456 | Diseases of the Spinal Cord |
Diseases of the spinal cord are frequently devastating. They produce quadriplegia, paraplegia, and sensory deficits far beyond the damage they would inflict elsewhere in the nervous system because the spinal cord contains, in a small cross-sectional area, almost the entire motor output and sensory input of the trunk and limbs. Many spinal cord diseases are reversible if recognized and treated at an early stage (Table 456-1); thus, they are among the most critical of neurologic emergencies. The efficient use of diagnostic procedures, guided by knowledge of the anatomy and the clinical features of spinal cord diseases, is required to maximize the likelihood of a successful outcome.
TREATABLE SPINAL CORD DISORDERS |
Abbreviations: CMV, cytomegalovirus; HSV, herpes simplex virus; HTLV, human T cell lymphotropic virus; VZV, varicella-zoster virus.
APPROACH TO THE PATIENT:
Spinal Cord Disease
SPINAL CORD ANATOMY RELEVANT TO CLINICAL SIGNS
The spinal cord is a thin, tubular extension of the central nervous system contained within the bony spinal canal. It originates at the medulla and continues caudally to the conus medullaris at the lumbar level; its fibrous extension, the filum terminale, terminates at the coccyx. The adult spinal cord is ~46 cm (18 in.) long, oval in shape, and enlarged in the cervical and lumbar regions, where neurons that innervate the upper and lower extremities, respectively, are located. The white matter tracts containing ascending sensory and descending motor pathways are located peripherally, whereas nerve cell bodies are clustered in an inner region of gray matter shaped like a four-leaf clover that surrounds the central canal (anatomically an extension of the fourth ventricle). The membranes that cover the spinal cord—the pia, arachnoid, and dura—are continuous with those of the brain, and the cerebrospinal fluid is contained within the subarachnoid space between the pia and arachnoid.
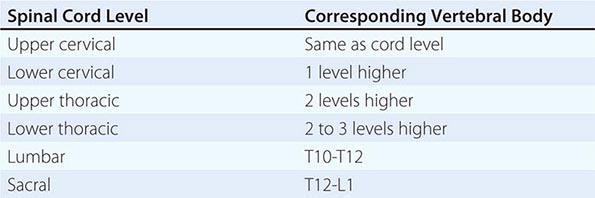
The spinal cord has 31 segments, each defined by an exiting ventral motor root and entering dorsal sensory root. During embryologic development, growth of the cord lags behind that of the vertebral column, and the mature spinal cord ends at approximately the first lumbar vertebral body. The lower spinal nerves take an increasingly downward course to exit via intervertebral foramina. The first seven pairs of cervical spinal nerves exit above the same-numbered vertebral bodies, whereas all the subsequent nerves exit below the same-numbered vertebral bodies because of the presence of eight cervical spinal cord segments but only seven cervical vertebrae. The relationship between spinal cord segments and the corresponding vertebral bodies is shown in Table 456-2. These relationships assume particular importance for localization of lesions that cause spinal cord compression. Sensory loss below the circumferential level of the umbilicus, for example, corresponds to the T10 cord segment but indicates involvement of the cord adjacent to the seventh or eighth thoracic vertebral body (see Figs. 31-2 and 31-3). In addition, at every level, the main ascending and descending tracts are somatotopically organized with a laminated distribution that reflects the origin or destination of nerve fibers.
SPINAL CORD LEVELS RELATIVE TO THE VERTEBRAL BODIES |
Determining the Level of the Lesion The presence of a horizontally defined level below which sensory, motor, and autonomic function is impaired is a hallmark of a lesion of the spinal cord. This sensory level is sought by asking the patient to identify a pinprick or cold stimulus applied to the proximal legs and lower trunk and successively moved up toward the neck on each side. Sensory loss below this level is the result of damage to the spinothalamic tract on the opposite side, one to two segments higher in the case of a unilateral spinal cord lesion, and at the level of a bilateral lesion. The discrepancy in the level of a unilateral lesion is the result of the course of the second-order sensory fibers, which originate in the dorsal horn, and ascend for one or two levels as they cross anterior to the central canal to join the opposite spinothalamic tract. Lesions that transect the descending corticospinal and other motor tracts cause paraplegia or quadriplegia with heightened deep tendon reflexes, Babinski signs, and eventual spasticity (the upper motor neuron syndrome). Transverse damage to the cord also produces autonomic disturbances consisting of absent sweating below the implicated cord level and bladder, bowel, and sexual dysfunction.
The uppermost level of a spinal cord lesion can also be localized by attention to the segmental signs corresponding to disturbed motor or sensory innervation by an individual cord segment. A band of altered sensation (hyperalgesia or hyperpathia) at the upper end of the sensory disturbance, fasciculations or atrophy in muscles innervated by one or several segments, or a muted or absent deep tendon reflex may be noted at this level. These signs also can occur with focal root or peripheral nerve disorders; thus, they are most useful when they occur together with signs of long tract damage. With severe and acute transverse lesions, the limbs initially may be flaccid rather than spastic. This state of “spinal shock” lasts for several days, rarely for weeks, and may be mistaken for extensive damage to the anterior horn cells over many segments of the cord or for an acute polyneuropathy.
The main features of transverse damage at each level of the spinal cord are summarized below.
CERVICAL CORD Upper cervical cord lesions produce quadriplegia and weakness of the diaphragm. The uppermost level of weakness and reflex loss with lesions at C5-C6 is in the biceps; at C7, in finger and wrist extensors and triceps; and at C8, finger and wrist flexion. Horner’s syndrome (miosis, ptosis, and facial hypohidrosis) may accompany a cervical cord lesion at any level.
THORACIC CORD Lesions here are localized by the sensory level on the trunk and, if present, by the site of midline back pain. Useful markers of the sensory level on the trunk are the nipples (T4) and umbilicus (T10). Leg weakness and disturbances of bladder and bowel function accompany the paralysis. Lesions at T9-T10 paralyze the lower—but not the upper—abdominal muscles, resulting in upward movement of the umbilicus when the abdominal wall contracts (Beevor’s sign).
LUMBAR CORD Lesions at the L2-L4 spinal cord levels paralyze flexion and adduction of the thigh, weaken leg extension at the knee, and abolish the patellar reflex. Lesions at L5-S1 paralyze only movements of the foot and ankle, flexion at the knee, and extension of the thigh, and abolish the ankle jerks (S1).
SACRAL CORD/CONUS MEDULLARIS The conus medullaris is the tapered caudal termination of the spinal cord, comprising the sacral and single coccygeal segments. The distinctive conus syndrome consists of bilateral saddle anesthesia (S3-S5), prominent bladder and bowel dysfunction (urinary retention and incontinence with lax anal tone), and impotence. The bulbocavernosus (S2-S4) and anal (S4-S5) reflexes are absent (Chap. 437). Muscle strength is largely preserved. By contrast, lesions of the cauda equina, the nerve roots derived from the lower cord, are characterized by low back and radicular pain, asymmetric leg weakness and sensory loss, variable areflexia in the lower extremities, and relative sparing of bowel and bladder function. Mass lesions in the lower spinal canal often produce a mixed clinical picture with elements of both cauda equina and conus medullaris syndromes. Cauda equina syndromes are also discussed in Chap. 22.
Special Patterns of Spinal Cord Disease The location of the major ascending and descending pathways of the spinal cord are shown in Fig. 456-1. Most fiber tracts—including the posterior columns and the spinocerebellar and pyramidal tracts—are situated on the side of the body they innervate. However, afferent fibers mediating pain and temperature sensation ascend in the spinothalamic tract contralateral to the side they supply. The anatomic configurations of these tracts produce characteristic syndromes that provide clues to the underlying disease process.
