462e | Muscular Dystrophies and Other Muscle Diseases |
Skeletal muscle diseases, or myopathies, are disorders with structural changes or functional impairment of muscle. These conditions can be differentiated from other diseases of the motor unit (e.g., lower motor neuron or neuromuscular junction pathologies) by characteristic clinical and laboratory findings.
Myasthenia gravis and related disorders are discussed in Chap. 461; dermatomyositis, polymyositis, and inclusion body myositis are discussed in Chap. 388.
CLINICAL FEATURES
Most myopathies present with proximal, symmetric limb weakness (arms or legs) with preserved reflexes and sensation. However, asymmetric and predominantly distal weakness can be seen in some myopathies. An associated sensory loss suggests injury to a peripheral nerve or the central nervous system (CNS) rather than myopathy. On occasion, disorders affecting the motor nerve cell bodies in the spinal cord (anterior horn cell disease), the neuromuscular junction, or peripheral nerves can mimic findings of myopathy.
Muscle Weakness Symptoms of muscle weakness can be either intermittent or persistent. Disorders causing intermittent weakness (Fig. 462e-1) include myasthenia gravis, periodic paralyses (hypokalemic, hyperkalemic, and paramyotonia congenita), and metabolic energy deficiencies of glycolysis (especially myophosphorylase deficiency), fatty acid utilization (carnitine palmitoyltransferase deficiency), and some mitochondrial myopathies. The states of energy deficiency cause activity-related muscle breakdown accompanied by myoglobinuria, appearing as light-brown- to dark-brown-colored urine.
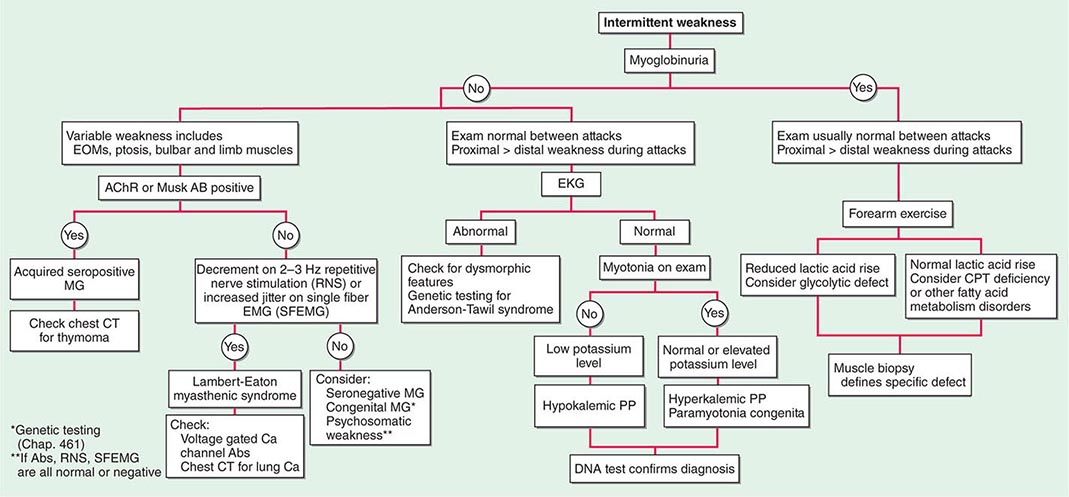
FIGURE 462e-1 Diagnostic evaluation of intermittent weakness. AChR AB, acetylcholine receptor antibody; CPT, carnitine palmitoyltransferase; EOMs, extraocular muscles; MG, myasthenia gravis; PP, periodic paralysis.
Most muscle disorders cause persistent weakness (Fig. 462e-2). In the majority of these, including most types of muscular dystrophy, polymyositis, and dermatomyositis, the proximal muscles are weaker than the distal and are symmetrically affected, and the facial muscles are spared, a pattern referred to as limb-girdle. The differential diagnosis is more restricted for other patterns of weakness. Facial weakness (difficulty with eye closure and impaired smile) and scapular winging (Fig. 462e-3) are characteristic of facioscapulohumeral dystrophy (FSHD). Facial and distal limb weakness associated with hand grip myotonia is virtually diagnostic of myotonic dystrophy type 1. When other cranial nerve muscles are weak, causing ptosis or extraocular muscle weakness, the most important disorders to consider include neuromuscular junction disorders, oculopharyngeal muscular dystrophy, mitochondrial myopathies, or some of the congenital myopathies (Table 462e-1). A pathognomonic pattern characteristic of inclusion body myositis is atrophy and weakness of the flexor forearm (e.g., wrist and finger flexors) and quadriceps muscles that is often asymmetric. Less frequently, but important diagnostically, is the presence of a dropped head syndrome indicative of selective neck extensor muscle weakness. The most important neuromuscular diseases associated with this pattern of weakness include myasthenia gravis, amyotrophic lateral sclerosis, late-onset nemaline myopathy, hyperparathyroidism, focal myositis, and some forms of inclusion body myopathy. A final pattern, recognized because of preferential distal extremity weakness, is typical of a unique category of muscular dystrophy, the distal myopathies.
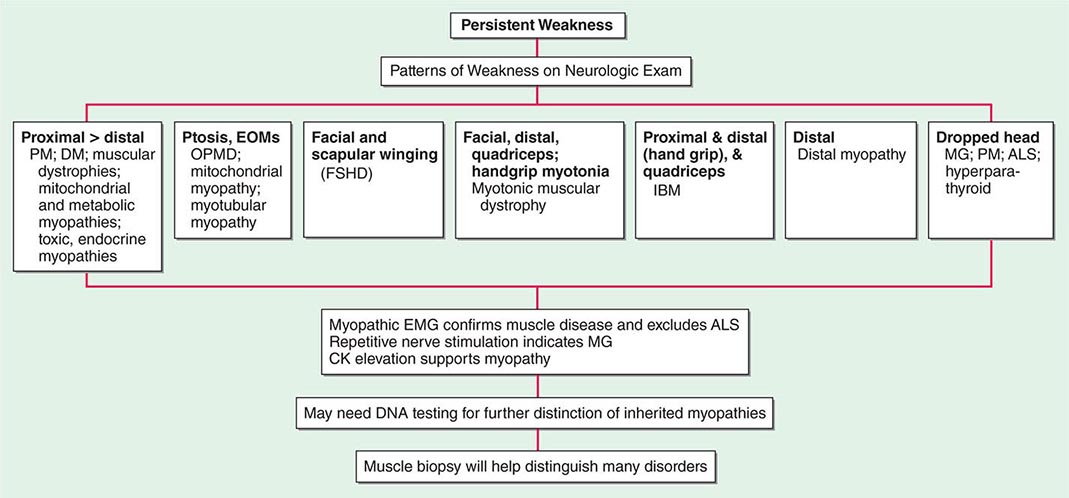
FIGURE 462e-2 Diagnostic evaluation of persistent weakness. Examination reveals one of seven patterns of weakness. The pattern of weakness in combination with the laboratory evaluation leads to a diagnosis. ALS, amyotrophic lateral sclerosis; CK, creatine kinase; DM, dermatomyositis; EMG, electromyography; EOMs, extraocular muscles; FSHD, facioscapulohumeral dystrophy; IBM, inclusion body myositis; MG, myasthenia gravis; OPMD, oculopharyngeal muscular dystrophy; PM, polymyositis.
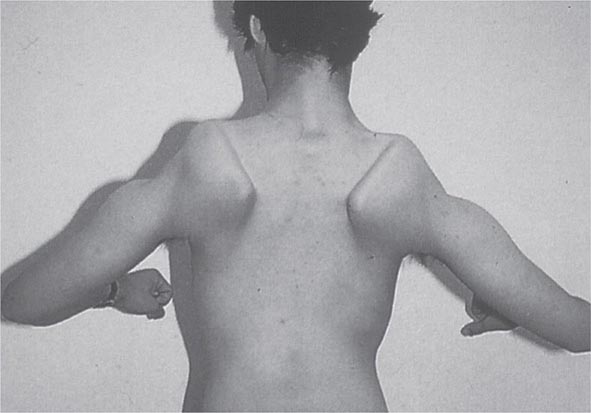
FIGURE 462e-3 Facioscapulohumeral dystrophy with prominent scapular winging.
NEUROMUSCULAR CAUSES OF PTOSIS OR OPHTHALMOPLEGIA |
It is important to examine functional capabilities to help disclose certain patterns of weakness (Table 462e-2). The Gowers’ sign (Fig. 462e-4) is particularly useful. Observing the gait of an individual may disclose a lordotic posture caused by combined trunk and hip weakness, frequently exaggerated by toe walking (Fig. 462e-5). A waddling gait is caused by the inability of weak hip muscles to prevent hip drop or hip dip. Hyperextension of the knee (genu recurvatum or back-kneeing) is characteristic of quadriceps muscle weakness; and a steppage gait, due to footdrop, accompanies distal weakness.
OBSERVATIONS ON EXAMINATION THAT DISCLOSE MUSCLE WEAKNESS |
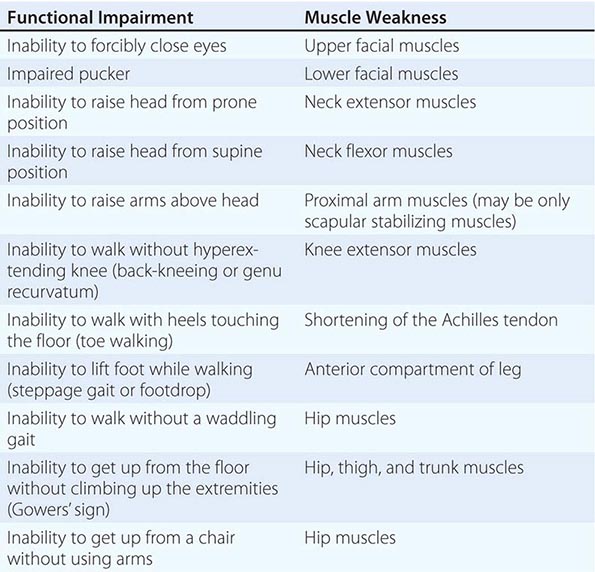
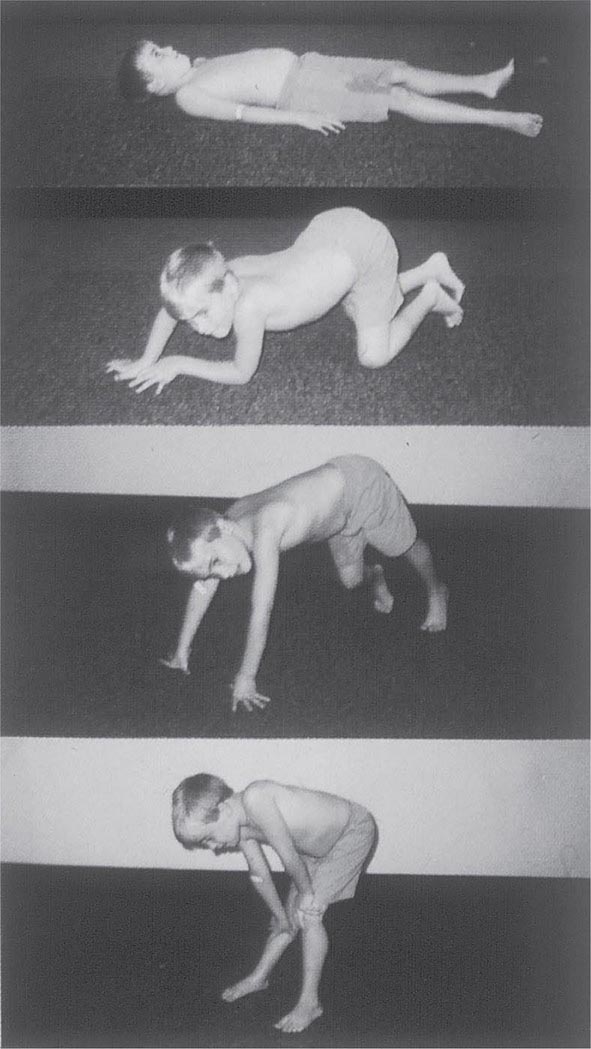
FIGURE 462e-4 Gowers’ sign showing a patient using his arms to climb up the legs in attempting to get up from the floor.
