Molecular spectroscopy and excited-state dynamics: Diatomics
In our treatment of atomic spectroscopy we have already introduced the fundamentals of all spectroscopy: spectra are made up of lines from bound–bound transitions and continuous regions from bound–free transitions. Lines have widths determined by lifetimes, collisions and any other relaxation or broadening mechanisms. Conservation of energy leads to the resonance condition in which the photon energy in either absorption or emission must exactly match the energy difference between the states involved in the transition. Conservation of angular momentum enforces selection rules on the states that can be connected by different types of transitions.
In atomic spectroscopy all transitions were between different electronic states. In molecules there are additional degrees of freedom: rotations and vibrations. There is also the possibility for photodissociation – the breaking of bonds caused by photon absorption. Molecules also exhibit characteristic symmetries. Molecular symmetry is important in determining new sets of selection rules. These are the details that we will investigate in more depth in this chapter. Here, we will concentrate on diatomic molecules. Brief reference is made to the spectroscopy of polyatomic molecules, which are discussed in more detail in Chapter 26.
25.1 Introduction to molecular spectroscopy
In the study of statistical mechanics we found that the energy of a molecule is distributed between translational energy ϵt and internal energy ϵi. There are three components to the internal energy: rotational energy ϵr, vibrational energy ϵv and electronic energy ϵe. To a first approximation we can consider these four contributions independent of each other; hence

Here, we neglect the nuclear degrees of freedom. We will add these as and when we need them, but we will neglect them as a first approximation. The independence of the four contributions allows us to factor the partition function Z for the molecule. We have already seen that the effect of the translation degree of freedom is observed in the linewidth of a transition through the Doppler effect. Otherwise, we can neglect translations as we consider the spectroscopy of internal degrees of freedom. In our introduction to quantum theory we also found that spin is another degree of freedom. Spin, which is not related to spatial coordinates, is quite important in spectroscopy. We will return to its effects later, but for now we concentrate on the influence of the spatial degrees of freedom on rotational, vibrational and electronic excitations.
The ability to factor the partition function extends to the wavefunctions. To a first approximation, the internal degrees of freedom do not interact and we can factor the wavefunction into separate rotational, vibrational and electronic contributions,

This means we can separate the discussion of rotational, vibrational and electronic spectroscopy, at least initially. The approximation that the electrons will always be able to find the lowest energy configuration as the nuclear coordinates change, for example as a result of vibration, is known as the Born–Oppenheimer approximation.1 We used it in the last chapter to construct potential energy curves for each electronic state. Its impact resonates throughout spectroscopy as well. Importantly, the Born–Oppenheimer approximation allows us to treat the nuclear coordinates R as a parameter that can be separated from the electronic coordinates r. The electrons are much lighter and faster than the nuclei. They respond almost instantaneously to changes in the nuclear coordinates.
Each electronic state has an associated potential energy curve, which if the state is bound usually has a shape similar to that shown in Fig. 25.1. Each bound electronic state contains several vibrational levels with energy spacings typically on the order of several tens of cm−1 up to 4000 cm−1 (in the infrared). On top of the vibrational levels (at an even finer energy scale) are superimposed the rotational levels, with energies in the microwave range. A dissociative state, also called a repulsive state, has no potential well and does not support bound vibrational levels such as those shown in Fig. 25.1. A repulsive potential decreases monotonically with increasing internuclear separation.

Figure 25.1 A potential energy curve representative of the ground electronic state of H2. Each electronic state has such a curve associated with it. The vibrational levels of a Morse oscillator designated by the vibrational quantum number v converge on the dissociation limit, which is positioned at the energetic origin in this diagram. The minimum potential occurs at Re. At an even finer level of energy resolution rotational states are associated with each vibrational level.
The nucleus has a spin. When the value of nuclear spin is nonzero, further energy level splittings arise. This also leads to the spectroscopy of nuclear magnetic resonance (NMR) in the radio wave region. The nucleus has a shell structure much like that of the electrons. Excited nuclear states are usually associated with gamma rays (high-energy excitations).
25.2 Pure rotational spectra of molecules
25.2.1 Diatomic molecules
For a rotational transition to occur by the interaction of light with a molecule, there must be an oscillating dipole moment in the molecule. This can only happen if the molecule has a permanent dipole. Only molecules with a permanent dipole moment exhibit pure rotational transitions. Therefore, homonuclear diatomic molecules do not have pure rotational spectra.
The pre-condition for a pure rotational transition is that the molecule must have a permanent dipole moment.
The first-order approximation for the rotational motion of a diatomic molecule is that of a rigid rotor: a massless rod of length R rotating about its center of mass, as shown in Fig. 25.2. The two atoms with masses m1 and m2 reside at either end of the rod. The rotational dynamics of this body are the same as the particle on a ring system that we solved in Chapter 21. Thus, the rotational energy of a diatomic molecule as modeled by a rigid rotor is given by

