Polyatomic molecules and group theory
Concentrating first on atoms and diatomic molecules allowed us to focus on the fundamental physics of poly-electron and molecular systems. Increasing the number of atoms in a molecule introduces not only increased complexity but also new phenomena. Two of the most important concepts that need to be developed more fully are those of delocalization and symmetry. In diatomic molecules, bonding is necessarily two-centered. In molecules, this need not be the case as we have seen in extended π-bonding systems. Just as for clusters, which were discussed in Chapter 5, coupled systems develop band structure in both electronic and vibrational states as the number of atoms increases. The development of band structure strongly influences energy relaxation dynamics, charge transport and the absorption and emission spectra of molecules.
The symmetry of diatomic molecules is also quite straightforward. There are heteronuclear and homonuclear diatomics. The former require a rotation of 2π radians to transform into a symmetry equivalent configuration. The latter require only a rotation of π radians. The introduction of more atoms into molecules introduces much more intricate symmetries. We have already seen that molecular symmetry is important in determining selection rules for spectroscopic transitions, and we need to develop that in more detail for polyatomics. The symmetry of molecules is important for determining numerous properties such as dipole moments and optical activity, in addition to their spectroscopic properties and selection rules.
26.1 Absorption and emission by polyatomics
Recall, as shown in Fig. 26.1, that two molecular energy levels are connected radiatively in three ways: (i) absorption; (ii) stimulated emission; and (iii) spontaneous emission. Stimulated emission, which is essential for the operation of lasers, will be discussed in Chapter 27. Here, we concentrate on absorption and spontaneous emission. Spontaneous emission is also called fluorescence or phosphorescence, and will be discussed later in more detail.

Figure 26.1 Radiative transitions between levels i and k can be stimulated by a radiation field (absorption and stimulated emission) or occur spontaneously by relaxation from the excited state k to the lower state i (spontaneous emission). Each process has a characteristic rate expression to describe the transfer of population from one state to the other.
Just as for atoms and diatomics, the transition rates between levels are determined by the magnitude of Einstein coefficients and transition dipole moment matrix elements. As discussed in more detail in Chapter 23, we assume that the transitions observed are electric dipole transitions. Thus, the transition rate for absorption is given by

where I(νki) is the intensity of the radiation at the frequency of the transition and Bki is the Einstein B coefficient,1
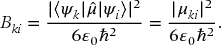
The intensity of radiation is proportional to the square of the electric field strength. The energy density of radiation ρ(νki) is related to the intensity by

Equation (26.1) is a special case of Fermi’s golden rule. It tells us the rate of change of the probability of being in one state or another for a molecule starting in state i and transitioning to state k. For the rate of change of the population of state i, we need to multiply by the number of molecules in state i.
The transition from upper state back to the lower state can occur in two ways. Spontaneous emission is governed by the Einstein A coefficient

from which we make the identification that the probability decay rate is, in fact, the Einstein A coefficient for the transition

Aik has units of s−1. Its meaning is two-fold. The Einstein A coefficient represents the rate of probability decay. It is also the rate constant for population decay of the upper state. The kinetics of population decay follows a first-order rate law whenever spontaneous emission is the only means of relaxation of the excited state.
The ratio of the Einstein A and B coefficients is

For two nondegenerate states i and k, Bik = Bki. The Einstein B coefficient is determined by the magnitude of the transition dipole moment matrix element according to

As discussed in Chapter 23, each transition has an intrinsic linewidth (also called natural linewidth), which for an isolated molecule in the gas phase in the absence of all other line broadening mechanisms is a Lorentzian lineshape. A Lorentzian peak profile centered at a wavenumber  is described by the equation
is described by the equation

Because all atoms in a particular excited state experience the same broadening from the effect of excited state lifetime, this is an example of homogeneous broadening. The Lorentzian linewidth parameter γ is related to the excited state lifetime τ by

and the full-width at half-maximum, FWHM, of a spectral line by

The natural linewidth of a transition involving nondissociating excited electronic states is much less than 1 cm−1, usually <0.01 cm−1. Because of the ν3ki dependence of the Einstein A coefficient, radiative lifetimes increase and linewidths decrease even further for transitions involving only rovibrational and rotational states. The absorption and emission of photons from two levels are connected by the Einstein coefficients. Just as an emission transition has a natural linewidth, so too does an absorption transition. However, as a consequence of Eq. (26.1) the linewidth of an absorption feature is a convolution of the natural linewidth and the linewidth of the excitation source. This is stated mathematically as