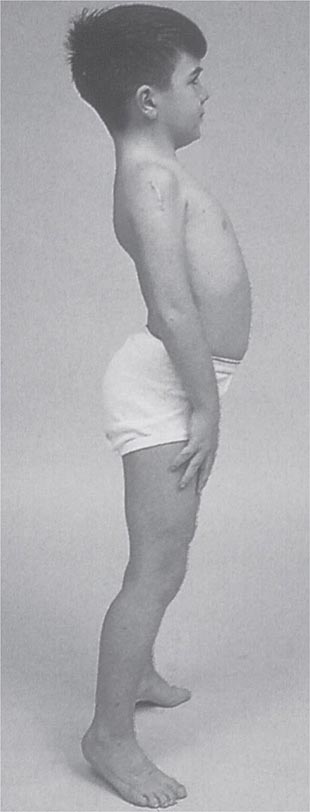
FIGURE 462e-5 Lordotic posture, exaggerated by standing on toes, associated with trunk and hip weakness.
Any disorder causing muscle weakness may be accompanied by fatigue, referring to an inability to maintain or sustain a force (pathologic fatigability). This condition must be differentiated from asthenia, a type of fatigue caused by excess tiredness or lack of energy. Associated symptoms may help differentiate asthenia and pathologic fatigability. Asthenia is often accompanied by a tendency to avoid physical activities, complaints of daytime sleepiness, necessity for frequent naps, and difficulty concentrating on activities such as reading. There may be feelings of overwhelming stress and depression. Thus, asthenia is not a myopathy. In contrast, pathologic fatigability occurs in disorders of neuromuscular transmission and in disorders altering energy production, including defects in glycolysis, lipid metabolism, or mitochondrial energy production. Pathologic fatigability also occurs in chronic myopathies because of difficulty accomplishing a task with less muscle. Pathologic fatigability is accompanied by abnormal clinical or laboratory findings. Fatigue without those supportive features almost never indicates a primary muscle disease.
Muscle Pain (Myalgias), Cramps, and Stiffness Muscle pain can be associated with cramps, spasms, contractures, and stiff or rigid muscles. In distinction, true myalgia (muscle aching), which can be localized or generalized, may be accompanied by weakness, tenderness to palpation, or swelling. Certain drugs cause true myalgia (Table 462e-3).
DRUGS THAT CAUSE TRUE MYALGIA |
There are two painful muscle conditions of particular importance, neither of which is associated with muscle weakness. Fibromyalgia is a common, yet poorly understood, type of myofascial pain syndrome. Patients complain of severe muscle pain and tenderness and have specific painful trigger points, sleep disturbances, and easy fatigability. Serum creatine kinase (CK), erythrocyte sedimentation rate (ESR), electromyography (EMG), and muscle biopsy are normal (Chap. 396). Polymyalgia rheumatica occurs mainly in patients >50 years and is characterized by stiffness and pain in the shoulders, lower back, hips, and thighs (Chap. 385). The ESR is elevated, while serum CK, EMG, and muscle biopsy are normal. Temporal arteritis, an inflammatory disorder of medium- and large-sized arteries, usually involving one or more branches of the carotid artery, may accompany polymyalgia rheumatica. Vision is threatened by ischemic optic neuritis. Glucocorticoids can relieve the myalgias and protect against visual loss.
Localized muscle pain is most often traumatic. A common cause of sudden abrupt-onset pain is a ruptured tendon, which leaves the muscle belly appearing rounded and shorter in appearance compared to the normal side. The biceps brachii and Achilles tendons are particularly vulnerable to rupture. Infection or neoplastic infiltration of the muscle is a rare cause of localized muscle pain.
A muscle cramp or spasm is a painful, involuntary, localized, muscle contraction with a visible or palpable hardening of the muscle. Cramps are abrupt in onset, short in duration, and may cause abnormal posturing of the joint. The EMG shows firing of motor units, reflecting an origin from spontaneous neural discharge. Muscle cramps often occur in neurogenic disorders, especially motor neuron disease (Chap. 452), radiculopathies, and polyneuropathies (Chap. 459), but are not a feature of most primary muscle diseases. Duchenne muscular dystrophy is an exception because calf muscle complaints are a common complaint. Muscle cramps are also common during pregnancy.
A muscle contracture is different from a muscle cramp. In both conditions, the muscle becomes hard, but a contracture is associated with energy failure in glycolytic disorders. The muscle is unable to relax after an active muscle contraction. The EMG shows electrical silence. Confusion is created because contracture also refers to a muscle that cannot be passively stretched to its proper length (fixed contracture) because of fibrosis. In some muscle disorders, especially in Emery-Dreifuss muscular dystrophy and Bethlem myopathy, fixed contractures occur early and represent distinctive features of the disease.
Muscle stiffness can refer to different phenomena. Some patients with inflammation of joints and periarticular surfaces feel stiff. This condition is different from the disorders of hyperexcitable motor nerves causing stiff or rigid muscles. In stiff-person syndrome, spontaneous discharges of the motor neurons of the spinal cord cause involuntary muscle contractions mainly involving the axial (trunk) and proximal lower extremity muscles. The gait becomes stiff and labored, with hyperlordosis of the lumbar spine. Superimposed episodic muscle spasms are precipitated by sudden movements, unexpected noises, and emotional upset. The muscles relax during sleep. Serum antibodies against glutamic acid decarboxylase are present in approximately two-thirds of cases. In neuromyotonia (Isaacs’ syndrome), there is hyperexcitability of the peripheral nerves manifesting as continuous muscle fiber activity. Myokymia (groups of fasciculations associated with continuous undulations of muscle) and impaired muscle relaxation are the result. Muscles of the leg are stiff, and the constant contractions of the muscle cause increased sweating of the extremities. This peripheral nerve hyperexcitability is mediated by antibodies that target voltage-gated potassium channels. The site of origin of the spontaneous nerve discharges is principally in the distal portion of the motor nerves.
Myotonia is a condition of prolonged muscle contraction followed by slow muscle relaxation. It always follows muscle activation (action myotonia), usually voluntary, but may be elicited by mechanical stimulation (percussion myotonia) of the muscle. Myotonia typically causes difficulty in releasing objects after a firm grasp. In myotonic muscular dystrophy type 1 (DM1), distal weakness usually accompanies myotonia, whereas in DM2, proximal muscles are more affected; thus the related term proximal myotonic myopathy (PROMM) is used to describe this condition. Myotonia also occurs with myotonia congenita (a chloride channel disorder), but in this condition muscle weakness is not prominent. Myotonia may also be seen in individuals with sodium channel mutations (hyperkalemic periodic paralysis or potassium-sensitive myotonia). Another sodium channelopathy, paramyotonia congenita, also is associated with muscle stiffness. In contrast to other disorders associated with myotonia in which the myotonia is eased by repetitive activity, paramyotonia congenita is named for a paradoxical phenomenon whereby the myotonia worsens with repetitive activity.
Muscle Enlargement and Atrophy In most myopathies muscle tissue is replaced by fat and connective tissue, but the size of the muscle is usually not affected. However, in many limb-girdle muscular dystrophies (and particularly the dystrophinopathies), enlarged calf muscles are typical. The enlargement represents true muscle hypertrophy; thus the term pseudohypertrophy should be avoided when referring to these patients. The calf muscles remain very strong even late in the course of these disorders. Muscle enlargement can also result from infiltration by sarcoid granulomas, amyloid deposits, bacterial and parasitic infections, and focal myositis. In contrast, muscle atrophy is characteristic of other myopathies. In dysferlinopathies (LGMD2B) and anoctaminopathies (LGMD2L), there is a predilection for early atrophy of the gastrocnemius muscles, particularly the medial aspect. Atrophy of the humeral muscles is characteristic of FSHD.
LABORATORY EVALUATION
A limited battery of tests can be used to evaluate a suspected myopathy. Nearly all patients require serum enzyme level measurements and electrodiagnostic studies as screening tools to differentiate muscle disorders from other motor unit diseases. The other tests described—DNA studies, the forearm exercise test, and muscle biopsy—are used to diagnose specific types of myopathies.
Serum Enzymes CK is the preferred muscle enzyme to measure in the evaluation of myopathies. Damage to muscle causes the CK to leak from the muscle fiber to the serum. The MM isoenzyme predominates in skeletal muscle, whereas creatine kinase-myocardial bound (CK-MB) is the marker for cardiac muscle. Serum CK can be elevated in normal individuals without provocation, presumably on a genetic basis or after strenuous activity, minor trauma (including the EMG needle), a prolonged muscle cramp, or a generalized seizure. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), aldolase, and lactic dehydrogenase (LDH) are enzymes sharing an origin in both muscle and liver. Problems arise when the levels of these enzymes are found to be elevated in a routine screening battery, leading to the erroneous assumption that liver disease is present when in fact muscle could be the cause. An elevated γ-glutamyl transferase (GGT) helps to establish a liver origin because this enzyme is not found in muscle.
Electrodiagnostic Studies EMG, repetitive nerve stimulation, and nerve conduction studies (Chap. 442e) are essential methods for evaluation of the patient with suspected muscle disease. In combination, they provide the information necessary to differentiate myopathies from neuropathies and neuromuscular junction diseases. Routine nerve conduction studies are typically normal in myopathies but reduced amplitudes of compound muscle action potentials may be seen in atrophied muscles. The needle EMG may reveal irritability on needle placement suggestive of a necrotizing myopathy (inflammatory myopathies, dystrophies, toxic myopathies, myotonic myopathies), whereas a lack of irritability is characteristic of long-standing myopathic disorders (muscular dystrophies, endocrine myopathies, disuse atrophy, and many of the metabolic myopathies). In addition, the EMG may demonstrate myotonic discharges that will narrow the differential diagnosis (Table 462e-4). Another important EMG finding is the presence of short-duration, small-amplitude, polyphasic motor unit action potentials (MUAPs). Such MUAPs can be seen in both myopathic and neuropathic disorders; however, the recruitment or firing pattern is different. In myopathies, the MUAPs fire early but at a normal rate to compensate for the loss of individual muscle fibers, whereas in neurogenic disorders the MUAPs fire faster. The EMG is usually normal in steroid or disuse myopathy, both of which are associated with type 2 fiber atrophy; this is because the EMG preferentially assesses the physiologic function of type 1 fibers. The EMG can also be invaluable in helping to choose an appropriately affected muscle to sample for biopsy.
MYOTONIC DISORDERS |
aAssociated with myotonic discharges on electromyography but no clinical myotonia.
DNA Analysis This serves as an important tool for the definitive diagnosis of many muscle disorders. Nevertheless, there are a number of limitations in currently available molecular diagnostics. For example, in Duchenne and Becker dystrophies, two-thirds of patients have deletion or duplication mutations in the dystrophin gene that are easy to detect, while the remainder have point mutations that are much more difficult to find. For patients without identifiable gene defects, the muscle biopsy remains the main diagnostic tool.
Forearm Exercise Test In myopathies with intermittent symptoms, and especially those associated with myoglobinuria, there may be a defect in glycolysis. Many variations of the forearm exercise test exist. For safety, the test should not be performed under ischemic conditions to avoid an unnecessary insult to the muscle, causing rhabdomyolysis. The test is performed by placing a small indwelling catheter into an antecubital vein. A baseline blood sample is obtained for lactic acid and ammonia. The forearm muscles are exercised by asking the patient to vigorously open and close the hand for 1 min. Blood is then obtained at intervals of 1, 2, 4, 6, and 10 min for comparison with the baseline sample. A three- to fourfold rise of lactic acid is typical. The simultaneous measurement of ammonia serves as a control, because it should also rise with exercise. In patients with myophosphorylase deficiency or other glycolytic defects, the lactic acid rise will be absent or below normal, while the rise in ammonia will reach control values. If there is lack of effort, neither lactic acid nor ammonia will rise. Patients with selective failure to increase ammonia may have myoadenylate deaminase deficiency. This condition has been reported to be a cause of myoglobinuria, but deficiency of this enzyme in asymptomatic individuals makes interpretation controversial.