where

is the moment of inertia, μ is the reduced mass

J the rotational quantum number and B the rotational constant,


Figure 25.2 In the rigid rotor model of a diatomic molecule, two masses m1 and m2 are held together by a massless rod. The length of the rod is fixed such that the distance between the centers of mass of the two atoms is R. The rotational axis J is perpendicular to the internuclear axis.
The associated energy level structure is given in Fig. 25.3. When expressed in wavenumbers, the energy of a rotational level is known as the rotational term F(J)


Figure 25.3 Rotational levels of a rigid rotor are spaced in a pattern. Different molecules have characteristic values of the rotational constant B, which leads to decidedly different spacings between rotational levels. H2 with B = 60.9 cm−1 has very few levels, while the levels of I2 with B = 0.037 cm−1 look like a quasi-continuum at this resolution. Centrifugal distortion shifts the energy of the H2 states compared to the prediction of the rigid rotor model. The shift increases with increasing rotational quantum number J.
The magnitude of the rotational angular momentum is given by the expectation value of the rotational angular momentum operator 

To each rotational quantum number J, there are 2J + 1 degenerate rotational states that are designated by the MJ quantum number. J is a positive integer but MJ ranges in units steps from –J to +J. Here, we are dealing only with rotation. Because it is unambiguously associated just with J, we will refer to MJ as M to simplify the notation. These degenerate states arise from space quantization. The projection of the rotational angular momentum is determined by the quantum number M,

The degeneracy of these levels can be lifted by an electric field, which is known as the Stark effect. In other words, when a rotating molecule is placed in an electric field, the field direction defines the orientation of the z-axis in the laboratory frame. The 2J + 1 different projections along the z axis of the rotational angular momentum of the states with different values of M cause the states to have different energies.
Each rotational quantum state can be identified by the JM quantum numbers, and to determine the selection rules we have to evaluate the matrix element of the transition dipole moment operator (as with atomic spectroscopy, we are assuming all transitions to be electric dipole transition unless we specify otherwise)

Spectroscopic notation by convention denotes the upper state with a single prime and the lower state with a double prime. In other words, we have to evaluate the integral