L(ν) is the linewidth of the transition and ρ(ν) is the energy density profile of the excitation source – that is, the energy density across the bandwidth of the laser. To calculate the exact absorption rate we need to take account of the vector nature of the transition dipole moment and the electric field. More specifically, we need to consider the polarization of the excitation source and how it lines up with the transition dipole vector. This need not concern us here. Conventional light sources suffer from a direct relationship between decreased linewidth and decreasing intensity. On the other hand, lasers can exhibit extraordinarily high intensity in extremely narrow bandwidth.
Analogously, conventional emission spectroscopy – in which a broadband source (or other means of excitation such as electrical discharge) excites a sample and then a spectrophotometer disperses and detects the emitted light – only obtains high resolution at the expense of signal intensity. With the use of laser excitation, we can perform high-resolution spectroscopy based on the absorption of photons with a photon source that is intense but also has a narrow bandwidth. As discussed further in Chapter 27, these techniques include laser-induced fluorescence (LIF), fluorescence excitation spectroscopy (FES) and resonance-enhanced multiphoton ionization (REMPI). Here, we focus on an example of FES. The principle behind FES is that a laser with a bandwidth smaller than the spacing between spectral lines (indeed, the laser may have a bandwidth even narrower than the natural linewidth) excites a molecule quantum-state specifically. Fluorescence usually involves excitation to an excited electronic state. The structure of the excited electronic state can be probed by state-specific excitation, followed by measuring the intensity of fluorescence as a function of the excitation wavelength. As the excitation light is tuned, different transitions are probed. Fitting the spectrum acquired in this manner to a Hamiltonian that includes sufficient interactions allows us to determine intimates details of the structure of the ground and excited states.
Consider the example of 9,10-dihydrophenanthrene (DHPH) with molecular formula C14H12. The structure of DHPH is shown in Fig. 26.2. An extreme example of the ultrahigh resolution afforded by laser spectroscopy is displayed in Fig. 26.3. Because this molecule contains 26 atoms, it has an incredibly high density of rovibrational states. When observed in the gas phase at room temperature, its absorption spectrum consists of only broad bands, as is typical of sufficiently large polyatomics (this point is discussed in more detail in subsequent sections). When the molecule is expanded in a supersonic molecular beam, its rotational temperature is cooled to roughly 5 K, which means that very few states are populated initially. A laser is used to excite the molecule from the ground electronic state to the first singlet electronically excited state. This is denoted the S0 → S1 transition. Figure 26.3(a) shows that transitions from the ground electronic state to a number of different vibrational states in the S1 state are resolved with the use of a tunable pulsed dye laser that engenders excitation in the UV with a spectral resolution of ~0.6 cm−1. This narrow (but easily obtainable) bandwidth is sufficient to resolve vibrational transitions, but not transitions involving individual rotational levels. To do so requires a more elaborate continuous wave dye laser with an effective spectral resolution of ~1 MHz, which corresponds to ~3 × 10−5 cm−1 in this spectral range. Lasers and their characteristics are discussed in detail in Chapter 27.

Figure 26.2 The structure of 9,10-dihydrophenanthrene (DHPH).