Muscle Biopsy Muscle biopsy is an important step in establishing the diagnosis of a suspected myopathy. The biopsy is usually obtained from a quadriceps or biceps brachii muscle, less commonly from a deltoid muscle. Evaluation includes a combination of techniques—light microscopy, histochemistry, immunocytochemistry with a battery of antibodies, and electron microscopy. Not all techniques are needed for every case. A specific diagnosis can be established in many disorders. Endomysial inflammatory cells surrounding and invading muscle fibers are seen in polymyositis; similar endomysial infiltrates associated with muscle fibers containing rimmed vacuoles and amyloid deposits consisting of SMI-31-, p62-, and TDP-43-positive inclusions within fibers are characteristic of inclusion body myositis; and perivascular, perimysial inflammation associated with perifascicular atrophy is a feature of dermatomyositis. In addition, the congenital myopathies have distinctive light and electron microscopy features essential for diagnosis. Mitochondrial and metabolic (e.g., glycogen and lipid storage diseases) myopathies also demonstrate distinctive histochemical and electron-microscopic profiles. Biopsied muscle tissue can be sent for metabolic enzyme or mitochondrial DNA analyses. A battery of antibodies is available for the identification of abnormal proteins to help diagnose specific types of muscular dystrophies. Western blot analysis on muscle specimens can be performed to determine whether specific muscle proteins are reduced in quantity or are of abnormal size.
HEREDITARY MYOPATHIES
Muscular dystrophy refers to a group of hereditary progressive diseases each with unique phenotypic and genetic features (Tables 462e-5, 462e-6, and 462e-7).
PROGRESSIVE MUSCULAR DYSTROPHIES |
AUTOSOMAL DOMINANT LIMB-GIRDLE MUSCULAR DYSTROPHIES (LGMDs) |
AUTOSOMAL RECESSIVE LIMB-GIRDLE MUSCULAR DYSTROPHIES (LGMDs) |
DUCHENNE MUSCULAR DYSTROPHY
This X-linked recessive disorder, sometimes also called pseudohypertrophic muscular dystrophy, has an incidence of ~1 per 5200 live-born males.
Clinical Features Duchenne dystrophy is present at birth, but the disorder usually becomes apparent between ages 3 and 5 years. The boys fall frequently and have difficulty keeping up with friends when playing. Running, jumping, and hopping are invariably abnormal. By age 5 years, muscle weakness is obvious by muscle testing. On getting up from the floor, the patient uses his hands to climb up himself (Gowers’ maneuver [Fig. 462e-4]). Contractures of the heel cords and iliotibial bands become apparent by age 6 years, when toe walking is associated with a lordotic posture. Loss of muscle strength is progressive, with predilection for proximal limb muscles and the neck flexors; leg involvement is more severe than arm involvement. Between ages 8 and 10 years, walking may require the use of braces; joint contractures and limitations of hip flexion, knee, elbow, and wrist extension are made worse by prolonged sitting. Prior to the use of glucocorticoids, most boys became wheelchair dependent by 12 years of age. Contractures become fixed, and a progressive scoliosis often develops that may be associated with pain. The chest deformity with scoliosis impairs pulmonary function, which is already diminished by muscle weakness. By age 16–18 years, patients are predisposed to serious, sometimes fatal pulmonary infections. Other causes of death include aspiration of food and acute gastric dilation.
A cardiac cause of death is uncommon despite the presence of a cardiomyopathy in almost all patients. Congestive heart failure seldom occurs except with severe stress such as pneumonia. Cardiac arrhythmias are rare. The typical electrocardiogram (ECG) shows an increased net RS in lead V1; deep, narrow Q waves in the precordial leads; and tall right precordial R waves in V1. Intellectual impairment in Duchenne dystrophy is common; the average intelligence quotient (IQ) is ~1 standard deviation (SD) below the mean. Impairment of intellectual function appears to be nonprogressive and affects verbal ability more than performance.
Laboratory Features Serum CK levels are invariably elevated to between 20 and 100 times normal. The levels are abnormal at birth but decline late in the disease because of inactivity and loss of muscle mass. EMG demonstrates features typical of myopathy. The muscle biopsy shows muscle fibers of varying size as well as small groups of necrotic and regenerating fibers. Connective tissue and fat replace lost muscle fibers. A definitive diagnosis of Duchenne dystrophy can be established on the basis of dystrophin deficiency in a biopsy of muscle tissue or mutation analysis on peripheral blood leukocytes, as discussed below.
Duchenne dystrophy is caused by a mutation of the gene that encodes dystrophin, a 427-kDa protein localized to the inner surface of the sarcolemma of the muscle fiber. The dystrophin gene is >2000 kb in size and thus is one of the largest identified human genes. It is localized to the short arm of the × chromosome at Xp21. The most common gene mutation is a deletion. The size varies but does not correlate with disease severity. Deletions are not uniformly distributed over the gene but rather are most common near the beginning (5′ end) and middle of the gene. Less often, Duchenne dystrophy is caused by a gene duplication or point mutation. Identification of a specific mutation allows for an unequivocal diagnosis, makes possible accurate testing of potential carriers, and is useful for prenatal diagnosis.
A diagnosis of Duchenne dystrophy can also be made by Western blot analysis of muscle biopsy specimens, revealing abnormalities on the quantity and molecular weight of dystrophin protein. In addition, immunocytochemical staining of muscle with dystrophin antibodies can be used to demonstrate absence or deficiency of dystrophin localizing to the sarcolemmal membrane. Carriers of the disease may demonstrate a mosaic pattern, but dystrophin analysis of muscle biopsy specimens for carrier detection is not reliable.
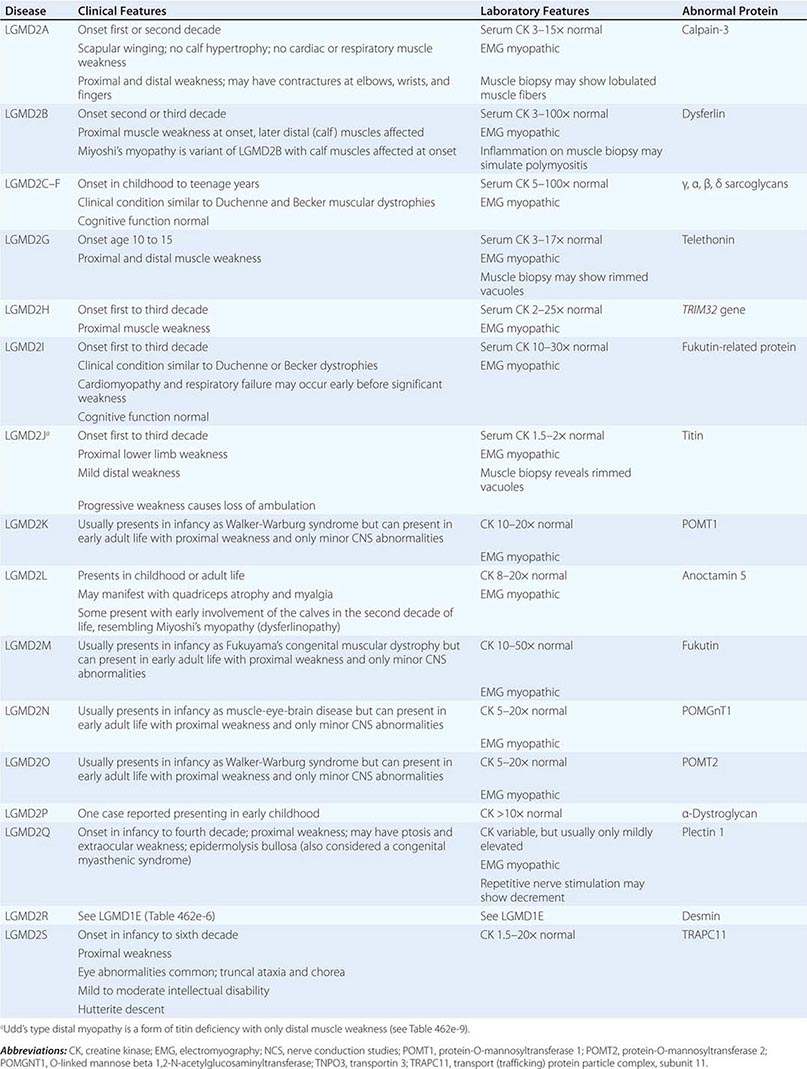
Pathogenesis Dystrophin is part of a large complex of sarcolemmal proteins and glycoproteins (Fig. 462e-6). Dystrophin binds to F-actin at its amino terminus and to β-dystroglycan at the carboxyl terminus. β-Dystroglycan complexes to α-dystroglycan, which binds to laminin in the extracellular matrix (ECM). Laminin has a heterotrimeric molecular structure arranged in the shape of a cross with one heavy chain and two light chains, β1 and γ1. The laminin heavy chain of skeletal muscle is designated laminin α2. Collagen proteins IV and VI are also found in the ECM. Like β-dystroglycan, the transmembrane sarcoglycan proteins also bind to dystrophin; these five proteins (designated α- through ε-sarcoglycan) complex tightly with each other. More recently, other membrane proteins implicated in muscular dystrophy have been found to be loosely affiliated with constituents of the dystrophin complex. These include caveolin-3, α7 integrin, and collagen VI.
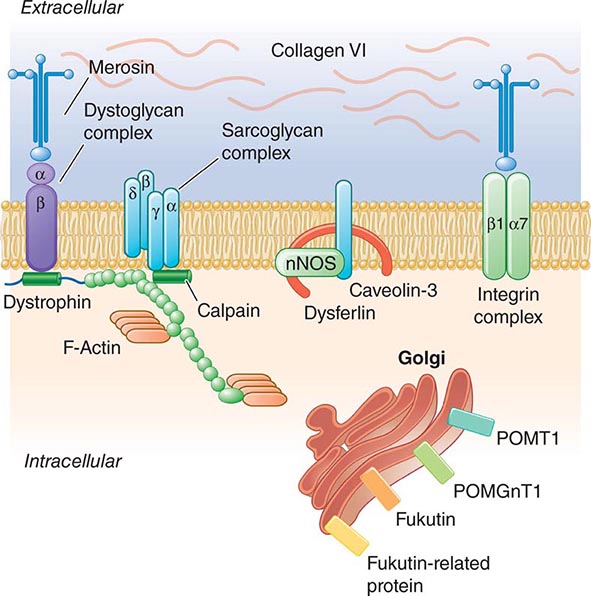
FIGURE 462e-6 Selected muscular dystrophy–associated proteins in the cell membrane and Golgi complex.
Dystrophin localizes to the cytoplasmic face of the muscle cell membrane. It complexes with two transmembrane protein complexes, the dystroglycans and the sarcoglycans. The dystroglycans bind to the extracellular matrix protein merosin, which is also complexed with β1 and α7 integrins (Tables 462e-5, 462e-6, and 462e-7). Dysferlin complexes with caveolin-3 (which binds to neuronal nitric oxide synthase, or nNOS) but not with the dystrophin-associated proteins or the integrins. In some of the congenital dystrophies and limb-girdle muscular dystrophies (LGMDs), there is loss of function of different enzymes that glycosylate α-dystroglycan, which thereby inhibits proper binding to merosin: POMT1, POMT2, POMGnT1, Fukutin, Fukutin-related protein, and LARGE.
The dystrophin-glycoprotein complex appears to confer stability to the sarcolemma, although the function of each individual component of the complex is incompletely understood. Deficiency of one member of the complex may cause abnormalities in other components. For example, a primary deficiency of dystrophin (Duchenne dystrophy) may lead to secondary loss of the sarcoglycans and dystroglycan. The primary loss of a single sarcoglycan (see “Limb-Girdle Muscular Dystrophy,” below) results in a secondary loss of other sarcoglycans in the membrane without uniformly affecting dystrophin. In either instance, disruption of the dystrophin-glycoprotein complexes weakens the sarcolemma, causing membrane tears and a cascade of events leading to muscle fiber necrosis. This sequence of events occurs repeatedly during the life of a patient with muscular dystrophy.