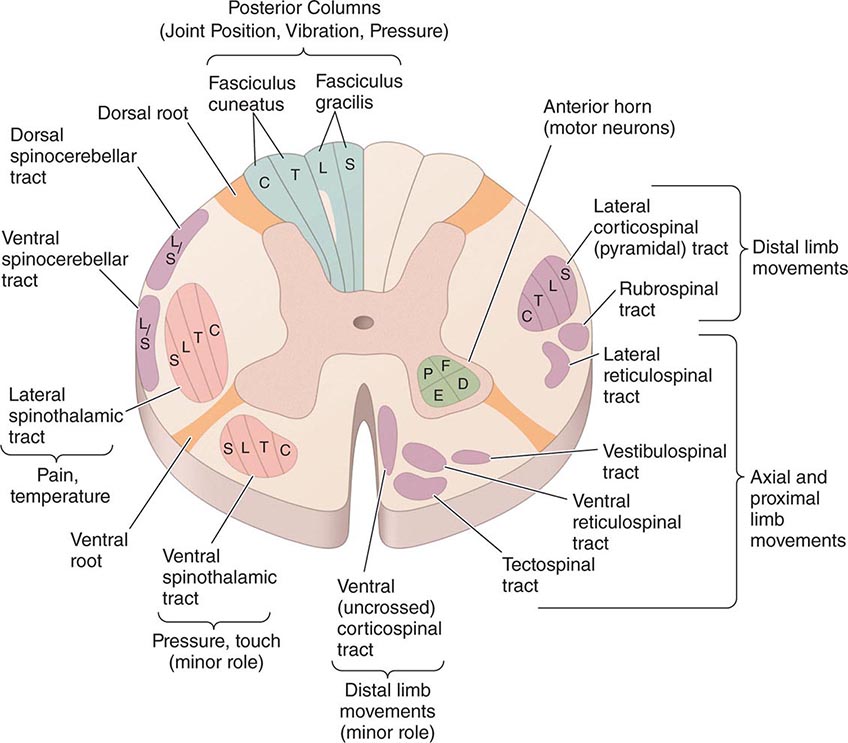
FIGURE 456-1 Transverse section through the spinal cord, composite representation, illustrating the principal ascending (left) and descending (right) pathways. The lateral and ventral spinothalamic tracts ascend contralateral to the side of the body that is innervated. C, cervical; D, distal; E, extensors; F, flexors; L, lumbar; P, proximal; S, sacral; T, thoracic.
BROWN-SEQUARD HEMICORD SYNDROME This consists of ipsilateral weakness (corticospinal tract) and loss of joint position and vibratory sense (posterior column), with contralateral loss of pain and temperature sense (spinothalamic tract) one or two levels below the lesion. Segmental signs, such as radicular pain, muscle atrophy, or loss of a deep tendon reflex, are unilateral. Partial forms are more common than the fully developed syndrome.
CENTRAL CORD SYNDROME This syndrome results from selective damage to the gray matter nerve cells and crossing spinothalamic tracts surrounding the central canal. In the cervical cord, the central cord syndrome produces arm weakness out of proportion to leg weakness and a “dissociated” sensory loss, meaning loss of pain and temperature sensations over the shoulders, lower neck, and upper trunk (cape distribution), in contrast to preservation of light touch, joint position, and vibration sense in these regions. Spinal trauma, syringomyelia, and intrinsic cord tumors are the main causes.
ANTERIOR SPINAL ARTERY SYNDROME Infarction of the cord is generally the result of occlusion or diminished flow in this artery. The result is bilateral tissue destruction at several contiguous levels that spares the posterior columns. All spinal cord functions—motor, sensory, and autonomic—are lost below the level of the lesion, with the striking exception of retained vibration and position sensation.
FORAMEN MAGNUM SYNDROME Lesions in this area interrupt decussating pyramidal tract fibers destined for the legs, which cross caudal to those of the arms, resulting in weakness of the legs (crural paresis). Compressive lesions near the foramen magnum may produce weakness of the ipsilateral shoulder and arm followed by weakness of the ipsilateral leg, then the contralateral leg, and finally the contralateral arm, an “around the clock” pattern that may begin in any of the four limbs. There is typically suboccipital pain spreading to the neck and shoulders.
INTRAMEDULLARY AND EXTRAMEDULLARY SYNDROMES It is useful to differentiate intramedullary processes, arising within the substance of the cord, from extramedullary ones that lie outside the cord and compress the spinal cord or its vascular supply. The differentiating features are only relative and serve as clinical guides. With extramedullary lesions, radicular pain is often prominent, and there is early sacral sensory loss and spastic weakness in the legs with incontinence due to the superficial location of the corresponding sensory and motor fibers in the spinothalamic and corticospinal tracts (Fig. 456-1). Intramedullary lesions tend to produce poorly localized burning pain rather than radicular pain and to spare sensation in the perineal and sacral areas (“sacral sparing”), reflecting the laminated configuration of the spinothalamic tract with sacral fibers outermost; corticospinal tract signs appear later. Regarding extramedullary lesions, a further distinction is made between extradural and intradural masses, as the former are generally malignant and the latter benign (neurofibroma being a common cause). Consequently, a long duration of symptoms favors an intradural origin.
ACUTE AND SUBACUTE SPINAL CORD DISEASES
The initial symptoms of structural diseases of the cord that evolve over days or weeks are focal neck or back pain, followed by various combinations of paresthesias, sensory loss, motor weakness, and sphincter disturbance. There may be only mild sensory symptoms or a devastating functional transection of the cord. Partial lesions selectively involve the posterior columns or anterior spinothalamic tracts or are limited to one side of the cord. Paresthesias or numbness typically begins in the feet and ascend symmetrically or asymmetrically. These symptoms simulate a polyneuropathy, but a sharply demarcated spinal cord level indicates the myelopathic nature of the process.
In severe and abrupt cases, areflexia reflecting spinal shock may be present, but hyperreflexia supervenes over days or weeks; persistent areflexic paralysis with a sensory level usually indicates necrosis over multiple segments of the spinal cord.
COMPRESSIVE MYELOPATHIES
Neoplastic Spinal Cord Compression In adults, most neoplasms are epidural in origin, resulting from metastases to the adjacent vertebral column. The propensity of solid tumors to metastasize to the vertebral column probably reflects the high proportion of bone marrow located in the axial skeleton. Almost any malignant tumor can metastasize to the spinal column, with breast, lung, prostate, kidney, lymphoma, and myeloma being particularly frequent. The thoracic spinal column is most commonly involved; exceptions are metastases from prostate and ovarian cancer, which occur disproportionately in the sacral and lumbar vertebrae, probably from spread through Batson’s plexus, a network of veins along the anterior epidural space. Retroperitoneal neoplasms (especially lymphomas or sarcomas) enter the spinal canal laterally through the intervertebral foramina and produce radicular pain with signs of weakness that corresponds to the level of involved nerve roots.
Pain is usually the initial symptom of spinal metastasis; it may be aching and localized or sharp and radiating in quality and typically worsens with movement, coughing, or sneezing and characteristically awakens patients at night. A recent onset of persistent back pain, particularly if in the thoracic spine (which is uncommonly involved by spondylosis), should prompt consideration of vertebral metastasis. Rarely, pain is mild or absent. Plain radiographs of the spine and radionuclide bone scans have a limited role in diagnosis because they do not identify 15–20% of metastatic vertebral lesions and fail to detect paravertebral masses that reach the epidural space through the intervertebral foramina. MRI provides excellent anatomic resolution of the extent of spinal tumors (Fig. 456-2) and is able to distinguish between malignant lesions and other masses—epidural abscess, tuberculoma, lipoma, or epidural hemorrhage, among others—that present in a similar fashion. Vertebral metastases are usually hypointense relative to a normal bone marrow signal on T1-weighted MRI; after the administration of gadolinium, contrast enhancement may deceptively “normalize” the appearance of the tumor by increasing its intensity to that of normal bone marrow. Infections of the spinal column (osteomyelitis and related disorders) are distinctive in that, unlike tumor, they often cross the disk space to involve the adjacent vertebral body.
