases Ca2+ absorption from the gut. Second, the kidney is a site of action of parathyroid hormone (PTH), resulting in Ca2+ retention and phosphate wasting in the urine. Further discussion of the role of the kidney in Ca2+ and phosphate homeostasis is presented in Chapter 17.
The kidney is the main site of production of the hormone erythropoietin, which stimulates bone marrow production and maturation of red blood cells. The signal for erythropoietin production is thought to be the level of blood oxygenation, which is monitored in the kidney. With progressive renal insufficiency, the capacity to produce erythropoietin becomes impaired and anemia can develop. Anemia typically begins to occur when the glomerular filtration rate (GFR) has fallen to 30–45 mL/min or less, and it is nearly universally observed in patients with end-stage renal disease. The primary management of the anemia of chronic kidney disease is hormone replacement therapy with a recombinant analog of erythropoietin. Additional discussion of the role of erythropoietin in the regulation of red blood cell mass is presented in Chapter 6.
There are a variety of physical, hormonal, and neural mechanisms by which the functions of the kidney are controlled. Vasopressin, together with the physics of the countercurrent multiplier in the loop of Henle and the hypertonic medullary interstitium, makes it possible to concentrate the urine under normal circumstances. This confers on the healthy kidney the ability to maintain fluid homeostasis under widely diverse conditions (by generating either a concentrated or dilute urine, depending on whether the body needs to conserve or excrete salt and water).
Tubuloglomerular feedback refers to the ability of the kidney to regulate the GFR in response to the solute concentration in the distal renal tubule. When an excessive concentration of Na+ in the tubular fluid is sensed by the macula densa, afferent arteriolar vasoconstriction is triggered. This diminishes the GFR so that the renal tubule has a smaller solute load per unit time, allowing Na+ to be more efficiently reclaimed from tubular fluid. A variety of vasoactive substances, including adenosine, prostaglandins, nitric oxide, and peptides such as endothelin and bradykinin, contribute to the humoral control of tubuloglomerular feedback.
Another important challenge for the kidney is regulation of renal cortical versus medullary blood flow. Renal cortical blood flow needs to be sufficient to maintain a GFR high enough to clear renally excreted wastes efficiently without exceeding the capacity of the renal tubules for solute reabsorption. Likewise, medullary blood flow must be closely regulated. Excessive medullary blood flow can disrupt the osmolar gradient achieved by the countercurrent exchange mechanism. Insufficient medullary blood flow can result in anoxic injury to the renal tubule. From the perspective of individual nephrons, redistribution of blood flow from cortex to medulla involves preferentially supplying blood (and, therefore, oxygen) to those nephrons with long loops of Henle that dip down into the inner medulla.
Adaptations of the kidney to injury can also be thought of as a form of regulation. Thus, loss of nephrons results in compensatory glomerular hyperfiltration (increased GFR per nephron) and renal hypertrophy. Although hyperfiltration may be adaptive in the short term, allowing maintenance of the total renal GFR, it has been implicated as a common inciting event in further nephron destruction from a variety of causes.
There are other clinically important adaptations to injury. Poor renal perfusion from any cause results in responses that improve perfusion through afferent arteriolar vasodilation and efferent arteriolar vasoconstriction in response to hormonal and neural cues. These regulatory effects are reinforced by inputs sensing Na+ balance. Alteration of Na+ balance is another way to influence blood pressure and hence renal perfusion pressure. Sympathetic innervation by the renal nerves influences renin release. Renal prostaglandins play an important role in vasodilation, especially in patients with chronically poor renal perfusion.
Checkpoint
3. What are the parts of the nephron, and what role does each part play in renal function?
4. How is renal function regulated?
5. What are the nonexcretory functions of the kidney?
6. What are the relationships, if any, between each nonexcretory function named previously and the kidney’s role in fluid, electrolyte, and blood pressure regulation?
Overview of Renal Disease
Renal disease can be categorized either by the site of the lesion (eg, glomerulopathy vs. tubulointerstitial disease) or by the nature of the factors that have led to kidney disease (eg, immunologic, metabolic, infiltrative, infectious, hemodynamic, or toxic).