BECKER MUSCULAR DYSTROPHY
This less severe form of X-linked recessive muscular dystrophy results from allelic defects of the same gene responsible for Duchenne dystrophy. Becker muscular dystrophy is ~10 times less frequent than Duchenne.
Clinical Features The pattern of muscle wasting in Becker muscular dystrophy closely resembles that seen in Duchenne. Proximal muscles, especially of the lower extremities, are prominently involved. As the disease progresses, weakness becomes more generalized. Significant facial muscle weakness is not a feature. Hypertrophy of muscles, particularly in the calves, is an early and prominent finding.
Most patients with Becker dystrophy first experience difficulties between ages 5 and 15 years, although onset in the third or fourth decade or even later can occur. By definition, patients with Becker dystrophy walk beyond age 15, whereas patients with Duchenne dystrophy are typically in a wheelchair by the age of 12. Patients with Becker dystrophy have a reduced life expectancy, but most survive into the fourth or fifth decade.
Mental retardation may occur in Becker dystrophy, but it is not as common as in Duchenne. Cardiac involvement occurs in Becker dystrophy and may result in heart failure; some patients manifest with only heart failure. Other less common presentations are asymptomatic hyper-CK-emia, myalgias without weakness, and myoglobinuria.
Laboratory Features Serum CK levels, results of EMG, and muscle biopsy findings closely resemble those in Duchenne dystrophy. The diagnosis of Becker muscular dystrophy requires Western blot analysis of muscle biopsy samples, demonstrating a reduced amount or abnormal size of dystrophin or mutation analysis of DNA from peripheral blood leukocytes. Genetic testing reveals deletions or duplications of the dystrophin gene in 65% of patients with Becker dystrophy, approximately the same percentage as in Duchenne dystrophy. In both Becker and Duchenne dystrophies, the size of the DNA deletion does not predict clinical severity; however, in ~95% of patients with Becker dystrophy, the DNA deletion does not alter the translational reading frame of messenger RNA. These “in-frame” mutations allow for production of some dystrophin, which accounts for the presence of altered rather than absent dystrophin on Western blot analysis.
LIMB-GIRDLE MUSCULAR DYSTROPHY
The syndrome of LGMD represents more than one disorder. Both males and females are affected, with onset ranging from late in the first decade to the fourth decade. The LGMDs typically manifest with progressive weakness of pelvic and shoulder girdle musculature. Respiratory insufficiency from weakness of the diaphragm may occur, as may cardiomyopathy.
A systematic classification of LGMD is based on autosomal dominant (LGMD1) and autosomal recessive (LGMD2) inheritance. Superimposed on the backbone of LGMD1 and LGMD2, the classification uses a sequential alphabetical lettering system (LGMD1A, LGMD2A, etc.). Disorders receive letters in the order in which they are found to have chromosomal linkage. This results in an ever-expanding list of conditions summarized in Tables 462e-6 and 462e-7. None of the conditions is as common as the dystrophinopathies; however, prevalence data for the LGMDs have not been systematically gathered for any large heterogeneous population. In referral-based clinical populations, Fukutin-related protein (FKRP) deficiency (LGMD2I), calpainopathy (LGMD2A), anoctaminopathy (LGMD2L), and to a lesser extent dysferlinopathy (LGMD2B) have emerged as the most common disorders.
EMERY-DREIFUSS MUSCULAR DYSTROPHY
There are at least five genetically distinct forms of Emery-Dreifuss muscular dystrophy (EDMD). Emerin mutations are the most common cause of X-linked EDMD, although mutations in FHL1 may also be associated with a similar phenotype, which is X-linked as well. Mutations involving the gene for lamin A/C are the most common cause of autosomal dominant EDMD (also known as LGMD1B) and are also a common cause of hereditary cardiomyopathy. Less commonly, autosomal dominant EDMD has been reported with mutations in nesprin-1, nesprin-2, and TMEM43.
Clinical Features Prominent contractures can be recognized in early childhood and teenage years, often preceding muscle weakness. The contractures persist throughout the course of the disease and are present at the elbows, ankles, and neck. Muscle weakness affects humeral and peroneal muscles at first and later spreads to a limb-girdle distribution. The cardiomyopathy is potentially life threatening and may result in sudden death. A spectrum of atrial rhythm and conduction defects includes atrial fibrillation and paralysis and atrioventricular heart block. Some patients have a dilated cardiomyopathy. Female carriers of the X-linked variant may have cardiac manifestations that become clinically significant.
Laboratory Features Serum CK may be elevated two- to tenfold. EMG is myopathic. Muscle biopsy usually shows nonspecific dystrophic features, although cases associated with FHL1 mutations have features of myofibrillar myopathy. Immunohistochemistry reveals absent emerin staining of myonuclei in X-linked EDMD due to emerin mutations. ECGs demonstrate atrial and atrioventricular rhythm disturbances.
X-linked EDMD usually arises from defects in the emerin gene encoding a nuclear envelope protein. FHL1 mutations are also a cause of X-linked scapuloperoneal dystrophy, but can also present with an X-linked form of EDMD. The autosomal dominant disease can be caused by mutations in the LMNA gene encoding lamin A and C; in the synaptic nuclear envelope protein 1 (SYNE1) or 2 (SYNE2) encoding nesprin-1 and nesprin-2, respectively; and most recently in TMEM43 encoding LUMA. These proteins are essential components of the filamentous network underlying the inner nuclear membrane. Loss of structural integrity of the nuclear envelope from defects in emerin, lamin A/C, nesprin-1, nesprin-2, and LUMA accounts for overlapping phenotypes (Fig. 462e-7).
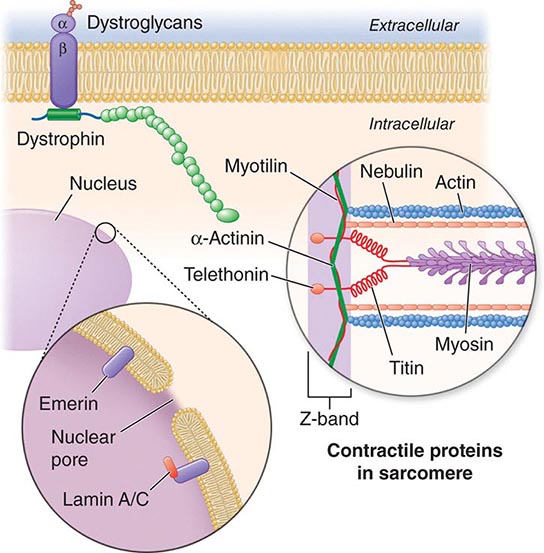
FIGURE 462e-7 Selected muscular dystrophy–associated proteins in the nuclear membrane and sarcomere. As shown in the exploded view, emerin and lamin A/C are constituents of the inner nuclear membrane. Several dystrophy-associated proteins are represented in the sarcomere including titin, nebulin, calpain, telethonin, actinin, and myotilin. The position of the dystrophin-dystroglycan complex is also illustrated.
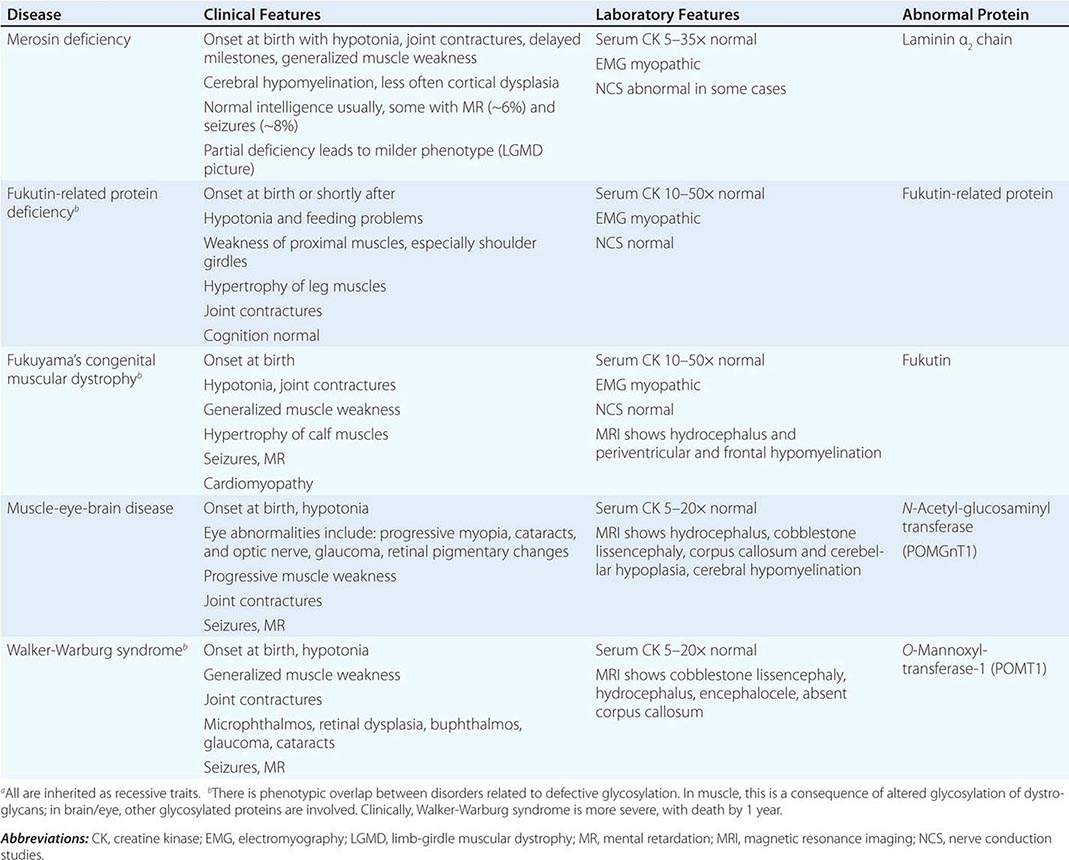
CONGENITAL MUSCULAR DYSTROPHY (CMD)
This is not one entity but rather a group of disorders with varying degrees of muscle weakness, CNS impairment, and eye abnormalities.
Clinical Features As a group, CMDs present at birth or in the first few months of life with hypotonia and proximal or generalized muscle weakness. Calf muscle hypertrophy is seen in some patients. Facial muscles may be weak, but other cranial nerve–innervated muscles are spared (e.g., extraocular muscles are normal). Most patients have joint contractures of varying degrees at elbows, hips, knees, and ankles. Contractures present at birth are referred to as arthrogryposis. Respiratory failure may be seen in some cases.
The CNS is affected in some forms of CMD. In merosin and FKRP deficiency, cerebral hypomyelination may be seen by magnetic resonance imaging (MRI), although only a small number of patients have mental retardation and seizures. Three forms of congenital muscular dystrophy have severe brain impairment. These include Fukuyama’s congenital muscular dystrophy (FCMD), muscle-eye-brain (MEB) disease, and Walker-Warburg syndrome (WWS). Patients are severely disabled in all three of these conditions. In MEB disease and WWS, but not in FCMD, ocular abnormalities impair vision. WWS is the most severe congenital muscular dystrophy, causing death by 1 year of age.
Laboratory Features Serum CK is markedly elevated in all of these conditions. The EMG is myopathic and muscle biopsies show nonspecific dystrophic features. Merosin, or laminin α2 chain (a basal lamina protein), is deficient in surrounding muscle fibers in merosin deficiency. Skin biopsies can also demonstrate defects in laminin α2 chain. In the other disorders (FKRP deficiency, FCMD, MEB disease, WWS), there is abnormal α-dystroglycan staining in muscle. In merosin deficiency, cerebral hypomyelination is common, and a host of brain malformations are seen in FCMD, MEB disease, and WWS.