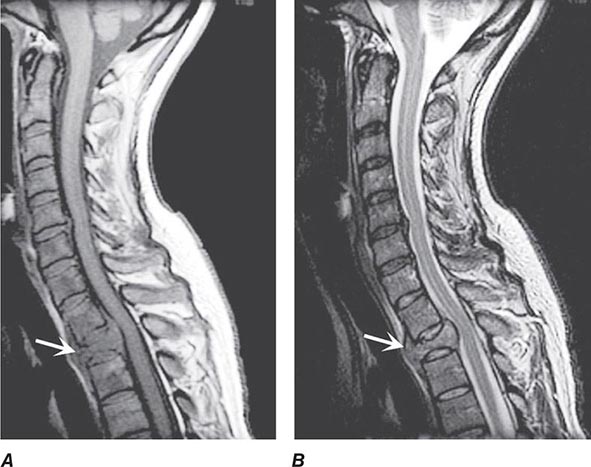
FIGURE 456-2 Epidural spinal cord compression due to breast carcinoma. Sagittal T1-weighted (A) and T2-weighted (B) magnetic resonance imaging scans through the cervicothoracic junction reveal an infiltrated and collapsed second thoracic vertebral body with posterior displacement and compression of the upper thoracic spinal cord. The low-intensity bone marrow signal in A signifies replacement by tumor.
If spinal cord compression is suspected, imaging should be obtained promptly. If there are radicular symptoms but no evidence of myelopathy, it may be safe to defer imaging for 24–48 h. Up to 40% of patients who present with cord compression at one level are found to have asymptomatic epidural metastases elsewhere; thus, the length of the spine is often imaged when epidural malignancy is in question.
In contrast to tumors of the epidural space, most intradural mass lesions are slow-growing and benign. Meningiomas and neurofibromas account for most of these, with occasional cases caused by chordoma, lipoma, dermoid, or sarcoma. Meningiomas (Fig. 456-3) are often located posterior to the thoracic cord or near the foramen magnum, although they can arise from the meninges anywhere along the spinal canal. Neurofibromas are benign tumors of the nerve sheath that typically arise from the posterior root; when multiple, neurofibromatosis is the likely etiology. Symptoms usually begin with radicular sensory symptoms followed by an asymmetric, progressive spinal cord syndrome. Therapy is surgical resection.
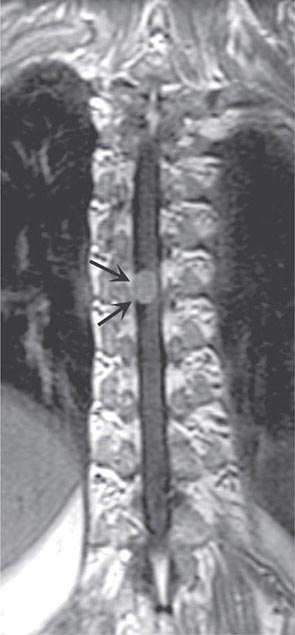
FIGURE 456-3 Magnetic resonance imaging of a thoracic meningioma. Coronal T1-weighted postcontrast image through the thoracic spinal cord demonstrates intense and uniform enhancement of a well-circumscribed extramedullary mass (arrows) that displaces the spinal cord to the left.
Primary intramedullary tumors of the spinal cord are uncommon. They present as central cord or hemicord syndromes, often in the cervical region. There may be poorly localized burning pain in the extremities and sparing of sacral sensation. In adults, these lesions are ependymomas, hemangioblastomas, or low-grade astrocytomas (Fig. 456-4). Complete resection of an intramedullary ependymoma is often possible with microsurgical techniques. Debulking of an intramedullary astrocytoma can also be helpful, as these are often slowly growing lesions; the value of adjunctive radiotherapy and chemotherapy is uncertain. Secondary (metastatic) intramedullary tumors also occur, especially in patients with advanced metastatic disease (Chap. 118), although these are not nearly as frequent as brain metastases.
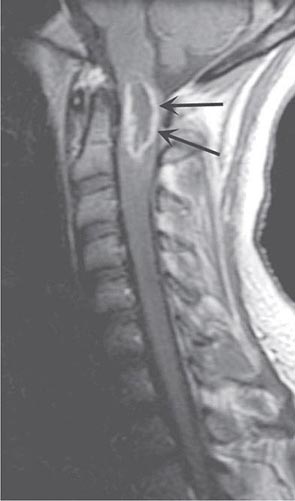
FIGURE 456-4 Magnetic resonance imaging of an intramedullary astrocytoma. Sagittal T1-weighted postcontrast image through the cervical spine demonstrates expansion of the upper cervical spine by a mass lesion emanating from within the spinal cord at the cervicomedullary junction. Irregular peripheral enhancement occurs within the mass (arrows).
Spinal Epidural Abscess Spinal epidural abscess presents with midline back or neck pain, fever, and progressive limb weakness. Prompt recognition of this distinctive process may prevent permanent sequelae. Aching pain is almost always present, either over the spine or in a radicular pattern. The duration of pain prior to presentation is generally ≤2 weeks but may on occasion be several months or longer. Fever is typically but not invariably present, accompanied by elevated white blood cell count, sedimentation rate, and C-reactive protein. As the abscess expands, further spinal cord damage results from venous congestion and thrombosis. Once weakness and other signs of myelopathy appear, progression may be rapid and irreversible. A more chronic sterile granulomatous form of abscess is also known, usually after treatment of an acute epidural infection.
Risk factors include an impaired immune status (HIV, diabetes mellitus, renal failure, alcoholism, malignancy), intravenous drug abuse, and infections of the skin or other tissues. Two-thirds of epidural infections result from hematogenous spread of bacteria from the skin (furunculosis), soft tissue (pharyngeal or dental abscesses; sinusitis), or deep viscera (bacterial endocarditis). The remainder arises from direct extension of a local infection to the subdural space; examples of local predisposing conditions are vertebral osteomyelitis, decubitus ulcers, lumbar puncture, epidural anesthesia, or spinal surgery. Most cases are due to Staphylococcus aureus; gram-negative bacilli, Streptococcus, anaerobes, and fungi can also cause epidural abscesses. Tuberculosis from an adjacent vertebral source (Pott’s disease) remains an important cause in the developing world.
MRI (Fig. 456-5) localizes the abscess and excludes other causes of myelopathy. Blood cultures are positive in more than half of cases, but direct aspiration of the abscess at surgery is often required for a microbiologic diagnosis. Lumbar puncture is only required if encephalopathy or other clinical signs raise the question of associated meningitis, a feature that is found in <25% of cases. The level of the puncture should be planned to minimize the risk of meningitis due to passage of the needle through infected tissue. A high cervical tap is sometimes the safest approach. Cerebrospinal fluid (CSF) abnormalities in epidural and subdural abscess consist of pleocytosis with a preponderance of polymorphonuclear cells, an elevated protein level, and a reduced glucose level, but the responsible organism is not cultured unless there is associated meningitis.