where dΩ = sin θ dθ dφ. The wavefunction for rotation is a spherical harmonic YJM(θ, φ). It does not depend on the radial coordinate.  is the dipole moment vector. The R indicates that it is directed along the bond. To determine which transitions are allowed, we argue just as we did for atoms. The angular momentum of the photon is one, and angular momentum must be conserved during absorption or emission of the photon. Thus, rotational angular momentum must change by ΔJ = ±1 while ΔM = 0, ±1 is possible. Most molecules have a ground electronic state that does not possess an electronic angular momentum. In Table 25.1 it can be seen that an exception (the one with a Π state instead of a Σ state) is NO. Molecules such as NO that do not have a Σ ground state exhibit ΔJ = 0 pure rotational transitions.
is the dipole moment vector. The R indicates that it is directed along the bond. To determine which transitions are allowed, we argue just as we did for atoms. The angular momentum of the photon is one, and angular momentum must be conserved during absorption or emission of the photon. Thus, rotational angular momentum must change by ΔJ = ±1 while ΔM = 0, ±1 is possible. Most molecules have a ground electronic state that does not possess an electronic angular momentum. In Table 25.1 it can be seen that an exception (the one with a Π state instead of a Σ state) is NO. Molecules such as NO that do not have a Σ ground state exhibit ΔJ = 0 pure rotational transitions.
Table 25.1 Ground-state spectroscopic constants for selected diatomic molecules taken from the 96th edition of the CRC Handbook of Chemistry and Physics. All values are in units of cm−1 except the centrifugal distortion constant D, which is in units of 10−6 cm−1, and re in units of Å. The traditional notation from Herzberg and Huber is to denote the fundamental wavenumber ωe, the harmonic bond length re, and the centrifugal distortion constant De. The vibration–rotation interaction constant is αe and xe is the anharmonicity constant
| Molecule | Ground state |  / cm−1 / cm−1 | ωexe / cm−1 | Be / cm−1 | αe / cm−1 | D / 10−6 cm−1 | Re = re Å |
|---|---|---|---|---|---|---|---|
| 1H2 | 1Σ+g | 4401.21 | 121.34 | 60.853 | 3.062 | 47100 | 0.74144 |
| 2H2 | 1Σ+g | 3115.50 | 61.82 | 30.444 | 1.0786 | 11410 | 0.74152 |
| 1H19F | 1Σ+ | 4138.39 | 89.94 | 20.953712 | 0.7933704 | 2150 | 0.91685 |
| 1H35Cl | 1Σ+ | 2990.92 | 52.80 | 10.5933002 | 0.3069985 | 531.94 | 1.27456 |
| 1H127I | 1Σ+ | 2309.01 | 39.64 | 6.4263650 | 0.1689 | 206.9 | 1.60916 |
| 19F2 | 1Σ+g | 916.93 | 11.32 | 0.889294 | 0.0125952 | 3.3 | 1.41264 |
| 35Cl2 | 1Σ+g | 559.75 | 2.69 | 0.24415 | 0.00152 | 0.186 | 1.9872 |
| 79Br2 | 1Σ+g | 325.32 | 1.08 | 0.082107 | 0.0003187 | 0.02092 | 2.2811 |
| 127I2 | 1Σ+g | 214.50 | 0.61 | 0.03737 | 0.000114 | 0.0043 | 2.666 |
| 12C16O | 1Σ+ | 2169.81 | 13.29 | 1.931280985 | 0.01750439 | 6.1216 | 1.12832 |
| 14N2 | 1Σ+g | 2358.56 | 14.32 | 1.998236 | 0.017310 | 5.737 | 1.09769 |
| 14N16O | 2Π1/2 | 1904.20 | 14.07 | 1.67195 | 0.0171 | 0.5 | 1.15077 |
| 16O2 | 3Σ−g | 1580.19 | 11.98 | 1.445622 | 0.015933 | 4.839 | 1.20752 |
| 23Na2 | 1Σ+g | 159.09 | 0.71 | 0.15473537 | 0.0086375 | 0.58 | 3.07858 |
| 138Ba127I | 2Σ+ | 152.14 | 0.27 | 0.02680587 | 0.00006634 | 0.00333 | 3.08476 |
The energy of a transition between rotational levels differing by one unit of J is given by

The wavenumber of the transition between these two states is given by the difference in the rotational terms

Therefore, the pure rotational spectrum is composed of equally spaced lines with a spacing of 2B between them.
The above equation is strictly true for a rigid rotor – that is, one in which the bond length R does not change. If a diatomic molecule were a simple harmonic oscillator, each vibrational level would have exactly the same bond length. A diatomic molecule is not a perfect harmonic oscillator, and anharmonicity leads to a lengthening of bond length with increasing vibrational quantum number v. Therefore, each vibrational level has its own value of rotational constant, which is denoted Bv because each vibrational level has its own value of equilibrium bond length Rv. Rotational excitation also stretches the bond. This lengthening of the bond as the rotational quantum number increases is known as centrifugal distortion. Accounting for the vibrational state dependence of the rotational constant and centrifugal distortion leads to the addition of a term that is second order in J(J + 1) multiplied by the centrifugal distortion constant D,

The effects of centrifugal distortion are shown in Fig. 25.4 for three molecules – H2, CO and I2 – with much different rotational constants. Some molecules have been characterized well enough that a vibrational-state-specific value Dv = De + corrections is used in place of a constant value, but we will not use De in this context so as to avoid confusion with the dissociation energy measured from the bottom of the well. The value of D can be approximated from the harmonic oscillator in terms of the ground vibrational state values of rotational constant and fundamental wavenumber as


Figure 25.4 Centrifugal distortion leads to increasingly large deviations from the predictions of the rigid rotor energy levels as shown here for H2, CO and I2. (a) The deviation is plotted versus the rotational quantum number. (b) The deviation is plotted against rotational energy. It is clear that H2 is much less rigid even when compared at equal levels of rotational energy.
A third-order correction to the rotational term that is multiplied by a constant Hv is known for some molecules. The vibration–rotation interaction constant αe is used to calculate the value of vibrational-level-specific value Bv from