All forms of CMD are inherited as autosomal recessive disorders. Chromosomal linkage and specific gene defects are presented in Table 462e-8. With the exception of merosin, the other gene defects affect posttranslational glycosylation of α-dystroglycan. This abnormality is thought to impair binding with merosin and leads to weakening of the dystrophin-glycoprotein complex, instability of the muscle membrane, and/or abnormalities in muscle contraction. CMDs with brain and eye phenotypes probably involve defective glycosylation of additional proteins, accounting for the more extensive phenotypes.
CONGENITAL MUSCULAR DYSTROPHIESa |
MYOTONIC DYSTROPHY
Myotonic dystrophy is also known as dystrophia myotonica (DM). The condition is composed of at least two clinical disorders with overlapping phenotypes and distinct molecular genetic defects: myotonic dystrophy type 1 (DM1), the classic disease originally described by Steinert, and myotonic dystrophy type 2 (DM2), also called proximal myotonic myopathy (PROMM).
Clinical Features The clinical expression of DM1 varies widely and involves many systems other than muscle. Affected patients have a typical “hatchet-faced” appearance due to temporalis, masseter, and facial muscle atrophy and weakness. Frontal baldness is also characteristic of the disease. Neck muscles, including flexors and sternocleidomastoids, and distal limb muscles are involved early. Weakness of wrist extensors, finger extensors, and intrinsic hand muscles impairs function. Ankle dorsiflexor weakness may cause footdrop. Proximal muscles remain stronger throughout the course, although preferential atrophy and weakness of quadriceps muscles occur in many patients. Palatal, pharyngeal, and tongue involvement produce a dysarthric speech, nasal voice, and swallowing problems. Some patients have diaphragm and intercostal muscle weakness, resulting in respiratory insufficiency.
Myotonia, which usually appears by age 5 years, is demonstrable by percussion of the thenar eminence, the tongue, and wrist extensor muscles. Myotonia causes a slow relaxation of hand grip after a forced voluntary closure. Advanced muscle wasting makes myotonia more difficult to detect.
Cardiac disturbances occur commonly in patients with DM1. ECG abnormalities include first-degree heart block and more extensive conduction system involvement. Complete heart block and sudden death can occur. Congestive heart failure occurs infrequently but may result from cor pulmonale secondary to respiratory failure. Mitral valve prolapse also occurs commonly. Other associated features include intellectual impairment, hypersomnia, posterior subcapsular cataracts, gonadal atrophy, insulin resistance, and decreased esophageal and colonic motility.
Congenital myotonic dystrophy is a more severe form of DM1 and occurs in ~25% of infants of affected mothers. It is characterized by severe facial and bulbar weakness, transient neonatal respiratory insufficiency, and mental retardation.
DM2, or PROMM, has a distinct pattern of muscle weakness affecting mainly proximal muscles. Other features of the disease overlap with DM1, including cataracts, testicular atrophy, insulin resistance, constipation, hypersomnia, and cognitive defects. Cardiac conduction defects occur but are less common, and the hatchet face and frontal baldness are less consistent features. A very striking difference is the failure to clearly identify a congenital form of DM2.
Laboratory Features The diagnosis of myotonic dystrophy can usually be made on the basis of clinical findings. Serum CK levels may be normal or mildly elevated. EMG evidence of myotonia is present in most cases of DM1 but may be more patchy in DM2. Muscle biopsy shows muscle atrophy, which selectively involves type 1 fibers in 50% of cases, and ringed fibers in DM1 but not in DM2. Typically, numerous internalized nuclei can be seen in individual muscle fibers as well as atrophic fibers with pyknotic nuclear clumps in both DM1 and DM2. Necrosis of muscle fibers and increased connective tissue, common in other muscular dystrophies, are less apparent in myotonic dystrophy.
DM1 and DM2 are both autosomal dominant disorders. New mutations do not appear to contribute to the pool of affected individuals. DM1 is transmitted by an intronic mutation consisting of an unstable expansion of a CTG trinucleotide repeat in a serine-threonine protein kinase gene (named DMPK) on chromosome 19q13.3. An increase in the severity of the disease phenotype in successive generations (genetic anticipation) is accompanied by an increase in the number of trinucleotide repeats. A similar type of mutation has been identified in fragile × syndrome (Chap. 451e). The unstable triplet repeat in myotonic dystrophy can be used for prenatal diagnosis. Congenital disease occurs almost exclusively in infants born to affected mothers; it is possible that sperm with greatly expanded triplet repeats do not function well.
DM2 is caused by a DNA expansion mutation consisting of a CCTG repeat in intron 1 of the ZNF9 gene located at chromosome 3q13.3-q24. The gene is believed to encode an RNA-binding protein expressed in many different tissues, including skeletal and cardiac muscle.
The DNA expansions in DM1 and DM2 almost certainly impair muscle function by a toxic gain of function of the mutant mRNA. In both DM1 and DM2, the mutant RNA appears to form intranuclear inclusions composed of aberrant RNA. These RNA inclusions sequester RNA-binding proteins essential for proper splicing of a variety of other mRNAs. This leads to abnormal transcription of multiple proteins in a variety of tissues/organ systems, in turn causing the systemic manifestations of DM1 and DM2.
FACIOSCAPULOHUMERAL (FSH) MUSCULAR DYSTROPHY
This form of muscular dystrophy has a prevalence of ~1 in 20,000. There are two forms of FSHD that have similar pathogenesis, as will be discussed. Most patients have FSHD type 1 (95%), whereas approximately 5% have FSHD2. FSHD1 and FSHD2 are clinically and histopathologically identical. FSHD is not to be confused with the genetically distinct scapuloperoneal dystrophies.
Clinical Features The condition typically has an onset in childhood or young adulthood. In most cases, facial weakness is the initial manifestation, appearing as an inability to smile, whistle, or fully close the eyes. Weakness of the shoulder girdles, rather than the facial muscles, usually brings the patient to medical attention. Loss of scapular stabilizer muscles makes arm elevation difficult. Scapular winging (Fig. 462e-3) becomes apparent with attempts at abduction and forward movement of the arms. Biceps and triceps muscles may be severely affected, with relative sparing of the deltoid muscles. Weakness is invariably worse for wrist extension than for wrist flexion, and weakness of the anterior compartment muscles of the legs may lead to footdrop.
In most patients, the weakness remains restricted to facial, upper extremity, and distal lower extremity muscles. In 20% of patients, weakness progresses to involve the pelvic girdle muscles, and severe functional impairment and possible wheelchair dependency result.
Characteristically, patients with FSHD do not have involvement of other organ systems, although labile hypertension is common, and there is an increased incidence of nerve deafness. Coats’ disease, a disorder consisting of telangiectasia, exudation, and retinal detachment, also occurs.
Laboratory Features The serum CK level may be normal or mildly elevated. EMG usually indicates a myopathic pattern. The muscle biopsy shows nonspecific features of a myopathy. A prominent inflammatory infiltrate, which is often multifocal in distribution, is present in some biopsy samples. The cause or significance of this finding is unknown.
An autosomal dominant inheritance pattern with almost complete penetrance has been established, but each family member should be examined for the presence of the disease, since ~30% of those affected are unaware of involvement. FSHD1 is associated with deletions of tandem 3.3-kb repeats at 4q35. The deletion reduces the number of repeats to a fragment of <35 kb in most patients. Within these repeats lies the DUX4 gene, which usually is not expressed. In patients with FSHD1 these deletions in the setting of a specific polymorphism leads to hypomethylation of the region and toxic expression of the DUX4 gene. In patients with FSHD2, there is no deletion, but a mutation in SMCHD1. Interestingly, in the setting of the same polymorphism, there again is seen hypomethylation of the region and the permissive expression of the DUX4 gene. In both FSHD1 and FSHD2, there is overexpression of the DUX4 transcript.
OCULOPHARYNGEAL DYSTROPHY
This form of muscular dystrophy represents one of several disorders characterized by progressive external ophthalmoplegia, which consists of slowly progressive ptosis and limitation of eye movements with sparing of pupillary reactions for light and accommodation. Patients usually do not complain of diplopia, in contrast to patients having conditions with a more acute onset of ocular muscle weakness (e.g., myasthenia gravis).
Clinical Features Oculopharyngeal muscular dystrophy has a late onset; it usually presents in the fourth to sixth decade with ptosis and/or dysphagia. The extraocular muscle impairment is less prominent in the early phase but may be severe later. The swallowing problem may become debilitating and result in pooling of secretions and repeated episodes of aspiration. Mild weakness of the neck and extremities also occurs.
Laboratory Features The serum CK level may be two to three times normal. Myopathic EMG findings are typical. On biopsy, muscle fibers are found to contain rimmed vacuoles, which by electron microscopy are shown to contain membranous whorls, accumulation of glycogen, and other nonspecific debris related to lysosomes. A distinct feature of oculopharyngeal dystrophy is the presence of tubular filaments, 8.5 nm in diameter, in muscle cell nuclei.
Oculopharyngeal dystrophy has an autosomal dominant inheritance pattern with complete penetrance. The incidence is high in French-Canadians and in Spanish-American families of the southwestern United States. Large kindreds of Italian and of eastern European Jewish descent have been reported. The molecular defect in oculopharyngeal muscular dystrophy is a subtle expansion of a modest polyalanine repeat tract in a poly-RNA-binding protein (PABP2) in muscle.
DISTAL MYOPATHIES
A group of muscle diseases, the distal myopathies, are notable for their preferential distal distribution of muscle weakness in contrast to most muscle conditions associated with proximal weakness. The major distal myopathies are summarized in Table 462e-9.
DISTAL MYOPATHIES |
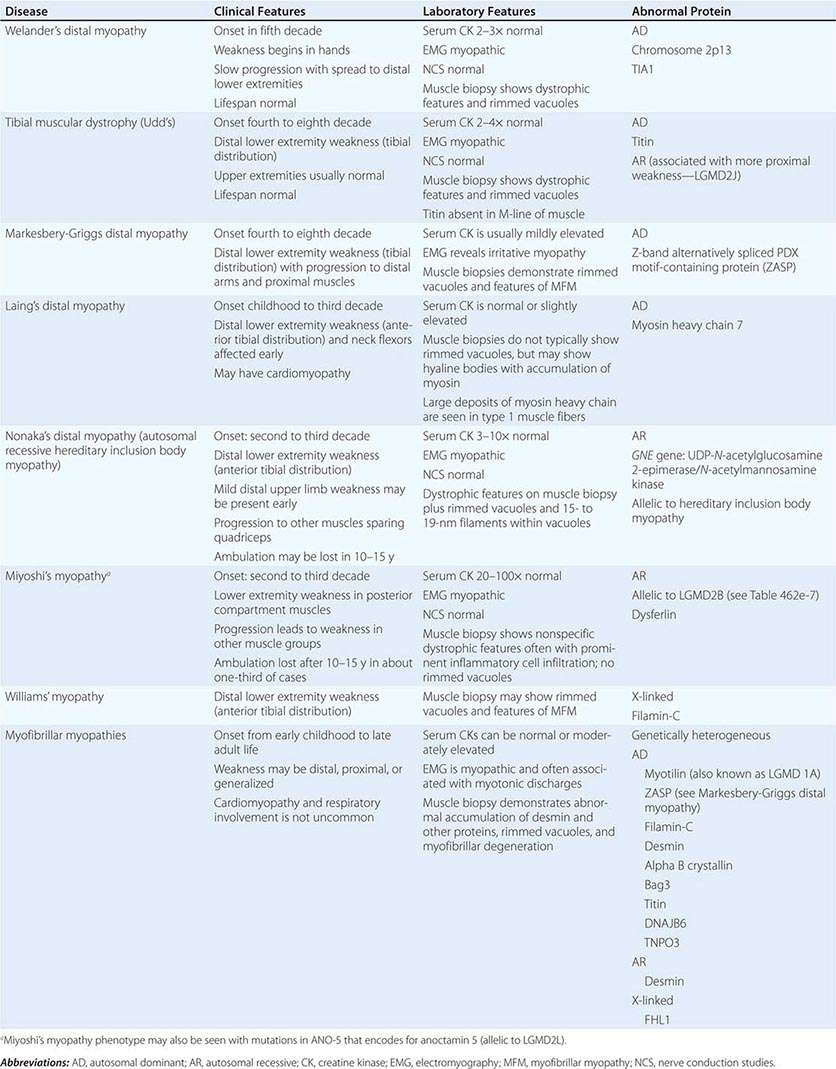
Clinical Features Welander’s, Udd’s, and Markesbery-Griggs type distal myopathies are all late-onset, dominantly inherited disorders of distal limb muscles, usually beginning after age 40 years. Welander’s distal myopathy preferentially involves the wrist and finger extensors, whereas the others are associated with anterior tibial weakness leading to progressive footdrop. Laing’s distal myopathy is also a dominantly inherited disorder heralded by tibial weakness; however, it is distinguished by onset in childhood or early adult life. Nonaka’s distal myopathy and Miyoshi’s myopathy are distinguished by autosomal recessive inheritance and onset in the late teens or twenties. Nonaka’s and Williams’ myopathy entails anterior tibial weakness, whereas Miyoshi’s myopathy is unique in that gastrocnemius muscles are preferentially affected at onset. Finally, the myofibrillar myopathies (MFMs) are a clinically and genetically heterogeneous group of disorders that can be associated with prominent distal weakness; they can be inherited in an autosomal dominant or recessive pattern. Of note, Markesbery-Griggs myopathy (caused by mutations in ZASP) and LGMD1B (caused by mutations in myotilin) are in fact subtypes of myofibrillar myopathy.