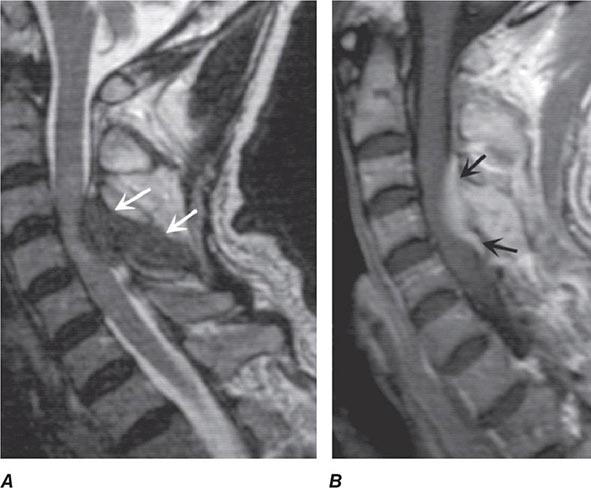
FIGURE 456-5 Magnetic resonance (MR) imaging of a spinal epidural abscess due to tuberculosis. A. Sagittal T2-weighted free spin-echo MR sequence. A hypointense mass replaces the posterior elements of C3 and extends epidurally to compress the spinal cord (arrows). B. Sagittal T1-weighted image after contrast administration reveals a diffuse enhancement of the epidural process (arrows) with extension into the epidural space.
Spinal Epidural Hematoma Hemorrhage into the epidural (or subdural) space causes acute focal or radicular pain followed by variable signs of a spinal cord or conus medullaris disorder. Therapeutic anticoagulation, trauma, tumor, or blood dyscrasias are predisposing conditions. Rare cases complicate lumbar puncture or epidural anesthesia. MRI and computed tomography (CT) confirm the clinical suspicion and can delineate the extent of the bleeding. Treatment consists of prompt reversal of any underlying clotting disorder and surgical decompression. Surgery may be followed by substantial recovery, especially in patients with some preservation of motor function preoperatively. Because of the risk of hemorrhage, lumbar puncture should be avoided whenever possible in patients with severe thrombocytopenia or other coagulopathies.
Hematomyelia Hemorrhage into the substance of the spinal cord is a rare result of trauma, intraparenchymal vascular malformation (see below), vasculitis due to polyarteritis nodosa or systemic lupus erythematosus (SLE), bleeding disorders, or a spinal cord neoplasm. Hematomyelia presents as an acute painful transverse myelopathy. With large lesions, extension into the subarachnoid space results in subarachnoid hemorrhage (Chap. 330). Diagnosis is by MRI or CT. Therapy is supportive, and surgical intervention is generally not useful. An exception is hematomyelia due to an underlying vascular malformation, for which spinal angiography and endovascular occlusion may be indicated, or surgery to evacuate the clot and remove the underlying vascular lesion.
NONCOMPRESSIVE MYELOPATHIES
The most frequent causes of noncompressive acute transverse myelopathy are spinal cord infarction; systemic inflammatory disorders, including SLE and sarcoidosis; demyelinating diseases, including multiple sclerosis (MS); neuromyelitis optica (NMO); postinfectious or idiopathic transverse myelitis, which is presumed to be an immune condition related to acute disseminated encephalomyelitis (Chap. 458); and infectious (primarily viral) causes. After spinal cord compression is excluded, the evaluation generally requires a lumbar puncture and a search for underlying systemic disease (Table 456-3).
EVALUATION OF ACUTE TRANSVERSE MYELOPATHY |
Abbreviations: ANA, antinuclear antibodies; CMV, cytomegalovirus; CSF, cerebrospinal fluid; CT, computed tomography; EBV, Epstein-Barr virus; ENA, epithelial neutrophil-activating peptide; ESR, erythrocyte sedimentation rate; HHV, human herpes virus; HSV, herpes simplex virus; HTLV, human T cell leukemia/lymphoma virus; MRI, magnetic resonance imaging; O&P, ova and parasites; p-ANCA, perinuclear antineutrophilic cytoplasmic antibodies; PCR, polymerase chain reaction; RPR, rapid plasma reagin (test); VDRL, Venereal Disease Research Laboratory; VZV, varicella-zoster virus.
Spinal Cord Infarction The cord is supplied by three arteries that course vertically over its surface: a single anterior spinal artery and paired posterior spinal arteries. The anterior spinal artery originates in paired branches of the vertebral arteries at the cranciocervical junction and is fed by additional radicular vessels that arise at C6, at an upper thoracic level, and, most consistently, at T11-L2 (artery of Adamkiewicz). At each spinal cord segment, paired penetrating vessels branch from the anterior spinal artery to supply the anterior two-thirds of the cord; the posterior spinal arteries, which often become less distinct below the midthoracic level, supply the posterior columns.
Spinal cord ischemia can occur at any level; however, the presence of the artery of Adamkiewicz below, and the anterior spinal artery circulation above, creates a region of marginal blood flow in the upper thoracic segments. With hypotension or cross-clamping of the aorta, cord infarction typically occurs at the level of T3-T4, and also at boundary zones between the anterior and posterior spinal artery territories. The latter may result in a rapidly progressive syndrome over hours of weakness and spasticity with little sensory change.
Acute infarction in the territory of the anterior spinal artery produces paraplegia or quadriplegia, dissociated sensory loss affecting pain and temperature sense but sparing vibration and position sense, and loss of sphincter control (“anterior cord syndrome”). Onset may be sudden but more typically is progressive over minutes or a few hours, quite unlike stroke in the cerebral hemispheres. Sharp midline or radiating back pain localized to the area of ischemia is frequent. Areflexia due to spinal shock is often present initially; with time, hyperreflexia and spasticity appear. Less common is infarction in the territory of the posterior spinal arteries, resulting in loss of posterior column function either on one side or bilaterally.
Causes of spinal cord infarction include aortic atherosclerosis, dissecting aortic aneurysm, vertebral artery occlusion or dissection in the neck, aortic surgery, or profound hypotension from any cause. A “surfer’s myelopathy” in the cervical region is probably vascular in origin. Cardiogenic emboli, vasculitis (Chap. 385), and collagen vascular disease (particularly SLE [Chap. 378], Sjögren’s syndrome [Chap. 383], and the antiphospholipid antibody syndrome [Chap. 379]) are other etiologies. Occasional cases develop from embolism of nucleus pulposus material into spinal vessels, usually from local spine trauma. In a substantial number of cases, no cause can be found, and thromboembolism in arterial feeders is suspected. MRI may fail to demonstrate infarctions of the cord, especially in the first day, but often the imaging becomes abnormal at the affected level.
In cord infarction due to presumed thromboembolism, acute anticoagulation is not indicated, with the possible exception of the unusual transient ischemic attack or incomplete infarction with a stuttering or progressive course. The antiphospholipid antibody syndrome is treated with anticoagulation (Chap. 379). Lumbar drainage of spinal fluid has reportedly been successful in some cases of cord infarction and has been used prophylactically during aortic surgery, but it has not been studied systematically.
Inflammatory and Immune Myelopathies (Myelitis) This broad category includes the demyelinating conditions MS, NMO, and postinfectious myelitis, as well as sarcoidosis and systemic autoimmune disease. In approximately one-quarter of cases of myelitis, no underlying cause can be identified. Some will later manifest additional symptoms of an immune-mediated disease. Recurrent episodes of myelitis are usually due to one of the immune-mediated diseases or to infection with herpes simplex virus (HSV) type 2 (below).