The constant Be is the rotational constant of a hypothetical harmonic state that exists at the minimum of the potential energy curve. However, as we have seen, the Heisenberg uncertainty principle leads to the presence of a zeropoint energy. Thus, B0 is ever so slightly shifted from Be.
Selected values of the spectroscopic constants of diatomics are given in Table 25.1. Note that because of the reduced mass dependence in Eq. (25.6), the rotational constant is extremely sensitive to the mass combination. Therefore, pure rotational spectroscopy is quite sensitive to isotopic composition. Pure rotational spectra form uniquely characteristic and easily interpretable fingerprints of molecules. These spectra are the primary feature by which molecules are identified in astrochemical studies.2
Inspection of the values in Table 25.1 reveals some expected trends. The behavior of H2 is exceptional because of its low mass and small size. There is a change in the fourth decimal place in the bond length of the H2 molecule upon isotopic substitution of 2H for 1H, but there is a sizeable shift in all other constants. Hydrides have very high rotational constants. Heavy–heavy diatomics have very low rotational constants. Bond lengths increase with increasing atomic number, as shown dramatically by the hydrogen halides and halogens. As reflected in the ratios ωexe/ωe and D/Be, hydrides are also considerably more anharmonic and strongly affected by centrifugal distortion than are the other diatomics.
The intensity of rotational absorption peaks varies with the population in the initial rotational state of the transition. If the sample is described by a temperature T, then the population follows the Boltzmann distribution. The relative number density of molecules in state J compared to the number density in the ground state is

Nascent molecules produced in a chemical reaction might not be fully equilibrated. This means that the rotational state distribution might not be Boltzmann, or that even though the population follows a Boltzmann distribution the corresponding temperature need not equal the temperature of the container, or be the same as the vibrational or translational temperature. The degeneracy term (2J + 1) increases with J but the exponential term decreases with J since the rotation energy is increasing. This means that the population, and therefore line intensities, go through a maximum at a value of Jmax that is related to the temperature and rotational constant by

25.2.1.1 Directed practice
Why does centrifugal distortion lead to closer spacing of rotational states?
25.2.2 Polyatomic molecules
Linear molecules can be treated just like diatomic molecules because they also only have one rotational axis. Symmetric linear polyatomics such as O=C=O do not exhibit a pure rotational spectrum because they do not possess a permanent dipole moment. Asymmetric polyatomics such as O=C=S, which have a permanent dipole moment, exhibit a pure rotational spectrum. Because of centrifugal distortion, tetrahedral molecules such as CH4 and SiH4 exhibit similar pure rotational spectra.
Nonlinear molecules can rotate about three principal axes of inertia, some of which may or may not be equal. There are three types of polyatomic rotors, as defined in Table 25.2.
Table 25.2 Classification of rotors based on the magnitudes of their moments of inertia IA, Ib, and Ic.
| Type of rotor/molecule | Moments of inertia |
|---|---|
| Spherical top | Ia = Ib = Ic |
| Symmetric top | Ia = Ib ≠ Ic or Ia ≠ Ib = Ic |
| Asymmetric top | Ia ≠ Ib ≠ Ic |
The rotational structure of polyatomic molecules is complex. Whereas the spherical top requires two quantum numbers to describe it (J and M, in analogy to the diatomic case), other tops require three quantum numbers to describe them, namely J, M and K. The last is associated with the projection of J onto the symmetry axis of the molecule. To a first approximation we can think of the rotational spectrum of a polyatomic as the sum of J stacks and K stacks, independent ladders of the rotational levels for each vibrational state. However, as the number of atoms increases and the level of excitation increases, the density of states rapidly increases. This leads to significant coupling between different rotational levels and eventually different vibrational levels as well. This is discussed further in Chapter 26.
25.3 Rovibrational spectra of molecules
25.3.1 Diatomic molecules
Because vibrational excitations (at low levels of excitation) tend to have much larger energy spacings than rotations, we cannot independently excite a vibration without considering the change of rotational state at the same time. To a first approximation we take rotations and vibrations to be independent. That means that we can simply add rotational energies to vibrational energies to determine the energies of transitions involving the excitation of both. At high values of the rotational quantum number J and the vibrational quantum number v, we will find considerable vibration–rotation coupling. Modeling molecular vibrations as simple harmonic oscillators will allow us to establish the basic features of rovibrational transitions, which we can then modify to include deviations from ideal behavior.
As shown in Chapter 21, when modeled as a harmonic oscillator, the vibrational energy of a diatomic molecule is given by