Figure 26.3 Spectroscopic investigation of 9,10-dihydrophenanthrene (DHPH) that has been cooled rotationally to ~5 K. (a) A conventional high-resolution laser fluorescence excitation scheme resolves various vibrational transitions that accompany excitation from the ground electronic state to the first excited singlet state. (b, c) Ultrahigh-resolution fluorescence excitation spectroscopy resolves individual rotational transitions even in this molecule with 26 atoms (C14H12). Figure provided by David Pratt. Reproduced with permission from Alvarez-Valtierra, L. and Pratt, D.W. (2007) J. Chem. Phys., 126, 224308. © 2007 AIP Publishing LLC.
The integral that determines the value of the transition dipole moment (and therefore also the values of the Einstein coefficients) is dependent on the changes in electron distribution that occur when the molecule absorbs light. It can be evaluated explicitly only in the simplest cases (for atoms, diatomics and some small polyatomics). In order to interpret complex spectra, such as that for DHPH and other polyatomics, we need to develop means of evaluating the transition dipole moment in general and approximately. Fortunately, the symmetry of the wavefunctions and the transition operator determine whether or not the integral has a finite value, or if it vanishes. This motivates us to investigate the symmetry of molecules systematically (as shown in the next few sections). We will also apply the Born–Oppenheimer approximation to factor the Hamiltonian. This allows us to treat each degree of freedom independently in the first approximation. The Franck–Condon factor will again be introduced, as it was for diatomics.
26.2 Electronic and vibronic selection rules
To understand spectra such as those in Fig. 26.3 in detail, we need to develop our understanding of selection rules and rovibrational structure beyond what we know already for atoms and diatomics. Rovibrational structure is discussed in Section 26.7. Here, we focus on selection rules arising from the electronic and vibrational degrees of freedom.
Selection rules for atomic transitions are particularly simple. Atoms are characterized by a set of good quantum numbers that are sufficient to express all of the selection rules in terms of these. For diatomic and linear polyatomic molecules, the selection rules are framed in terms of the good quantum numbers. However, it is necessary to include one or two other symmetry properties to fully establish their selection rules. For homonuclear diatomics and symmetrical linear molecules, we need to add an indication of whether a state is even (gerade) or odd (ungerade) with respect to inversion. Inversion symmetry must change during a transition (g to u or u to g). For all diatomic and linear molecules in Σ states (for which the projection of electron orbital angular momentum along the internuclear axis is zero, that is, Λ = 0), we must also consider the symmetry of the molecule upon reflection through any plane containing the internuclear axis. This symmetry is called parity. If there is no change in sign upon reflection, the state is designated +, but if the sign changes the state is designated –. If the molecule lacks symmetry in such a plane, no sign is indicated. For Σ–Σ transitions, the parity must not change.
In the absence of strong spin–orbit coupling, spin changing transitions are forbidden in atoms and diatomics. This is the only quantum number-based selection rule that applies to polyatomics as well. In the absence of high-Z atoms the total electron spin quantum number S should not change,

Just as for diatomics, the Born–Oppenheimer approximation is our starting point for calculations involving the molecular wavefunction. We use this and the Franck–Condon principle to make our first approximations of how to treat the spectroscopy of polyatomics. We begin by factoring of the wavefunction,

The rotational part is relatively simple. It leads to a transition dipole moment component, denoted Mr, the absolute square of which is also called the Hönl–London factor. As discussed in more detail below, it is always zero unless ΔJ = 0, ± 1. This leads to the familiar P, Q and R branches.
The transition dipole moment is calculated for the electronic and vibronic parts from

Vibronic is used to differentiate the change in vibrational state that occurs during an electronic transition, as is the case here, from changes in vibrational state that occur in the absence of electronic excitation. First, we integrate over the electronic coordinates r to obtain

where Me is the electronic transition moment

The vibronic part of the wavefunction is independent of the electronic coordinates r. Thus, it factors out of the integral involving the nuclear coordinates. The Born–Oppenheimer approximation allows us to consider the nuclear coordinates as fixed with respect to electronic motion. Thus, Me is a constant that can be pulled out of the integral over the nuclear coordinates R, to yield

The transition dipole moment is composed of two terms. The first term Me, the electronic transition dipole moment, describes how well the two electronic states are coupled to one another by the radiation. The second term Mv = ⟨ψ′v|ψ″v⟩ describes the vibrational overlap of the two electronic states. Vibrational overlap compares whether the two vibronic wavefunctions have significant amplitude at the internuclear separation where the transition occurs, and is the basis of the Franck–Condon principle:
Electronic transitions are vertical. It is the excited state with the greatest vibrational overlap with the initial state that has the highest intensity.
The Franck–Condon principle holds for transitions with energies well into the extreme UV and even soft X-ray region. In a vertical transition the linear momentum transfer from the photon is negligible and does not change the positions of the nuclei during the transition. Thus, transitions between Born–Oppenheimer potential energy curves for the electronic states can be drawn as vertical lines.
A strong transition is said to have a good Franck–Condon factor as opposed to a weaker transition with a bad Franck–Condon factor. The Franck–Condon factor is quantified by the square of the vibrational overlap integral