Glomerular disease can be further categorized according to clinical presentation. Thus, some disorders present with profound proteinuria but no evidence of a cellular inflammatory reaction (nephrotic disorders), whereas others have variable degrees of proteinuria associated with red and white blood cells in the urine (nephritic disorders).
Nephrotic disorders typically show immune complex deposition at or under the epithelial cells, often with morphologic changes in the foot processes (Figure 16–4). This probably reflects damage to the selective nature of the glomerular filter (eg, by immune complex formation) or deposition of preformed complexes, in some cases with complement activation but without concomitant activation of a cellular immune response. Although the lack of a cellular immune response may limit the damage done, it also slows the resolution of the disorder, with proteinuria taking months or years to resolve even when the underlying disease has been brought under control.
Figure 16–4
Anatomy of a normal glomerular capillary is shown on the left. Note the fenestrated endothelium (EN), glomerular basement membrane (GBM), and the epithelium with its foot processes (EP). The mesangium is composed of mesangial cells (MC) surrounded by extracellular matrix (MM) in direct contact with the endothelium. Ultrafiltration occurs across the glomerular wall and through channels in the mesangial matrix into the urinary space (US). Typical localization of immune deposits and other pathologic changes is depicted on the right. (1) Uniform subepithelial deposits as in membranous nephropathy. (2) Large, irregular subepithelial deposits or “humps” seen in acute postinfectious glomerulonephritis. (3) Subendothelial deposits as in diffuse proliferative lupus glomerulonephritis. (4) Mesangial deposits characteristic of immunoglobulin A nephropathy. (5) Antibody binding to the glomerular basement membrane (as in Goodpasture syndrome) does not produce visible deposits, but a smooth linear pattern is seen on immunofluorescence. (6) Effacement of the epithelial foot processes is common in all forms of glomerular injury with proteinuria.
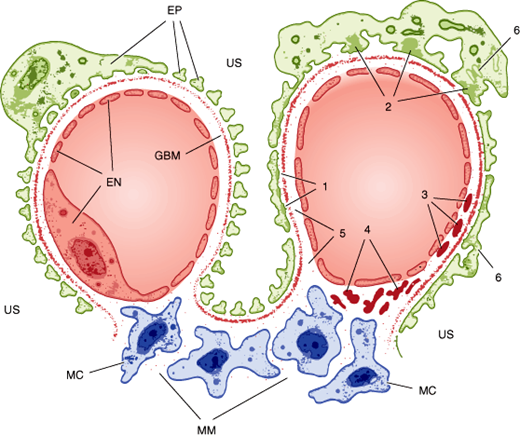
Nephritic disorders show immune complex deposits either in a subendothelial location or in the glomerular basement membrane or mesangium (Figure 16–4). The cellular immune system has ready access to all of these locations, and the resulting inflammatory reaction can be a “double-edged sword.” Thus, when the underlying process can be controlled, phagocytosis of the subendothelial deposits speeds recovery. On the other hand, an uncontrolled or prolonged inflammatory response can result in a greater degree of destruction of glomerular architecture, in part because of the local production and action of cytokines.
Specific regions of the kidney are particularly susceptible to certain kinds of injury: (1) Hemodynamic factors regulating blood flow have profound effects on the kidney both because the GFR, a primary determinant of renal function, depends on renal blood flow and because the kidney is susceptible to hypoxic injury; (2) the renal medulla is a low oxygen tension environment, which makes it very susceptible to ischemic injury; and (3) the glomerulus is the initial filter of blood entering the kidney and thus is a prominent site of injury related to immune complex deposition and complement fixation.