with the fundamental wavenumber given by

We will use wavenumber  to avoid confusion between the frequency ν (Greek nu) and the vibrational quantum number v (italicized v). In much of the vibrational spectroscopy literature, in which the nomenclature of Herzberg is used, ω is used as the symbol for wavenumber. This is reflected in Table 25.1 for the value ωe because this is the IUPAC recommended notation used in data tables. Here, we will use ω to represent the angular frequency with the only exception being ωe and ωexe. Thus,
to avoid confusion between the frequency ν (Greek nu) and the vibrational quantum number v (italicized v). In much of the vibrational spectroscopy literature, in which the nomenclature of Herzberg is used, ω is used as the symbol for wavenumber. This is reflected in Table 25.1 for the value ωe because this is the IUPAC recommended notation used in data tables. Here, we will use ω to represent the angular frequency with the only exception being ωe and ωexe. Thus,  .
.
When performed at low resolution (or in the liquid or solid phase), a broad peak corresponding to the vibrational energy level change is observed. When performed at higher resolution, a fine structure appears that corresponds to rotational level structure. The internal energy of a molecule is the sum of the rotational and vibrational energies

The vibrational term G(v) in wavenumbers is then

When molecules make transitions from one vibrational state to another, the change in rotational state must also be taken into account. The pre-condition for a vibrational transition is that there must be a change in the dipole moment during the transition. For a harmonic oscillator, the selection rules for the quantum numbers are

As always, the energy of a transition is given by the difference in energy of the states involved

Alternatively, the wavenumber of the transition is

The prime refers to the upper level; the double prime refers to the lower level.
As the resolution is increased, the lines will continue to get narrower until some limit is reached. The linewidth of a rovibrational peak is determined, just as for atomic spectroscopy (as discussed in Section 23.1.2) by several factors that include the Doppler effect and the lifetime of the state. Relaxation of the excited state can result from collisions with other molecules (including a solvent) or the walls of the container. Assuming that only lifetime broadening is significant, the lifetime τ (in ps) of a state is related to the linewidth in cm−1 by

The wavenumbers of rovibrational transitions are sensitive reporters of the local, dynamic environment of the molecule. They provide information on vibrational energy exchange and solvent restructuring as the system approaches equilibrium after reaction or excitation by collisions or irradiation.
25.3.1.1 Example
The vibrational lifetime of the CO stretch vibration is 33 ms in the gas phase,3 4.3 ms when physisorbed on NaCl,4 2.3 ns when chemisorbed on Si(100),5 and 2.2 ps when chemisorbed on Pt(111).6 Calculate the natural linewidths expected in each case if the only mechanism of line broadening is lifetime broadening due to population relaxation.
Substituting into Eq. (25.26) we obtain the results found in Table 25.3. The linewidth increases from values that are difficult, if not impossible, to measure directly due to experimental constraints for the gas phase and physisorbed CO, to values that are easily accessible in the chemisorbed phase. Note how the linewidth scales with the strength of interactions of CO with its environment. Isolated in the gas phase, CO only interacts with fluctuations in the vacuum field. CO interacts very weakly with the NaCl surface in the physisorbed phase, with neither its vibrational frequency nor lifetime changed by a large amount. However, it interacts much more strongly with Si and Pt when chemisorbed, which leads to both shifts in the vibrational frequency and reduction of the vibrational lifetime. The difference between Si and Pt can be traced back to their electronic structures. Si is a semiconductor with a band gap. Therefore, the primary means of vibrational relaxation is through the interaction of various vibrational modes. Pt is a metal. The absence of a band gap means that excitations of the electrons near the Fermi energy (so-called electron–hole pair formation) are also possible and quite efficient at inducing vibrational relaxation.
Table 25.3 Vibrational lifetimes and predicted natural linewidths for CO in various environments.
 |  | |
|---|---|---|
| CO(g) | 33 × 109 | 1.6 × 10−10 |
| CO(a)/NaCl | 4.3 × 109 | 1.2 × 10−9 |
| CO(a)/Si | 2300 | 2.3 × 10−3 |
| CO(a)/Pt | 2.2 | 2.41 |
25.3.2 P, Q, and R branches
The combination of rotational selection rules with vibrational selection rules leads to a spectrum that can be broken up into three regions known as the rotational branches. As illustrated in Fig. 25.5, the R branch corresponds to the ΔJ = +1 transitions, the P branch to ΔJ = –1 transitions. The Q branch corresponds to ΔJ = 0 transitions, which are forbidden for most diatomics, those with Σ ground states, but not necessarily for polyatomic molecules. The lettering scheme for rotational branches is easily remembered as follows. The Q branch is thought of as the origin (Quelle) of the band (also called the band center) that is associated with a particular v’–v” transition. Then assign letters in alphabetical order beginning with ΔJ = –1 (P), ΔJ = 0 (Q), then ΔJ = +1 (R). Should ΔJ = –2 be possible (as in Raman spectroscopy or two-photon transitions), then the lettering would go O, P, Q, R, S for the branches spanning from ΔJ = –2 to ΔJ = +2.