If the value of the equilibrium bond distance Re is the same in the lower and upper states, the Franck–Condon factor is greatest for the Δv = 0 transition and then drops off to higher values, mimicking the behavior of an anharmonic oscillator. When the Re values in the two states are displaced, there will be a maximum Franck–Condon factor at some value of Δv ≠ 0, the larger the displacement, the greater the value of the difference. The intensity of transitions on either side of this maximum decays to lower intensity.
In Section 26.1, we found that transition rates are proportional to the absolute square of the transition dipole moment, |μki|2. The value of the transition dipole moment is proportional to the product of the electronic transition moment and the Franck–Condon factor,

The electronic and vibronic selection rules for polyatomics are based on finding those circumstances under which the product in Eq. (26.19) has a finite value as opposed to zero. The integrals that define Me and Mv are integrals over symmetrical limits. Therefore, they only have finite values if the integrand is totally symmetric (in the cases of nondegenerate states) or contains a symmetric species (in the case of degenerate states). Whether or not the integral vanishes can be determined by looking at the symmetry representations, Γ, of the initial and final wavefunctions and the transition dipole operator that connects them.
The electronic transition moment is written in Dirac notation as

This integral only has a finite value if the cross-product of the symmetry representations of the final wavefunction with the operator and the initial wavefunction is totally symmetric

The designation A indicates the totally symmetric representation. Equation (26.21) holds for nondegenerate states. For degenerate states, the cross-product produces more than one representation. For a transition involving a degenerate state to be allowed, the cross-product must ‘contain’ a totally symmetric representation – that is, at least one of the representations generated must be totally symmetric,
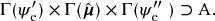
The electric dipole transition operator is a vector with components along the Cartesian axes x, y, and z. Therefore, the symmetry of each of these components is the same as a translation along the appropriate axis. We say that the components transform as the translations along these axes, which are labeled Tx, Ty and Tz, respectively. Labeling the components of the electronic transition moment Me,x, Me,y and Me,z, the absolute square of the total electronic transition moment is equal to the sum of the squares of the components

where the absolute squares of the components are defined analogously to Eq. (26.20) with the total operator replaced by the component operator. Because Eq. (26.23) contains a sum rather than a product, an electronic transition is allowed if any one of the components is non-zero. Written in terms of symmetry representations, this is



(for degenerate states replace = with ⊃). What these symmetry representations are and how we determine them are discussed in Section 26.3.
The product of two symmetry representations is totally symmetric when those two species are the same. Therefore, Eqs (26.24–26.25) are only satisfied when the cross-product of the representations of the wavefunctions is the same as the representations of one of the components of translation. Formally, we write this condition as

The cross-product of the symmetry species of the initial and final wavefunctions must transform as one of the translational axes. Just as for diatomics, most polyatomics have closed shells and a Σ ground state, which is totally symmetric. Because the product of any representation with A is equal to the same representation, the selection rule is expressed in the most common circumstance as

In other words, for any system with a totally symmetric ground state, the electronic transition is allowed as long as the excited state wavefunction transforms as one of the translational axes.
If vibrations are excited in either the initial state or the final state (or both), then we need to look at the symmetry of the full integral in Eq. (26.14) to determine its value. In Dirac notation this is

Using the same arguments as above, in the most common case of a Σ ground state, the selection rule is found from

The symmetry of the electronic vibronic state is given by the product
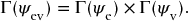
Consequently, the full selection rule is that the cross-product of the composite wavefunctions must transform as at least one of the components of translation along x, y or z,

If the same vibration is excited in the initial and final states, Γ(ψ′v) = Γ(ψv″). The selection rule is then the same as the electronic selection rule. If no vibration is excited (i.e., in the vibrational ground state) the representation of that vibrational component is totally symmetric.
What are these symmetry representations and how do we determine them? For atoms this is extremely simple. All atoms belong to point group Kh. Linear molecules (diatomic and polyatomics) belong to either D∞h or C∞v. Polyatomics in general can belong to a wide range of point groups. In the next section we investigate how to determine which symmetry elements pertain to a given molecule, and to which point group that molecule belongs.
26.3 Molecular symmetry
Figures 26.4 and 26.5 display a number of different molecules with characteristic shapes. The shapes demonstrate the types of transformations – called symmetry elements – that we need to define to treat molecular symmetries. Terms for these symmetry operations and their classification are given in the figure captions, and they are defined in the subsequent sections. A linear molecule such as CO2 has symmetry much like a homonuclear diatomic molecule. A linear molecule such as HCN has symmetry more akin to a heteronuclear diatomic. Categorizing and classifying molecules by symmetry facilitates the understanding of a number of molecular properties, such as any involving dipole moments, and molecular spectroscopy.