One useful organizing scheme that combines a consideration of both the site and the cause of renal disease in approaching patients with new renal failure is to first categorize the cause of the patient’s renal failure as prerenal, intrarenal, or postrenal and then to subdivide each of these categories according to specific causes and anatomic locations (Table 16–2).
|
|
|
|
|
|
Decreased kidney function leads to an accumulation of urea and an inability to maintain electrolyte, water, and acid-base balance. The failure to adequately excrete urea, manifested as progressive elevation of blood urea nitrogen (BUN), serum creatinine, and other poorly defined toxins, results in uremia (see Chronic Kidney Disease below). Uremia is a syndrome characterized by a unique set of symptoms, physical examination findings, and laboratory abnormalities (see Table 16–7), presumably caused by a buildup of one or more uncharacterized toxins. In the absence of adequate renal clearance, ingestion of excess amounts of Na+, K+, water, or acids results in electrolyte, volume, and acid-base abnormalities that can be life threatening. Furthermore, excess Na+ ingestion in a patient with renal insufficiency results in intravascular volume expansion, which in turn can lead to hypertension and heart failure.
Checkpoint
7. What characteristics of various parts of the nephron make it particularly susceptible to certain types of injury?
8. What are the features that distinguish prerenal, intrarenal, and postrenal causes of renal failure?
9. What are the major categories of complications of inadequate renal function?
Pathophysiology of Selected Renal Diseases
Acute kidney injury is produced by a heterogeneous group of disorders that have in common the rapid deterioration of renal function, resulting in accumulation in the blood of nitrogenous wastes that would normally be excreted in the urine. The patient presents with a rapidly rising BUN (ie, azotemia) and serum creatinine. Depending on the cause and when the patient comes to medical attention, there may be other presenting features as well (Table 16–3). Thus, diminished urine volume (oliguria) is commonly but not always seen. Urine volume may be normal early or indeed at any time in milder forms of acute kidney injury. Patients presenting relatively late may display any of the clinical manifestations described later.
| Syndrome | Important Clues to Diagnosis | Common Findings Not of Diagnostic Value |
|---|---|---|
| Acute or rapidly progressive renal failure | Anuria | Hypertension |
| Oliguria | Hematuria, proteinuria, pyuria, casts | |
| Documented recent decline in GFR | Edema | |
| Acute nephritis | Hematuria, red cell casts | Proteinuria, pyuria |
| Azotemia, oliguria | Circulatory congestion | |
| Edema, hypertension | ||
| Chronic renal failure | Azotemia for >3 months | Hematuria, proteinuria, casts |
| Prolonged symptoms or signs of uremia | Oliguria, polyuria, nocturia | |
| Symptoms or signs of renal osteodystrophy | Edema, hypertension | |
| Kidneys reduced in size bilaterally | Electrolyte disorders | |
| Broad casts in urinary sediment | ||
| Nephrotic syndrome | Proteinuria >3.5 g/1.73 m2 per 24 hours | Casts |
| Hypoalbuminemia | Edema | |
| Hyperlipidemia | ||
| Lipiduria | ||
| Asymptomatic urinary abnormalities | Hematuria | |
| Proteinuria (below nephrotic range) | ||
| Sterile pyuria, casts | ||
| Urinary tract infection | Bacteriuria >105 colonies/mL | Hematuria |
| Other infectious agent documented in urine | Mild azotemia | |
| Pyuria, leukocyte casts | Mild proteinuria | |
| Frequency, urgency | Fever | |
| Bladder tenderness, flank tenderness | ||
| Renal tubular defects | Electrolyte disorders | Hematuria |
| Polyuria, nocturia | Mild azotemia | |
| Symptoms or signs of renal osteodystrophy | Mild proteinuria | |
| Large kidneys | Fever | |
| Renal transport defects | ||
| Hypertension | Systolic/diastolic hypertension | Proteinuria |
| Casts | ||
| Azotemia | ||
| Nephrolithiasis | History of stone passage or removal | Hematuria |
| Stone seen by x-ray | Pyuria | |
| Renal colic | Frequency, urgency | |
| Urinary tract obstruction | Azotemia, oliguria, anuria | Hematuria |
| Polyuria, nocturia, urinary retention | Pyuria | |
| Slowing of urinary stream | Enuresis, dysuria | |
| Large prostate, large kidneys | ||
| Flank tenderness, full bladder after voiding |
The most widely accepted definition of acute kidney injury is a rise in serum creatinine of 0.3 mg/dL or more within a 48-hour period or a fall in urine output to less than 0.5 mL/kg/h for at least 6 hours.