Laboratory Features Serum CK level is particularly helpful in diagnosing Miyoshi’s myopathy because it is very elevated. In the other conditions, serum CK is only slightly increased. EMGs are myopathic. In the MFMs, myotonic or pseudomyotonic discharges are common. Muscle biopsy shows nonspecific dystrophic features and, with the exception of Laing’s and Miyoshi’s myopathies, often shows rimmed vacuoles. MFM is associated with the accumulation of dense inclusions, as well as amorphous material best seen on Gomori trichrome and myofibrillar disruption on electron microscopy. Immune staining sometimes demonstrates accumulation of desmin and other proteins in MFM, large deposits of myosin heavy chain in the subsarcolemmal region of type 1 muscle fibers in Laing’s myopathy, and reduced or absent dysferlin in Miyoshi’s myopathy.
The affected genes and their gene products are listed in Table 462e-9.
CONGENITAL MYOPATHIES
These rare disorders are distinguished from muscular dystrophies by the presence of specific histochemical and structural abnormalities in muscle. Although primarily disorders of infancy or childhood, three forms that may present in adulthood are described here: central core disease, nemaline (rod) myopathy, and centronuclear (myotubular) myopathy. Sarcotubular myopathy is caused by mutations in TRIM-32 and is identical to LGMD2H. Other types, such as minicore myopathy (multi-minicore disease), fingerprint body myopathy, and cap myopathy, are not discussed.
CENTRAL CORE DISEASE
Patients with central core disease may have decreased fetal movements and breech presentation. Hypotonia and delay in motor milestones, particularly in walking, are common. Later in childhood, patients develop problems with stair climbing, running, and getting up from the floor. On examination, there is mild facial, neck-flexor, and proximal-extremity muscle weakness. Legs are more affected than arms. Skeletal abnormalities include congenital hip dislocation, scoliosis, and pes cavus; clubbed feet also occur. Most cases are nonprogressive, but exceptions are well documented. Susceptibility to malignant hyperthermia must be considered as a potential risk factor for patients with central core disease. Recent series have demonstrated that many cases of late-onset axial myopathy in which patients manifest with bent spine (camptocormia) or neck extensor weakness (neck extensor myopathy) are caused by mutations in the ryanodine receptor gene (RYR1). This illustrates the interesting spectrum of RYR1 mutations.
The serum CK level is usually normal. Needle EMG demonstrates a myopathic pattern. Muscle biopsy shows fibers with single or multiple central or eccentric discrete zones (cores) devoid of oxidative enzymes. Cores occur preferentially in type 1 fibers and represent poorly aligned sarcomeres associated with Z disk streaming.
Autosomal dominant inheritance is characteristic; sporadic cases also occur. As alluded above, this myopathy is caused by point mutations of RYR1, encoding the calcium-release channel of the sarcoplasmic reticulum of skeletal muscle; mutations of this gene also account for some cases of inherited malignant hyperthermia (Chap. 23). Malignant hyperthermia is an allelic condition; C-terminal mutations of the RYR1 gene predispose to this complication.
Specific treatment is not required, but establishing a diagnosis of central core disease is extremely important because these patients have a known predisposition to malignant hyperthermia during anesthesia.
NEMALINE MYOPATHY
The term nemaline refers to the distinctive presence in muscle fibers of rods or threadlike structures (Greek nema, “thread”). Nemaline myopathy is clinically heterogeneous. A severe neonatal form presents with hypotonia and feeding and respiratory difficulties, leading to early death. Nemaline myopathy usually presents in infancy or childhood with delayed motor milestones. The course is nonprogressive or slowly progressive. The physical appearance is striking because of the long, narrow facies, high-arched palate, and open-mouthed appearance due to a prognathous jaw. Other skeletal abnormalities include pectus excavatum, kyphoscoliosis, pes cavus, and clubfoot deformities. Facial and generalized muscle weakness, including respiratory muscle weakness, is common. An adult-onset disorder with progressive proximal or distal weakness may be seen. Myocardial involvement is occasionally present in both the childhood and adult-onset forms. The serum CK level is usually normal or slightly elevated. The EMG demonstrates a myopathic pattern. Muscle biopsy shows clusters of small rods (nemaline bodies), which occur preferentially, but not exclusively, in the sarcoplasm of type 1 muscle fibers. Occasionally, the rods are also apparent in myonuclei. The muscle often shows type 1 muscle fiber predominance. Rods originate from the Z disk material of the muscle fiber.
Six genes have been associated with nemaline myopathy. Five of these code for thin filament–associated proteins, suggesting disturbed assembly or interplay of these structures as a pivotal mechanism. Mutations of the nebulin (NEB) gene account for most cases, including both severe neonatal and early childhood forms, inherited as autosomal recessive disorders. Neonatal and childhood cases, inherited as predominantly autosomal dominant disorders, are caused by mutations of the skeletal muscle a-actinin (ACTA1) gene. In milder forms of the disease with autosomal dominant inheritance, mutations have been identified in both the slow a-tropomyosin (TPM3) and β;-tropomyosin (TPM2) genes accounting for <3% of cases. Muscle troponin T (TNNT1) gene mutations appear to be limited to the Amish population in North America. Mutations may also be seen in NEM6 that encodes a putative BTB/Kelch protein. No specific treatment is available.
CENTRONUCLEAR (MYOTUBULAR) MYOPATHY
Three distinct variants of centronuclear myopathy occur. A neonatal form, also known as myotubular myopathy, presents with severe hypotonia and weakness at birth. The late infancy–early childhood form presents with delayed motor milestones. Later, difficulty with running and stair climbing becomes apparent. A marfanoid, slender body habitus, long narrow face, and high-arched palate are typical. Scoliosis and clubbed feet may be present. Most patients exhibit progressive weakness, some requiring wheelchairs. Progressive external ophthalmoplegia with ptosis and varying degrees of extraocular muscle impairment are characteristic of both the neonatal and the late-infantile forms. A third variant, the late childhood–adult form, has an onset in the second or third decade. Patients have full extraocular muscle movements and rarely exhibit ptosis. There is mild, slowly progressive limb weakness that may be distally predominant (some of these patients have been classified as having Charcot-Marie-Tooth disease type 2 [CMT2; Chap. 459]).
Normal or slightly elevated CK levels occur in each of the forms. Nerve conduction studies may reveal reduced amplitudes of distal compound muscle action potentials, in particular in adult-onset cases that resemble CMT2. EMG studies often give distinctive results, showing positive sharp waves and fibrillation potentials, complex and repetitive discharges, and rarely myotonic discharges. Muscle biopsy specimens in longitudinal section demonstrate rows of central nuclei, often surrounded by a halo. In transverse sections, central nuclei are found in 25–80% of muscle fibers.
A gene for the neonatal form of centronuclear myopathy has been localized to Xq28; this gene encodes myotubularin, a protein tyrosine phosphatase. Missense, frameshift, and splice-site mutations predict loss of myotubularin function in affected individuals. Carrier identification and prenatal diagnosis are possible. Autosomal recessive forms are caused by mutations in BIB1 that encodes for amphyphysin-2, whereas some autosomal dominant cases, which are allelic to a form of CMT2, are associated with mutations in the gene that encodes dynamin-2. No specific medical treatments are available at this time.
DISORDERS OF MUSCLE ENERGY METABOLISM
There are two principal sources of energy for skeletal muscle—fatty acids and glucose. Abnormalities in either glucose or lipid utilization can be associated with distinct clinical presentations that can range from an acute, painful syndrome with rhabdomyolysis and myoglobinuria to a chronic, progressive muscle weakness simulating muscular dystrophy.
GLYCOGEN STORAGE AND GLYCOLYTIC DEFECTS
Disorders of Glycogen Storage Causing Progressive Weakness • α-GLUCOSIDASE, OR ACID MALTASE, DEFICIENCY (POMPE’S DISEASE) Three clinical forms of α-glucosidase, or acid maltase, deficiency (type II glycogenosis) can be distinguished. The infantile form is the most common, with onset of symptoms in the first 3 months of life. Infants develop severe muscle weakness, cardiomegaly, hepatomegaly, and respiratory insufficiency. Glycogen accumulation in motor neurons of the spinal cord and brainstem contributes to muscle weakness. Death usually occurs by 1 year of age. In the childhood form, the picture resembles muscular dystrophy. Delayed motor milestones result from proximal limb muscle weakness and involvement of respiratory muscles. The heart may be involved, but the liver and brain are unaffected. The adult form usually begins in the third or fourth decade but can present as late as the seventh decade. Respiratory failure and diaphragmatic weakness are often initial manifestations, heralding progressive proximal muscle weakness. The heart and liver are not involved.
The serum CK level is 2–10 times normal in infantile or childhood-onset Pompe’s disease but can be normal in adult-onset cases. EMG examination demonstrates a myopathic pattern, but other features are especially distinctive, including myotonic discharges, trains of fibrillation and positive waves, and complex repetitive discharges. EMG discharges are very prominent in the paraspinal muscles. The muscle biopsy in infants typically reveals vacuoles containing glycogen and the lysosomal enzyme acid phosphatase. Electron microscopy reveals membrane-bound and free tissue glycogen. However, muscle biopsies in late-onset Pompe’s disease may demonstrate only nonspecific abnormalities. Enzyme analysis of dried blood spots is a sensitive technique to screen for Pompe’s disease. A definitive diagnosis is established by enzyme assay in muscle or cultured fibroblasts or by genetic testing.
Pompe’s disease is inherited as an autosomal recessive disorder caused by mutations of the α-glucosidase gene. Enzyme replacement therapy (ERT) with IV recombinant human α-glucosidase has been shown to be beneficial in infantile-onset Pompe’s disease. Clinical benefits in the infantile disease include reduced heart size, improved muscle function, reduced need for ventilatory support, and longer life. In late-onset cases, ERT has not been associated with the dramatic response that can be seen in classic infantile Pompe’s disease, yet it appears to stabilize the disease process.
OTHER GLYCOGEN STORAGE DISEASES WITH PROGRESSIVE WEAKNESS In de-branching enzyme deficiency (type III glycogenosis), a slowly progressive form of muscle weakness can develop after puberty. Rarely, myoglobinuria may be seen. Patients are usually diagnosed in infancy, however, because of hypotonia and delayed motor milestones, hepatomegaly, growth retardation, and hypoglycemia. Branching enzyme deficiency (type IV glycogenosis) is a rare and fatal glycogen storage disease characterized by failure to thrive and hepatomegaly. Hypotonia and muscle wasting may be present, but the skeletal muscle manifestations are minor compared to liver failure.