MULTIPLE SCLEROSIS MS may present with acute myelitis, particularly in individuals of Asian or African ancestry. In Caucasians, MS attacks rarely cause a transverse myelopathy (i.e., attacks of bilateral sensory disturbances, unilateral or bilateral weakness, and bladder or bowel symptoms), but it is among the most common causes of a partial cord syndrome. MRI findings in MS-associated myelitis typically consist of mild swelling of the cord and diffuse or multifocal “shoddy” areas of abnormal signal on T2-weighted sequences. Contrast enhancement, indicating disruption in the blood-brain barrier associated with inflammation, is present in many acute cases. A brain MRI is most helpful in gauging the likelihood that a case of myelitis represents an initial attack of MS. A normal scan indicates that the risk of evolution to MS is low, ~10–15% over 5 years; in contrast, the finding of multiple periventricular T2-bright lesions indicates a much higher risk, >50% over 5 years and >90% by 14 years. The CSF may be normal, but more often there is a mild mononuclear cell pleocytosis, with normal or mildly elevated CSF protein levels; the presence of oligoclonal bands is variable, but when they are found, a diagnosis of MS is more likely.
There are no adequate trials of therapy for MS-associated transverse myelitis. Intravenous methylprednisolone (500 mg qd for 3 days) followed by oral prednisone (1 mg/kg per day for several weeks, then gradual taper) has been used as initial treatment. A course of plasma exchange may be indicated for severe cases if glucocorticoids are ineffective. MS is discussed in Chap. 458.
NEUROMYELITIS OPTICA NMO is an immune-mediated demyelinating disorder consisting of a severe myelopathy that is typically longitudinally extensive, meaning that the lesion spans three or more vertebral segments. NMO is associated with optic neuritis that is often bilateral and may precede or follow myelitis by weeks or months, and also by brainstem and, in some cases, hypothalamic involvement. Recurrent myelitis without optic nerve involvemement can also occur in NMO; affected individuals are usually female and often of Asian ancestry. CSF studies reveal a variable mononuclear pleocytosis of up to several hundred cells per microliter; unlike MS, oligoclonal bands are generally absent. Diagnostic serum autoantibodies against the water channel protein aquaporin-4 are present in 60–70% of patients with NMO. NMO has also been associated with SLE and antiphospholipid antibodies (see below) as well as with other systemic autoimmune diseases; rare cases are paraneoplastic in origin. Treatment is with glucocorticoids and, for refractory cases, plasma exchange (as for MS, above). Preliminary studies suggest that treatment with azathioprine, mycophenylate, or anti-CD20 (anti–B cell) monoclonal antibody may protect against subsequent relapses; treatment for 5 years or longer is generally recommended. NMO is discussed in Chap. 458.
SYSTEMIC IMMUNE-MEDIATED DISORDERS Myelitis occurs in a small number of patients with SLE, many cases of which are associated with antibodies to antiphospholipids and/or to aquaporin-4. Patients with aquaporin-4 antibodies are likely to have longitudinally extensive myelitis by MRI, are considered to have an NMO-spectrum disorder, and are at high risk of developing future episodes of myelitis and/or optic neuritis. The CSF in SLE myelitis is usually normal or shows a mild lymphocytic pleocytosis; oligoclonal bands are a variable finding. Although there are no systematic trials of therapy for SLE myelitis, based on limited data, high-dose glucocorticoids followed by cyclophosphamide have been recommended. Acute severe episodes of transverse myelitis that do not initially respond to glucocorticoids are often treated with a course of plasma exchange. Sjögren’s syndrome (Chap. 383) can also be associated with NMO spectrum disorder and also with cases of acute transverse or chronic progressive myelopathy. Other immune-mediated myelitides include antiphospholipid antibody syndrome (Chap. 379), mixed connective tissue disease (Chap. 382), Behçet’s syndrome (Chap. 387), and vasculitis related to polyarteritis nodosa, perinuclear antineutrophilic cytoplasmic (p-ANCA) antibodies, or primary central nervous system vasculitis (Chap. 385).
Another important consideration in this group is sarcoid myelopathy that may present as a slowly progressive or relapsing disorder. MRI reveals an edematous swelling of the spinal cord that may mimic tumor; there is almost always gadolinium enhancement of active lesions and in some cases nodular enhancement of the adjacent surface of the cord; lesions may be single or multiple, and on axial images, enhancement of the central cord is usually present. The typical CSF profile consists of a mild lymphocytic pleocytosis and mildly elevated protein level; in a minority of cases, reduced glucose and oligoclonal bands are found. The diagnosis is particularly difficult when systemic manifestations of sarcoid are minor or absent (nearly 50% of cases) or when other typical neurologic manifestations of the disease—such as cranial neuropathy, hypothalamic involvement, or meningeal enhancement visualized by MRI—are lacking. A slit-lamp examination of the eye to search for uveitis, chest x-ray and CT to assess pulmonary involvement and mediastinal lymphadenopathy, serum or CSF angiotensin-converting enzyme (ACE; CSF values elevated in only a minority of cases), serum calcium, and a gallium scan may assist in the diagnosis. The usefulness of spinal fluid ACE is uncertain. Initial treatment is with oral glucocorticoids; immunosuppressant drugs, including the tumor necrosis factor α inhibitor infliximab, have been used for resistant cases. Sarcoidosis is discussed in Chap. 390.
POSTINFECTIOUS MYELITIS Many cases of myelitis, termed postinfectious or postvaccinal, follow an infection or vaccination. Numerous organisms have been implicated, including Epstein-Barr virus (EBV), cytomegalovirus (CMV), mycoplasma, influenza, measles, varicella, rubeola, and mumps. As in the related disorder acute disseminated encephalomyelitis (Chap. 458), postinfectious myelitis often begins as the patient appears to be recovering from an acute febrile infection, or in the subsequent days or weeks, but an infectious agent cannot be isolated from the nervous system or CSF. The presumption is that the myelitis represents an autoimmune disorder triggered by infection and is not due to direct infection of the spinal cord. No randomized controlled trials of therapy exist; treatment is usually with glucocorticoids or, in fulminant cases, plasma exchange.
ACUTE INFECTIOUS MYELITIS Many viruses have been associated with an acute myelitis that is infectious in nature rather than postinfectious. Nonetheless, the two processes are often difficult to distinguish. Herpes zoster is the best characterized viral myelitis, but HSV types 1 and 2, EBV, CMV, and rabies virus are other well-described causes. HSV-2 (and less commonly HSV-1) produces a distinctive syndrome of recurrent sacral cauda equina neuritis in association with outbreaks of genital herpes (Elsberg’s syndrome). Poliomyelitis is the prototypic viral myelitis, but it is more or less restricted to the anterior gray matter of the cord containing the spinal motoneurons. A polio-like syndrome can also be caused by a large number of enteroviruses (including enterovirus 71 and coxsackie), and with West Nile virus and other flaviviruses. Recently, cases of paralysis in children and adolescents were associated with enterovirus D-68 infection but a causal role for this virus has not been established. Chronic viral myelitic infections, such as those due to HIV or human T cell lymphotropic virus type 1 (HTLV-1), are discussed below.
Bacterial and mycobacterial myelitis (most are essentially abscesses) are less common than viral causes and much less frequent than cerebral bacterial abscess. Almost any pathogenic species may be responsible, including Borrelia burgdorferi (Lyme disease), Listeria monocytogenes, Mycobacterium tuberculosis, and Treponema pallidum (syphilis). Mycoplasma pneumoniae may be a cause of myelitis, but its status is uncertain because many cases are more properly classified as postinfectious.
Schistosomiasis (Chap. 259) is an important cause of parasitic myelitis in endemic areas. The process is intensely inflammatory and granulomatous, caused by a local response to tissue-digesting enzymes from the ova of the parasite, typically Schistosoma mansoni. Toxoplasmosis (Chap. 253) can occasionally cause a focal myelopathy, and this diagnosis should especially be considered in patients with AIDS (Chap. 226).