Figure 25.5 The three possible rotational branches arise from the restrictions on simultaneously combining the rotational and vibrational selection rules. The longer arrow of the R branch corresponds to higher wavenumber for the transition. The R branch falls on the high-energy side of the Q branch. The P branch falls on the low-energy side of the Q branch.
Under the assumption that B is independent of v and J, that is for a harmonic oscillator-rigid rotor, the wavenumber of a rovibrational transition is

For the R branch J′ = J′′ + 1 and the allowed wavenumbers are

For the P branch J′ = J′′ − 1 and the allowed wavenumbers are

When labeling the transitions with the notation R(J) and P(J) it is understood that J refers to the lower state value J”.
25.3.3 Anharmonicity and the morse oscillator
The vibrations of real molecules are close to harmonic near the ground state, but exhibit progressively larger deviations for higher vibrational levels, as shown in Figure 25.1. The harmonic oscillator has the obviously unphysical characteristic that it does not dissociate – a poor representation of a molecule if we are looking for a model that allows for chemistry to occur. A Morse oscillator more accurately describes the potential energy curve, in particular because it has a finite binding energy De measured from the bottom of the well. The Morse potential is defined by



where De is the classical dissociation energy (as opposed to Dv, the dissociation energy of the (v, J = 0) state). De differs from D0 by the zero point energy, which is half the fundamental wavenumber

The force constant k, which is defined as the curvature of the potential in the vicinity of the minimum at an internuclear separation Re, and α can be treated as fitting parameters to get the shape of the potential correct. To calculate representative Morse potentials requires the constants De, α, k and the equilibrium bond distance. Selected values of De, α and k are found in Table 25.4. The equilibrium bond distance is found in Table 25.1.
Table 25.4 Ground-state constants for selected diatomic molecules taken from the 96th edition of the CRC Handbook of Chemistry and Physics. The value of the Morse parameter α is calculated from Eq. (25.31) and classical dissociation energy De from Eq. (25.33). The value of the force constant k for BaI is calculated from ωe using Eq. (25.20).
| Molecule | De / eV | D0 / kJ mol−1 | α / Å−1 | k / N m−1 |
|---|---|---|---|---|
| 1H2 | 4.7508 | 435.78 | 1.943 | 575 |
| 2H2 | 4.7508 | 439.75 | 1.943 | 575 |
| 1H19F | 6.1223 | 569.68 | 2.219 | 966 |
| 1H35Cl | 4.6176 | 431.36 | 1.867 | 516 |
| 1H127I | 3.1958 | 298.26 | 1.751 | 314 |
| 19F2 | 1.6628 | 158.67 | 2.970 | 470 |
| 35Cl2 | 2.5131 | 242.85 | 2.003 | 323 |
| 79Br2 | 1.9908 | 193.86 | 1.964 | 246 |
| 127I2 | 1.5527 | 152.25 | 1.859 | 172 |
| 12C16O | 11.2545 | 1076.63 | 2.297 | 1902 |
| 14N2 | 9.9006 | 944.87 | 2.690 | 2295 |
| 14N16O | 6.6149 | 630.57 | 2.743 | 1595 |
| 16O2 | 5.2256 | 498.46 | 2.651 | 1177 |
| 23Na2 | 0.7466 | 74.8 | 0.843 | 17 |
| 138Ba127I | 3.2978 | 321. | 0.925 | 90.5 |
Anharmonicity leads to a decreasing spacing between levels as v increases. We account for this to a first approximation by adding a correction factor to the vibrational term
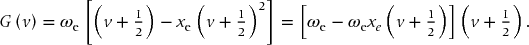
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