Disorders of Glycolysis Causing Exercise Intolerance Several glycolytic defects are associated with recurrent myoglobinuria: myophosphorylase deficiency (type V glycogenosis), phosphofructokinase deficiency (type VII glycogenosis), phosphoglycerate kinase deficiency, phosphorylase kinase deficiency (type IX glycogenosis), phosphoglycerate mutase deficiency (type × glycogenosis), lactate dehydrogenase deficiency (glycogenosis type XI), and β-enolase deficiency. Myophosphorylase deficiency, also known as McArdle’s disease, is by far the most common of the glycolytic defects associated with exercise intolerance. These glycolytic defects result in a common failure to support energy production at the initiation of exercise, although the exact site of energy failure remains controversial.
Clinical muscle manifestations in these conditions usually begin in adolescence. Symptoms are precipitated by brief bursts of high-intensity exercise such as running or lifting heavy objects. A history of myalgia and muscle stiffness usually precedes the intensely painful muscle contractures, which may be followed by myoglobinuria. Acute renal failure accompanies significant pigmenturia.
Certain features help distinguish some enzyme defects. In McArdle’s disease, exercise tolerance can be enhanced by a slow induction phase (warm-up) or brief periods of rest, allowing for the start of the “second-wind” phenomenon (switching to utilization of fatty acids). Varying degrees of hemolytic anemia accompany deficiencies of both phosphofructokinase (mild) and phosphoglycerate kinase (severe). In phosphoglycerate kinase deficiency, the usual clinical presentation is a seizure disorder associated with mental retardation; exercise intolerance is an infrequent manifestation.
In all of these conditions, the serum CK levels fluctuate widely and may be elevated even during symptom-free periods. CK levels >100 times normal are expected, accompanying myoglobinuria. All patients with suspected glycolytic defects leading to exercise intolerance should undergo a forearm exercise test. An impaired rise in venous lactate is highly indicative of a glycolytic defect. In lactate dehydrogenase deficiency, venous levels of lactate do not increase, but pyruvate rises to normal. A definitive diagnosis of glycolytic disease is made by muscle biopsy and subsequent enzyme analysis or by genetic testing.
Myophosphorylase deficiency, phosphofructokinase deficiency, and phosphoglycerate mutase deficiency are inherited as autosomal recessive disorders. Phosphoglycerate kinase deficiency is X-linked recessive. Mutations can be found in the respective genes encoding the abnormal proteins in each of these disorders.
Training may enhance exercise tolerance, perhaps by increasing perfusion to muscle. Dietary intake of free glucose or fructose prior to activity may improve function but care must be taken to avoid obesity from ingesting too many calories.
LIPID AS AN ENERGY SOURCE AND ASSOCIATED DEFECTS
Lipid is an important muscle energy source during rest and during prolonged, submaximal exercise. Fatty acids are derived from circulating very-low-density lipoprotein (VLDL) in the blood or from triglycerides stored in muscle fibers. Oxidation of fatty acids occurs in the mitochondria. To enter the mitochondria, a fatty acid must first be converted to an “activated fatty acid,” acyl-CoA. The acyl-CoA must be linked with carnitine by the enzyme carnitine palmitoyltransferase (CPT) I for transport into the mitochondria. CPT I is present on the inner side of the outer mitochondrial membrane. Carnitine is removed by CPT II, an enzyme attached to the inside of the inner mitochondrial membrane, allowing transport of acyl-CoA into the mitochondrial matrix for β-oxidation.
Carnitine Palmitoyltransferase Deficiency CPT II deficiency is the most common recognizable cause of recurrent myoglobinuria, more common than the glycolytic defects. Onset is usually in the teenage years or early twenties. Muscle pain and myoglobinuria typically occur after prolonged exercise but can also be precipitated by fasting or infections; up to 20% of patients do not exhibit myoglobinuria, however. Strength is normal between attacks. In contrast to disorders caused by defects in glycolysis, in which muscle cramps follow short, intense bursts of exercise, the muscle pain in CPT II deficiency does not occur until the limits of utilization have been exceeded and muscle breakdown has already begun. Episodes of rhabdomyolysis may produce severe weakness. In young children and newborns, CPT II deficiency can present with a very severe clinical picture including hypoketotic hypoglycemia, cardiomyopathy, liver failure, and sudden death.
Serum CK levels and EMG findings are both usually normal between episodes. A normal rise of venous lactate during forearm exercise distinguishes this condition from glycolytic defects, especially myophosphorylase deficiency. Muscle biopsy does not show lipid accumulation and is usually normal between attacks. The diagnosis requires direct measurement of muscle CPT or genetic testing.
CPT II deficiency is much more common in men than women (5:1); nevertheless, all evidence indicates autosomal recessive inheritance. A mutation in the gene for CPT II (chromosome 1p36) causes the disease in some individuals. Attempts to improve exercise tolerance with frequent meals and a low-fat, high-carbohydrate diet, or by substituting medium-chain triglycerides in the diet, have not proven to be beneficial.
Myoadenylate Deaminase Deficiency The muscle enzyme myoadenylate deaminase converts adenosine-5′-monophosphate (5′-AMP) to inosine monophosphate (IMP) with liberation of ammonia. Myoadenylate deaminase may play a role in regulating adenosine triphosphate (ATP) levels in muscles. Most individuals with myoadenylate deaminase deficiency have no symptoms. There have been a few reports of patients with this disorder who have exercise-exacerbated myalgia and myoglobinuria. Many questions have been raised about the clinical effects of myoadenylate deaminase deficiency, and, specifically, its relationship to exertional myalgia and fatigability, but there is no consensus.
MITOCHONDRIAL MYOPATHIES
In 1972, Olson and colleagues recognized that muscle fibers with significant numbers of abnormal mitochondria could be highlighted with the modified trichrome stain; the term ragged red fibers was coined. By electron microscopy, the mitochondria in ragged red fibers are enlarged and often bizarrely shaped and have crystalline inclusions. Since that seminal observation, the understanding of these disorders of muscle and other tissues has expanded (Chap. 82).
Mitochondria play a key role in energy production. Oxidation of the major nutrients derived from carbohydrate, fat, and protein leads to the generation of reducing equivalents. The latter are transported through the respiratory chain in the process known as oxidative phosphorylation. The energy generated by the oxidation-reduction reactions of the respiratory chain is stored in an electrochemical gradient coupled to ATP synthesis.
A novel feature of mitochondria is their genetic composition. Each mitochondrion possesses a DNA genome that is distinct from that of the nuclear DNA. Human mitochondrial DNA (mtDNA) consists of a double-strand, circular molecule comprising 16,569 base pairs. It codes for 22 transfer RNAs, 2 ribosomal RNAs, and 13 polypeptides of the respiratory chain enzymes. The genetics of mitochondrial diseases differ from the genetics of chromosomal disorders. The DNA of mitochondria is directly inherited from the cytoplasm of the gametes, mainly from the oocyte. The sperm contributes very little of its mitochondria to the offspring at the time of fertilization. Thus, mitochondrial genes are derived almost exclusively from the mother, accounting for maternal inheritance of some mitochondrial disorders.
Patients with mitochondrial myopathies have clinical manifestations that usually fall into three groups: chronic progressive external ophthalmoplegia (CPEO), skeletal muscle–CNS syndromes, and pure myopathy simulating muscular dystrophy or metabolic myopathy.
PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA SYNDROMES WITH RAGGED RED FIBERS
The single most common sign of a mitochondrial myopathy is CPEO, occurring in >50% of all mitochondrial myopathies. Varying degrees of ptosis and weakness of extraocular muscles are seen, usually in the absence of diplopia, a point of distinction from disorders with fluctuating eye weakness (e.g., myasthenia gravis).
KEARNS-SAYRE SYNDROME (KSS)
KSS is a widespread multiorgan system disorder with a defined triad of clinical findings: onset before age 20, CPEO, and pigmentary retinopathy, plus one or more of the following features: complete heart block, cerebrospinal fluid (CSF) protein >1 g/L (100 mg/dL), or cerebellar ataxia. Some patients with CPEO and ragged red fibers may not fulfill all of the criteria for KSS. The cardiac disease includes syncopal attacks and cardiac arrest related to the abnormalities in the cardiac conduction system: prolonged intraventricular conduction time, bundle branch block, and complete atrioventricular block. Death attributed to heart block occurs in ~20% of the patients. Varying degrees of progressive limb muscle weakness and easy fatigability affect activities of daily living. Endocrine abnormalities are common, including gonadal dysfunction in both sexes with delayed puberty, short stature, and infertility. Diabetes mellitus is a cardinal sign of mitochondrial disorders and is estimated to occur in 13% of KSS patients. Other less common endocrine disorders include thyroid disease, hyperaldosteronism, Addison’s disease, and hypoparathyroidism. Both mental retardation and dementia are common accompaniments to this disorder. Serum CK levels are normal or slightly elevated. Serum lactate and pyruvate levels may be elevated. EMG is myopathic. Nerve conduction studies may be abnormal related to an associated neuropathy. Muscle biopsies reveal ragged red fibers, highlighted in oxidative enzyme stains, many showing defects in cytochrome oxidase. By electron microscopy, there are increased numbers of mitochondria that often appear enlarged and contain paracrystalline inclusions.
KSS is a sporadic disorder. The disease is caused by single mtDNA deletions presumed to arise spontaneously in the ovum or zygote. The most common deletion, occurring in about one-third of patients, removes 4977 bp of contiguous mtDNA. Monitoring for cardiac conduction defects is critical. Prophylactic pacemaker implantation is indicated when ECGs demonstrate a bifascicular block. In KSS, no benefit has been shown for supplementary therapies, including multivitamins or coenzyme Q10. Of all the proposed options, exercise might be the most applicable but must be approached cautiously because of defects in the cardiac conduction system.
PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA (PEO)
This condition is caused by nuclear DNA mutations affecting mtDNA copy number and integrity and is thus inherited in a Mendelian fashion. Onset is usually after puberty. Fatigue, exercise intolerance, and complaints of muscle weakness are typical. Some patients notice swallowing problems. The neurologic examination confirms the ptosis and ophthalmoplegia, usually asymmetric in distribution. A sensorineural hearing loss may be encountered. Mild facial, neck flexor, and proximal weakness are typical. Rarely, respiratory muscles may be progressively affected and may be the direct cause of death. Serum CK is normal or mildly elevated. The resting lactate level is normal or slightly elevated but may rise excessively after exercise. CSF protein is normal. The EMG is myopathic, and nerve conduction studies are usually normal. Ragged red fibers are prominently displayed in the muscle biopsy. Southern blots of muscle reveal a normal mtDNA band at 16.6 kb and several additional mtDNA deletion bands with genomes varying from 0.5 to 10 kb.
This autosomal dominant form of CPEO has been linked to loci on three chromosomes: 4q35, 10q24, and 15q22–26. In the chromosome 4q-related form of disease, mutations of the gene encoding the heart and skeletal muscle–specific isoform of the adenine nucleotide translocator 1 (ANT1) gene are found. This highly abundant mitochondrial protein forms a homodimeric inner mitochondrial channel through which adenosine diphosphate (ADP) enters and ATP leaves the mitochondrial matrix. In the chromosome 10q–related disorder, mutations of the gene C10orf2 are found. Its gene product, twinkle, co-localizes with the mtDNA and is named for its punctate, starlike staining properties. The function of twinkle is presumed to be critical for lifetime maintenance of mitochondrial integrity. In the cases mapped to chromosome 15q, a mutation affects the gene encoding mtDNA polymerase (POLG), an enzyme important in mtDNA replication. Autosomal recessive PEO has also been described with mutations in the POLG gene. Point mutations have been identified within various mitochondrial tRNA (Leu, Ile, Asn, Trp) genes in families with maternal inheritance of PEO.
Exercise may improve function but will depend on the patient’s ability to participate.