In cases of suspected viral myelitis, it may be appropriate to begin specific therapy pending laboratory confirmation. Herpes zoster, HSV, and EBV myelitis are treated with intravenous acyclovir (10 mg/kg q8h) or oral valacyclovir (2 g tid) for 10–14 days; CMV is treated with ganciclovir (5 mg/kg IV bid) plus foscarnet (60 mg/kg IV tid) or cidofovir (5 mg/kg per week for 2 weeks).
High-Voltage Electrical Injury Spinal cord injuries are prominent following electrocution from lightning strikes or other accidental electrical exposures. The syndrome consists of transient weakness acutely (often with an altered sensorium and focal cerebral disturbances), followed several days or even weeks later by a myelopathy that can be severe and permanent. This is a rare injury type, and limited data incriminate a vascular pathology involving the anterior spinal artery and its branches in some cases. Therapy is supportive.
CHRONIC MYELOPATHIES
SPONDYLOTIC MYELOPATHY
Spondylotic myelopathy is one of the most common causes of chronic cord compression and of gait difficulty in the elderly. Neck and shoulder pain with stiffness are early symptoms; impingement of bone and soft tissue overgrowth on nerve roots results in radicular arm pain, most often in a C5 or C6 distribution. Compression of the cervical cord, which occurs in fewer than one-third of cases, produces a slowly progressive spastic paraparesis, at times asymmetric and often accompanied by paresthesias in the feet and hands. Vibratory sense is diminished in the legs, there is a Romberg sign, and occasionally there is a sensory level for vibration or pinprick on the upper thorax. In some cases, coughing or straining produces leg weakness or radiating arm or shoulder pain. Dermatomal sensory loss in the arms, atrophy of intrinsic hand muscles, increased deep-tendon reflexes in the legs, and extensor plantar responses are common. Urinary urgency or incontinence occurs in advanced cases, but there are many alternative causes of these problems in older individuals. A tendon reflex in the arms is often diminished at some level; most often at the biceps (C5-C6). In individual cases, radicular, myelopathic, or combined signs may predominate. The diagnosis should be considered in appropriate cases of progressive cervical myelopathy, paresthesias of the feet and hands, or wasting of the hands.
Diagnosis is usually made by MRI and may be suspected from CT images; plain x-rays are less helpful. Extrinsic cord compression and deformation are appreciated on axial MRI views, and T2-weighted sequences may reveal areas of high signal intensity within the cord adjacent to the site of compression. A cervical collar may be helpful in milder cases, but definitive therapy consists of surgical decompression. Posterior laminectomy or an anterior approach with resection of the protruded disk and bony material may be required. Cervical spondylosis and related degenerative diseases of the spine are discussed in Chap. 22.
VASCULAR MALFORMATIONS OF THE CORD AND DURA
Vascular malformations of the cord and overlying dura are treatable causes of progressive myelopathy. Most common are fistulas located within the dura or posteriorly along the surface of the cord. Most dural arteriovenous (AV) fistulas are located at or below the midthoracic level, usually consisting of a direct connection between a radicular feeding artery in the nerve root sleeve with dural veins. The typical presentation is a middle-aged man with a progressive myelopathy that worsens slowly or intermittently and may have periods of remission, sometimes mimicking MS. Acute deterioration due to hemorrhage into the spinal cord (hematomyelia) or subarachnoid space may also occur but is rare. A saltatory progression is most common and appears to be the result of local ischemia and edema from venous congestion. Most patients have incomplete sensory, motor, and bladder disturbances. The motor disorder may predominate and produce a mixture of upper and restricted lower motor neuron signs, simulating amyotrophic lateral sclerosis (ALS). Pain over the dorsal spine, dysesthesias, or radicular pain may be present. Other symptoms suggestive of AV malformation (AVM) or dural fistula include intermittent claudication; symptoms that change with posture, exertion, Valsalva maneuver, or menses; and fever.
Less commonly, AVM disorders are intramedullary rather than dural. One unusual disorder is a progressive thoracic myelopathy with paraparesis developing over weeks or months, characterized pathologically by abnormally thick, hyalinized vessels within the cord (subacute necrotic myelopathy, or Foix-Alajouanine syndrome).
Spinal bruits are infrequent but may be sought at rest and after exercise in suspected cases. A vascular nevus on the overlying skin may indicate an underlying vascular malformation as occurs with Klippel-Trenaunay-Weber syndrome. High-resolution MRI with contrast administration detects the draining vessels of many but not all AVMs (Fig. 456-6). An uncertain proportion may be visualized by CT myelography as enlarged vessels along the surface of the cord. Definitive diagnosis requires selective spinal angiography, which defines the feeding vessels and the extent of the malformation. Endovascular embolization of the major feeding vessels may stabilize a progressive neurologic deficit or allow for gradual recovery. Some lesions, especially small dural fistulas, can be resected surgically.
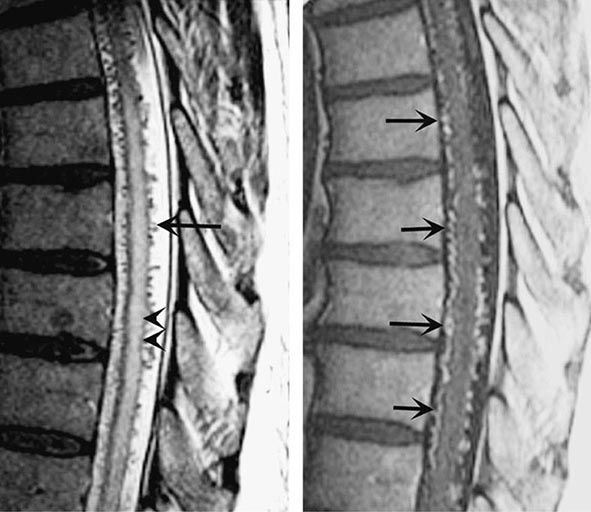
FIGURE 456-6 Arteriovenous malformation. Sagittal magnetic resonance scans of the thoracic spinal cord: T2 fast spin-echo technique (left) and T1 postcontrast image (right). On the T2-weighted image (left), abnormally high signal intensity is noted in the central aspect of the spinal cord (arrowheads). Numerous punctate flow voids indent the dorsal and ventral spinal cord (arrow). These represent the abnormally dilated venous plexus supplied by a dural arteriovenous fistula. After contrast administration (right), multiple, serpentine, enhancing veins (arrows) on the ventral and dorsal aspect of the thoracic spinal cord are visualized, diagnostic of arteriovenous malformation. This patient was a 54-year-old man with a 4-year history of progressive paraparesis.
RETROVIRUS-ASSOCIATED MYELOPATHIES
The myelopathy associated with HTLV-1, formerly called tropical spastic paraparesis, is a slowly progressive spastic syndrome with variable sensory and bladder disturbance. Approximately half of patients have mild back or leg pain. The neurologic signs may be asymmetric, often lacking a well-defined sensory level; the only sign in the arms may be hyperreflexia after several years of illness. The onset is insidious, and the illness is slowly progressive at a variable rate; most patients are unable to walk within 10 years of onset. This presentation may resemble primary progressive MS or a thoracic AVM. Diagnosis is made by demonstration of HTLV-1-specific antibody in serum by enzyme-linked immunosorbent assay (ELISA), confirmed by radioimmunoprecipitation or Western blot analysis. Especially in endemic areas, a finding of HTLV-1 seropositivity in a patient with myelopathy does not necessarily prove that HTLV-1 is causative. The CSF/serum antibody index may provide support by establishing intrathecal synthesis of antibodies favoring HTVL-1 myelopathy over asymptomatic carriage. Measuring proviral DNA by polymerase chain reaction (PCR) in serum and CSF cells can be useful as an ancillary part of diagnosis, because proviral DNA levels may be higher in patients with myelopathy. The myelopathy appears to result from an immune-mediated attack on the spinal cord rather than the result of direct viral infection. There is no effective treatment, but symptomatic therapy for spasticity and bladder symptoms may be helpful.