The major causes of acute kidney injury are presented in Table 16–4.
| Disorder | Examples |
|---|---|
| Hypovolemia | Volume loss via the skin, gastrointestinal tract, or kidney. Hemorrhage. Sequestration of extracellular fluid (burns, pancreatitis, peritonitis). |
| Cardiovascular failure | Impaired cardiac output (infarction, tamponade). Vascular pooling (anaphylaxis, sepsis, drugs). |
| Extrarenal obstruction | Urethral occlusion: vesical, pelvic, prostatic, or retroperitoneal neoplasms. Surgical accident. Medication. Calculi. Pus, blood clots. |
| Intrarenal obstruction | Crystals (uric acid, oxalic acid, sulfonamides, methotrexate). |
| Bladder rupture | Trauma. |
| Vascular diseases | Vasculitis. Malignant hypertension. Thrombotic thrombocytopenia purpura. Scleroderma. Arterial or venous occlusion. |
| Glomerulonephritis | Immune complex disease. Anti-GBM disease. |
| Interstitial nephritis | Drugs. Hypercalcemia. Infections. Idiopathic. |
| Postischemic | All conditions listed above under hypovolemia and cardiovascular failure. |
| Pigment-induced | Hemolysis (transfusion reaction, malaria). Rhabdomyolysis (trauma, muscle disease, coma, heat stroke, severe exercise, potassium or phosphate depletion). |
| Poison-induced | Antibiotics. Contrast material. Anesthetic agents. Heavy metals. Organic solvents. |
| Pregnancy-related | Septic abortion. Uterine hemorrhage. Eclampsia. |
As demonstrated by the Starling equation, filtration across a glomerulus is determined by the hydrostatic and oncotic pressures in both the glomerular capillary and its surrounding tubular lumen as described by the relationship:
filtration = Kf [Pc – Pt] – σ[πc – πt]
Kf and σ are constants determined by the permeability of a given glomerulus and the effective contribution of osmotic pressure, respectively; Pc = intracapillary hydrostatic pressure, πc = intracapillary oncotic pressure, Pt = intratubular hydrostatic pressure, and πt = intratubular oncotic pressure. Perturbations in any of the above factors may alter renal filtration. Of particular importance is the intracapillary hydrostatic pressure which is determined by relative blood flow into and out of the glomerular capillary. A normal kidney has the unique ability to autoregulate blood flow both in and out of the glomerular capillary through alterations in resistance of the afferent and efferent arterioles across a wide range of systemic blood pressure. Most capillary beds only possess the former. Lower relative flows into the glomerulus with decreased renal blood flow or afferent artery constriction may lower intracapillary hydrostatic pressure and diminish filtration. Likewise, higher relative flows out of the glomerulus with efferent artery dilation may also lower intracapillary hydrostatic pressure.
Despite the ability of the kidney to autoregulate and maintain the GFR, more advanced volume depletion can result in the development of azotemia. This can result from excessive volume losses (renal, GI, or cutaneous in origin), low fluid intake, or low effective circulating volume. An example of the latter is decompensated heart failure with poor cardiac output and diminished renal perfusion (termed the “cardiorenal syndrome”).
Drugs are another important cause of prerenal acute kidney injury. Some patients who are dependent on prostaglandin-mediated vasodilation to maintain renal perfusion can develop renal failure simply from ingestion of nonsteroidal anti-inflammatory drugs (NSAIDs). Similarly, patients with renal hypoperfusion (eg, renovascular disease) who are dependent on angiotensin II–mediated vasoconstriction of the efferent renal arterioles to maintain renal perfusion pressure may develop acute kidney injury on ingesting ACE inhibitors.
The intrarenal causes of acute kidney injury can be further divided into specific inflammatory diseases (eg, vasculitis, glomerulonephritis [GN], drug-induced injury) and acute tubular necrosis resulting from many causes (including ischemia and endogenous or exogenous toxic injury).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