MITOCHONDRIAL DNA SKELETAL MUSCLE–CENTRAL NERVOUS SYSTEM SYNDROMES
Myoclonic Epilepsy with Ragged Red Fibers (MERRF) The onset of MERRF is variable, ranging from late childhood to middle adult life. Characteristic features include myoclonic epilepsy, cerebellar ataxia, and progressive muscle weakness. The seizure disorder is an integral part of the disease and may be the initial symptom. Cerebellar ataxia precedes or accompanies epilepsy. It is slowly progressive and generalized. The third major feature of the disease is muscle weakness in a limb-girdle distribution. Other more variable features include dementia, peripheral neuropathy, optic atrophy, hearing loss, and diabetes mellitus.
Serum CK levels are normal or slightly increased. The serum lactate may be elevated. EMG is myopathic, and in some patients nerve conduction studies show a neuropathy. The electroencephalogram is abnormal, corroborating clinical findings of epilepsy. Typical ragged red fibers are seen on muscle biopsy. MERRF is caused by maternally inherited point mutations of mitochondrial tRNA genes. The most common mutation found in 80% of MERRF patients is an A to G substitution at nucleotide 8344 of tRNA lysine (A8344G tRNAlys). Other tRNAlys mutations include base-pair substitutions T8356C and G8363A. Only supportive treatment is possible, with special attention to epilepsy.
Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Strokelike Episodes (MELAS) MELAS is the most common mitochondrial encephalomyopathy. The term strokelike is appropriate because the cerebral lesions do not conform to a strictly vascular distribution. The onset in the majority of patients is before age 20. Seizures, usually partial motor or generalized, are common and may represent the first clearly recognizable sign of disease. The cerebral insults that resemble strokes cause hemiparesis, hemianopia, and cortical blindness. A presumptive stroke occurring before age 40 should place this mitochondrial encephalomyopathy high in the differential diagnosis. Associated conditions include hearing loss, diabetes mellitus, hypothalamic pituitary dysfunction causing growth hormone deficiency, hypothyroidism, and absence of secondary sexual characteristics. In its full expression, MELAS leads to dementia, a bedridden state, and a fatal outcome. Serum lactic acid is typically elevated. The CSF protein is also increased but is usually ≤1 g/L (100 mg/dL). Muscle biopsies show ragged red fibers. Neuroimaging demonstrates basal ganglia calcification in a high percentage of cases. Focal lesions that mimic infarction are present predominantly in the occipital and parietal lobes. Strict vascular territories are not respected, and cerebral angiography fails to demonstrate lesions of the major cerebral blood vessels.
MELAS is caused by maternally inherited point mutations of mitochondrial tRNA genes. Most of the tRNA mutations are lethal, accounting for the paucity of multigeneration families with this syndrome. The A3243G point mutation in tRNALeu(UUR) is the most common, occurring in ~80% of MELAS cases. About 10% of MELAS patients have other mutations of the tRNALeu(UUR) gene, including 3252G, 3256T, 3271C, and 3291C. Other tRNA gene mutations have also been reported in MELAS, including G583A tRNAPhe, G1642A tRNAVal, G4332A tRNAGlu, and T8316C tRNALys. Mutations have also been reported in mtDNA polypeptide-coding genes. Two mutations were found in the ND5 subunit of complex I of the respiratory chain. A missense mutation has been reported at mtDNA position 9957 in the gene for subunit III of cytochrome C oxidase. No specific treatment is available. Supportive treatment is essential for the strokelike episodes, seizures, and endocrinopathies.
PURE MYOPATHY SYNDROMES
Muscle weakness and fatigue can be the predominant manifestations of mtDNA mutations. When the condition affects exclusively muscle (pure myopathy), the disorder becomes difficult to recognize. Occasionally, mitochondrial myopathies can present with recurrent myoglobinuria without fixed weakness and thus resemble a glycogen storage disorder or CPT deficiency.
Mitochondrial DNA Depletion Syndromes Mitochondrial DNA depletion syndrome (MDS) is a heterogeneous group of disorders that are inherited in an autosomal recessive fashion and can present in infancy or adults. MDS can be caused by mutations in genes (TK2, DGUOK, RRM2B, TYMP, SUCLA1, and SUCLA2) that lead to depletion of pools of mitochondrial deoxyribonucleotide (dNTP) pools necessary for mtDNA replication The other major cause of MDS is a set of mutations in genes essential for mtDNA replication (e.g., POLG1 and C10orf2). The clinical phenotypes associated with MDS vary. Patients may develop a severe encephalopathy (e.g., Leigh’s syndrome), PEO, an isolated myopathy, myo-neuro-gastrointestinal-encephalopathy (MNGIE), and a sensory neuropathy with ataxia.
DISORDERS OF MUSCLE MEMBRANE EXCITABILITY
Muscle membrane excitability is affected in a group of disorders referred to as channelopathies. The heart may also be involved, resulting in life-threatening complications (Table 462e-10).
CLINICAL FEATURES OF PERIODIC PARALYSIS AND NONDYSTROPHIC MYOTONIAS |
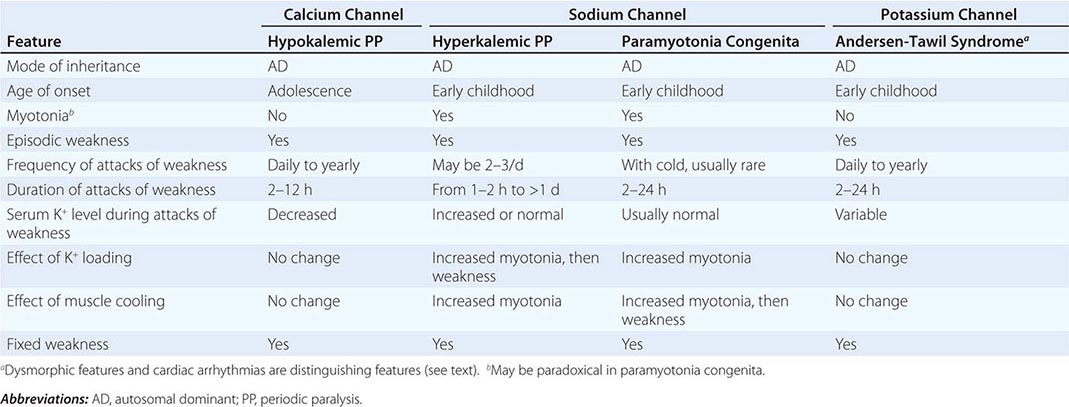
CALCIUM CHANNEL DISORDERS OF MUSCLE
Hypokalemic Periodic Paralysis (HypoKPP) Onset occurs at adolescence. Men are more often affected because of decreased penetrance in women. Episodic weakness with onset after age 25 is almost never due to periodic paralyses, with the exception of thyrotoxic periodic paralysis (see below). Attacks are often provoked by meals high in carbohydrates or sodium and may accompany rest following prolonged exercise. Weakness usually affects proximal limb muscles more than distal. Ocular and bulbar muscles are less likely to be affected. Respiratory muscles are usually spared, but when they are involved, the condition may prove fatal. Weakness may take as long as 24 h to resolve. Life-threatening cardiac arrhythmias related to hypokalemia may occur during attacks. As a late complication, patients commonly develop severe, disabling proximal lower extremity weakness.
Attacks of thyrotoxic periodic paralysis resemble those of primary HypoKPP. Despite a higher incidence of thyrotoxicosis in women, men, particularly those of Asian descent, are more likely to manifest this complication. Attacks abate with treatment of the underlying thyroid condition.
A low serum potassium level during an attack, excluding secondary causes, establishes the diagnosis. Interattack muscle biopsies show the presence of single or multiple centrally placed vacuoles or tubular aggregates. Provocative tests with glucose and insulin to establish a diagnosis are usually not necessary and are potentially hazardous.
In the midst of an attack of weakness, motor conduction studies may demonstrate reduced amplitudes, whereas EMG may show electrical silence in severely weak muscles. In between attacks, the EMG and routine nerve conduction studies are normal. However, a long exercise test may demonstrate a decrementing amplitude, and myopathic MUAPs may be seen on EMG in patients with fixed weakness.
HypoKPP is caused by mutations in either of two genes. HypoKPP type 1, the most common form, is inherited as an autosomal dominant disorder with incomplete penetrance. These patients have mutations in the voltage-sensitive, skeletal muscle calcium channel gene, CALCL1A3 (Fig. 462e-8). Approximately 10% of cases are HypoKPP type 2, arising from mutations in the voltage-sensitive sodium channel gene (SCN4A). In either instance, the mutations lead to an abnormal gating pore current that predisposes the muscle cell to depolarize when potassium levels are low. It is also now recognized that some cases of thyrotoxic HypoKPP are caused by genetic variants in a potassium channel (Kir 2.6), whose expression is regulated by thyroid hormone.
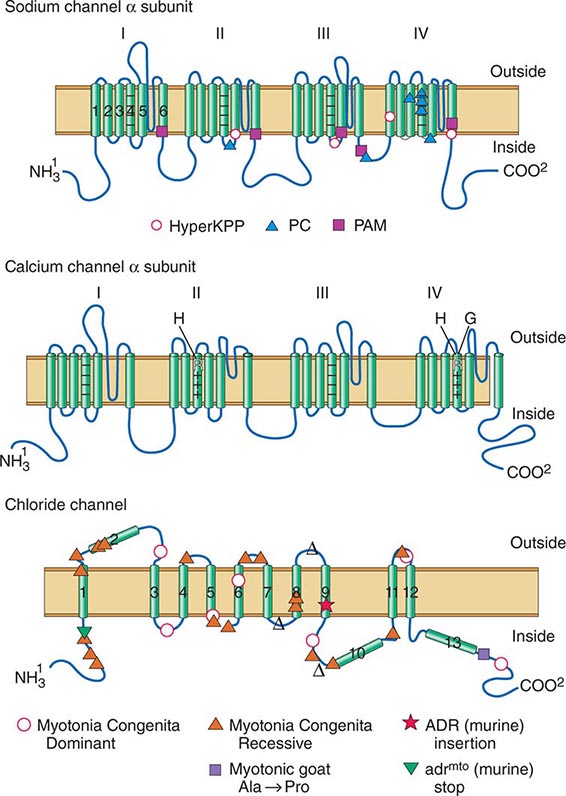
FIGURE 462e-8 The sodium and calcium channels are depicted here as containing four homologous domains, each with six membrane-spanning segments. The fourth segment of each domain bears positive charges and acts as the “voltage sensor” for the channel. The association of the four domains is thought to form a pore through which ions pass. Sodium channel mutations are shown along with the phenotype that they confer. HyperKPP, hyperkalemic periodic paralysis; PC, paramyotonia congenita; PAM, potassium-aggravated myotonia. See text for details.
The chloride channel is envisioned to have 10 membrane-spanning domains. The positions of mutations causing dominantly and recessively inherited myotonia congenita are indicated, along with mutations that cause this disease in mice and goats.
TREATMENT | HYPOKALEMIC PERIODIC PARALYSIS |
The acute paralysis improves after the administration of potassium. Muscle strength and ECG should be monitored. Oral KCl (0.2–0.4 mmol/kg) should be given every 30 min. Only rarely is IV therapy necessary (e.g., when swallowing problems or vomiting is present). Administration of potassium in a glucose solution should be avoided because it may further reduce serum potassium levels. Mannitol is the preferred vehicle for administration of IV potassium. The long-term goal of therapy is to avoid attacks. This may reduce late-onset, fixed weakness. Patients should be made aware of the importance of a low-carbohydrate, low-sodium diet and consequences of intense exercise. Prophylactic administration of acetazolamide (125–1000 mg/d in divided doses) reduces or may abolish attacks in HypoKPP type 1. Paradoxically the potassium is lowered, but this is offset by the beneficial effect of metabolic acidosis. If attacks persist on acetazolamide, oral KCl should be added. Some patients require treatment with triamterene (25–100 mg/d) or spironolactone (25–100 mg/d). However, in patients with HypoKPP type 2, attacks of weakness can be exacerbated with acetazolamide.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