A progressive myelopathy may also result from HIV infection (Chap. 226). It is characterized by vacuolar degeneration of the posterior and lateral tracts, resembling subacute combined degeneration (see below).
SYRINGOMYELIA
Syringomyelia is a developmental cavity of the cervical cord that may enlarge and produce progressive myelopathy or may remain asymptomatic. Symptoms begin insidiously in adolescence or early adulthood, progress irregularly, and may undergo spontaneous arrest for several years. Many young patients acquire a cervical-thoracic scoliosis. More than half of all cases are associated with Chiari type 1 malformations in which the cerebellar tonsils protrude through the foramen magnum and into the cervical spinal canal. The pathophysiology of syrinx expansion is controversial, but some interference with the normal flow of CSF seems likely, perhaps by the Chiari malformation. Acquired cavitations of the cord in areas of necrosis are also termed syrinx cavities; these follow trauma, myelitis, necrotic spinal cord tumors, and chronic arachnoiditis due to tuberculosis and other etiologies.
The presentation is a central cord syndrome consisting of a regional dissociated sensory loss (loss of pain and temperature sensation with sparing of touch and vibration) and areflexic weakness in the upper limbs. The sensory deficit has a distribution that is “suspended” over the nape of the neck, shoulders, and upper arms (cape distribution) or in the hands. Most cases begin asymmetrically with unilateral sensory loss in the hands that leads to injuries and burns that are not appreciated by the patient. Muscle wasting in the lower neck, shoulders, arms, and hands with asymmetric or absent reflexes in the arms reflects expansion of the cavity in the gray matter of the cord. As the cavity enlarges and compresses the long tracts, spasticity and weakness of the legs, bladder and bowel dysfunction, and a Horner’s syndrome appear. Some patients develop facial numbness and sensory loss from damage to the descending tract of the trigeminal nerve (C2 level or above). In cases with Chiari malformations, cough-induced headache and neck, arm, or facial pain may be reported. Extension of the syrinx into the medulla, syringobulbia, causes palatal or vocal cord paralysis, dysarthria, horizontal or vertical nystagmus, episodic dizziness or vertigo, and tongue weakness with atrophy.
MRI accurately identifies developmental and acquired syrinx cavities and their associated spinal cord enlargement (Fig. 456-7). Images of the brain and the entire spinal cord should be obtained to delineate the full longitudinal extent of the syrinx, assess posterior fossa structures for the Chiari malformation, and determine whether hydrocephalus is present.
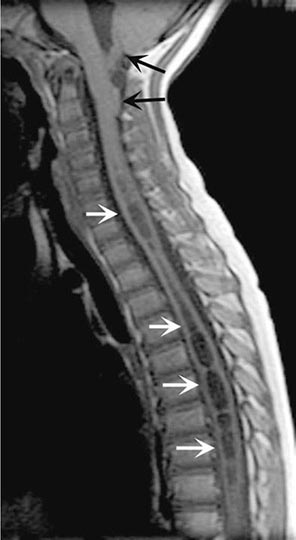
FIGURE 456-7 Magnetic resonance imaging of syringomyelia associated with a Chiari malformation. Sagittal T1-weighted image through the cervical and upper thoracic spine demonstrates descent of the cerebellar tonsils below the level of the foramen magnum (black arrows). Within the substance of the cervical and thoracic spinal cord, a cerebrospinal fluid collection dilates the central canal (white arrows).
CHRONIC MYELOPATHY OF MULTIPLE SCLEROSIS
A chronic progressive myelopathy is the most frequent cause of disability in both primary progressive and secondary progressive forms of MS. Involvement is typically bilateral but asymmetric and produces motor, sensory, and bladder/bowel disturbances. Fixed motor disability appears to result from extensive loss of axons in the corticospinal tracts. Diagnosis is facilitated by identification of earlier attacks such as optic neuritis. MRI, CSF, and evoked response testing are confirmatory. Disease-modifying therapy is indicated for patients with progressive myelopathy who also have coexisting MS relapses. Therapy is sometimes offered to patients who have a progressive course without relapses but with “active” MRI scans (e.g., the presence of new focal demyelinating lesions) despite the lack of evidence supporting the value of treatment in this setting. MS is discussed in Chap. 458.
SUBACUTE COMBINED DEGENERATION (VITAMIN B12 DEFICIENCY)
This treatable myelopathy presents with subacute paresthesias in the hands and feet, loss of vibration and position sensation, and a progressive spastic and ataxic weakness. Loss of reflexes due to an associated peripheral neuropathy in a patient who also has Babinski signs is an important diagnostic clue. Optic atrophy and irritability or other cognitive changes may be prominent in advanced cases and are occasionally the presenting symptoms. The myelopathy of subacute combined degeneration tends to be diffuse rather than focal; signs are generally symmetric and reflect predominant involvement of the posterior and lateral tracts, including Romberg’s sign. The diagnosis is confirmed by the finding of macrocytic red blood cells, a low serum B12 concentration, elevated serum levels of homocysteine and methylmalonic acid, and in uncertain cases, testing for anti–parietal cell antibodies and a Schilling test. Treatment is by replacement therapy, beginning with 1000 μg of intramuscular vitamin B12 repeated at regular intervals or by subsequent oral treatment (Chap. 128).
HYPOCUPRIC MYELOPATHY
This myelopathy is similar to subacute combined degeneration (described above), except there is no neuropathy, and explains cases with normal serum levels of B12. Low levels of serum copper are found, and often there is also a low level of serum ceruloplasmin. Some cases follow gastrointestinal procedures, particularly bariatric surgery, that result in impaired copper absorption; others have been associated with excess zinc from health food supplements or, until recently, zinc-containing denture creams, all of which impair copper absorption via induction of metallothionein, a copper-binding protein. Many cases are idiopathic. Improvement or at least stabilization may be expected with reconstitution of copper stores by oral supplementation. There is microcytic or macrocytic anemia. The pathophysiology and pathology of the idiopathic form are not known.
TABES DORSALIS
The classic syphilitic syndromes of tabes dorsalis and meningovascular inflammation of the spinal cord are now less frequent than in the past but must be considered in the differential diagnosis of spinal cord disorders. The characteristic symptoms of tabes are fleeting and repetitive lancinating pains, primarily in the legs or less often in the back, thorax, abdomen, arms, and face. Ataxia of the legs and gait due to loss of position sense occurs in half of patients. Paresthesias, bladder disturbances, and acute abdominal pain with vomiting (visceral crisis) occur in 15–30% of patients. The cardinal signs of tabes are loss of reflexes in the legs; impaired position and vibratory sense; Romberg’s sign; and, in almost all cases, bilateral Argyll Robertson pupils, which fail to constrict to light but accommodate. Diabetic polyradiculopathy may simulate tabes.
FAMILIAL SPASTIC PARAPLEGIA
Many cases of slowly progressive myelopathy are genetic in origin (Chap. 452). More than 30 different causative loci have been identified, including autosomal dominant, autosomal recessive, and X-linked forms. Especially for the recessive and X-linked forms, a family history of myelopathy may be lacking. Most patients present with almost imperceptibly progressive spasticity and weakness in the legs, usually but not always symmetrical. Sensory symptoms and signs are absent or mild, but sphincter disturbances may be present. In some families, additional neurologic signs are prominent, including nystagmus, ataxia, or optic atrophy. The onset may be as early as the first year of life or as late as middle adulthood. Only symptomatic therapies are available.
ADRENOMYELONEUROPATHY
This X-linked disorder is a variant of adrenoleukodystrophy. Most affected males have a history of adrenal insufficiency and then develop a progressive spastic (or ataxic) paraparesis beginning in early or sometimes middle adulthood; some patients also have a mild peripheral neuropathy. Female heterozygotes may develop a slower, insidiously progressive spastic myelopathy beginning later in adulthood and without adrenal insufficiency. Diagnosis is usually made by demonstration of elevated levels of very-long-chain fatty acids in plasma and in cultured fibroblasts. The responsible gene encodes the adrenoleukodystrophy protein (ADLP), a peroxisomal membrane transporter involved in carrying long-chain fatty acids to peroxisomes for degradation. Corticosteroid replacement is indicated if hypoadrenalism is present, and bone marrow transplantation and nutritional supplements have been attempted for this condition without clear evidence of efficacy.
OTHER CHRONIC MYELOPATHIES
Primary lateral sclerosis (Chap. 452) is a degenerative disorder characterized by progressive spasticity with weakness, eventually accompanied by dysarthria and dysphonia; bladder symptoms occur in approximately half of patients. Sensory function is spared. The disorder resembles ALS and is considered a variant of the motor neuron degenerations, but without the characteristic lower motor neuron disturbance. Some cases may represent familial spastic paraplegia, particularly autosomal recessive or X-linked varieties in which a family history may be absent.
Tethered cord syndrome is a developmental disorder of the lower spinal cord and nerve roots that rarely presents in adulthood as low back pain accompanied by a progressive lower spinal cord and/or nerve root syndrome. Some patients have a small leg or foot deformity indicating a long-standing process, and in others, a dimple, patch of hair, or sinus tract on the skin overlying the lower back is the clue to a congenital lesion. Diagnosis is made by MRI, which demonstrates a low-lying conus medullaris and thickened filum terminale. The MRI may also reveal diastematomyelia (division of the lower spinal cord into two halves), lipomas, cysts, or other congenital abnormalities of the lower spine coexisting with the tethered cord. Treatment is with surgical release.
There are a number of rare toxic causes of spastic myelopathy, including lathyrism due to ingestion of chickpeas containing the excitotoxin β-N-oxalylamino-L-alanine (BOAA), seen primarily in the developing world, and nitrous oxide inhalation producing a myelopathy identical to subacute combined degeneration. SLE, Sjögren’s syndrome, and sarcoidosis may each cause a myelopathy without overt evidence of systemic disease. Cancer-related causes of chronic myelopathy, besides the common neoplastic compressive myelopathy discussed earlier, include radiation injury (Chap. 118) and rare paraneoplastic myelopathies. The last of these are most often associated with lung or breast cancer and anti-Hu antibodies (Chap. 122) or with lymphoma that causes a syndrome of destruction of anterior horn cells; NMO (Chap. 458) can also rarely be paraneoplastic in origin. Metastases to the cord are probably more common than either of these in patients with cancer. Often, a cause of intrinsic myelopathy can be identified only through periodic reassessment.
REHABILITATION OF SPINAL CORD DISORDERS
The prospects for recovery from an acute destructive spinal cord lesion fade after ~6 months. There are currently no effective means to promote repair of injured spinal cord tissue; promising but entirely experimental approaches include the use of factors that influence reinnervation by axons of the corticospinal tract, nerve and neural sheath graft bridges, forms of electrical stimulation at the site of injury, and the local introduction of stem cells. The disability associated with irreversible spinal cord damage is determined primarily by the level of the lesion and by whether the disturbance in function is complete or incomplete (Table 456-4). Even a complete high cervical cord lesion may be compatible with a productive life. The primary goals are development of a rehabilitation plan framed by realistic expectations and attention to the neurologic, medical, and psychological complications that commonly arise.
EXPECTED NEUROLOGIC FUNCTION FOLLOWING COMPLETE CORD LESIONS |

Many of the usual symptoms associated with medical illnesses, especially somatic and visceral pain, may be lacking because of the destruction of afferent pain pathways. Unexplained fever, worsening of spasticity, or deterioration in neurologic function should prompt a search for infection, thrombophlebitis, or an intraabdominal pathology. The loss of normal thermoregulation and inability to maintain normal body temperature can produce recurrent fever (quadriplegic fever), although most episodes of fever are due to infection of the urinary tract, lung, skin, or bone.
Bladder dysfunction generally results from loss of supraspinal innervation of the detrusor muscle of the bladder wall and the sphincter musculature. Detrusor spasticity is treated with anticholinergic drugs (oxybutynin, 2.5–5 mg qid) or tricyclic antidepressants with anticholinergic properties (imipramine, 25–200 mg/d). Failure of the sphincter muscle to relax during bladder emptying (urinary dyssynergia) may be managed with the α-adrenergic blocking agent terazosin hydrochloride (1–2 mg tid or qid), with intermittent catheterization, or, if that is not feasible, by use of a condom catheter in men or a permanent indwelling catheter. Surgical options include the creation of an artificial bladder by isolating a segment of intestine that can be catheterized intermittently (enterocystoplasty) or can drain continuously to an external appliance (urinary conduit). Bladder areflexia due to acute spinal shock or conus lesions is best treated by catheterization. Bowel regimens and disimpaction are necessary in most patients to ensure at least biweekly evacuation and avoid colonic distention or obstruction.
Patients with acute cord injury are at risk for venous thrombosis and pulmonary embolism. Use of calf-compression devices and anticoagulation with low-molecular-weight heparin is recommended. In cases of persistent paralysis, anticoagulation should probably be continued for 3 months.
Prophylaxis against decubitus ulcers should involve frequent changes in position in a chair or bed, the use of special mattresses, and cushioning of areas where pressure sores often develop, such as the sacral prominence and heels. Early treatment of ulcers with careful cleansing, surgical or enzyme debridement of necrotic tissue, and appropriate dressing and drainage may prevent infection of adjacent soft tissue or bone.
Spasticity is aided by stretching exercises to maintain mobility of joints. Drug treatment is effective but may result in reduced function, as some patients depend on spasticity as an aid to stand, transfer, or walk. Baclofen (up to 240 mg/d in divided doses) is effective; it acts by facilitating γ-aminobutyric acid–mediated inhibition of motor reflex arcs. Diazepam acts by a similar mechanism and is useful for leg spasms that interrupt sleep (2–4 mg at bedtime). Tizanidine (2–8 mg tid), an α2 adrenergic agonist that increases presynaptic inhibition of motor neurons, is another option. For nonambulatory patients, the direct muscle inhibitor dantrolene (25–100 mg qid) may be used, but it is potentially hepatotoxic. In refractory cases, intrathecal baclofen administered via an implanted pump, botulinum toxin injections, or dorsal rhizotomy may be required to control spasticity.
Despite the loss of sensory function, many patients with spinal cord injury experience chronic pain sufficient to diminish their quality of life. Randomized controlled studies indicate that gabapentin or pregabalin is useful in this setting. Epidural electrical stimulation and intrathecal infusion of pain medications have been tried with some success. Management of chronic pain is discussed in Chap. 18.
A paroxysmal autonomic hyperreflexia may occur following lesions above the major splanchnic sympathetic outflow at T6. Headache, flushing, and diaphoresis above the level of the lesion, as well as hypertension with bradycardia or tachycardia, are the major symptoms. The trigger is typically a noxious stimulus—for example, bladder or bowel distention, a urinary tract infection, or a decubitus ulcer—below the level of the cord lesion. Treatment consists of removal of offending stimuli; ganglionic blocking agents (mecamylamine, 2.5–5 mg) or other short-acting antihypertensive drugs are useful in some patients.
Attention to these details allows longevity and a productive life for patients with complete transverse myelopathies.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