Figure 26.4 Molecules representative of a number of different symmetry classes. (a) CHFClBr, C1 with only a C1 axis of rotation, which is the same as doing nothing (identity symmetry element). (b) CO2, D∞h with a C∞ axis along the internuclear axis, C2 axis perpendicular to it, and vertical planes of reflection. (c) HCN, C∞v with C∞ and vertical planes. (d) H2O, C2v with a C2 axis and a vertical plane of reflection. (e) HDO, Cs with only a plane of symmetry (f) NH3, trigonal pyramid, C3v with a C3 axis and vertical planes. (g) CH4, tetrahedral, Td. (h) XeF4, square planar, D4h with C4 and C2 axes, vertical, horizontal, dihedral planes. (i) PCl5, trigonal bipyramidal, D3h with C3, C2 and S3 axes, vertical and horizontal planes. (j) SF6, octahedral, Oh. (k) Benzene, D6h, C6 axis, vertical and horizontal planes and much more.

Figure 26.5 Isomerization and its effect on symmetry. Dichloroethene, C2H2Cl2, exists in three forms with different symmetries. (a) 1,1-Dichloroethene, C2v. (b) cis-1,2-dichloroethene, C2v and (c) trans-1,2-dichloroethene, C2h. All three forms are planar (contain a horizontal plane of reflection) with a C2 axis (though along three different axes for the different molecules) but the trans (or E) isomer does not contain a vertical plane.
From geometrical studies it has been determined that five types of symmetry elements must be defined to describe the symmetry of molecules. These symmetry elements are listed, along with their systematic notation in terms of Schönflies symbols in Table 26.1.
Table 26.1 Symmetry operators in molecule-fixed coordinates, known as Schönflies symbols.
| Operation | Symbol |
|---|---|
| identity | E |
| rotation by 2π/n | Cn |
| reflection | σ, σv, σd, σdh |
| inversion | i |
| rotation-reflection | Sn (= Cnσh) |
26.3.1 Rotation – n-fold axis of symmetry, Cn
A molecule possesses an n-fold axis of symmetry if you can stick a fork into it, rotate it clockwise by 2π/n radians, and return the molecule to an orientation that looks indistinguishable from the initial geometry. The fork is called a rotational axis or, more accurately, an n-fold axis of symmetry, and is shown in Fig. 26.6. For instance, the CO2 molecule has a C2 axis perpendicular to the internuclear axis passing through the center of the C atom. Rotation by π radians (180°) leaves the central C atom where it was (atoms by themselves are spherically symmetric) and moves one O atom to where the other one was initially. Since the two O atoms are indistinguishable (assuming they correspond to the same isotope of O), the molecule has been transformed from an initial configuration into a symmetry equivalent configuration. As also shown in Fig. 26.6(b), placing an axis parallel to and centered upon the molecular axis, the molecule can be rotated by an arbitrary amount and it still looks the same. Such an axis is known as a C∞ axis. The value of n change in integer steps from n = 1, 2, 3, … ∞.

Figure 26.6 Clockwise rotation about a Cn axis looking down the z-axis transforms x and y by rotating them through 2π/n radians within the xy-plane. (a) A C4 rotation is shown. (b) CO2 possesses a C2 axis perpendicular to the internuclear axis and a C∞ axis along it.
Corresponding to every symmetry element (a property of the object) is a symmetry operation (the thing that is done to the object) with the same symbol. Thus, we speak of a CO2 molecule possessing a C2 axis and state that when transformed under a C2 symmetry operation by rotation of π radians about the C2 axis (or more simply, under C2), the CO2 molecule is symmetric. The highest-fold axis of a molecule defines the vertical axis, labeled as the z-axis. If a Cn operation is performed twice, we denote this by C2n and so forth for other multiples. Cn corresponds to a clockwise rotation by 2π/n radians, whereas C− 1n corresponds to a counterclockwise rotation by –2π/n radians,
Some molecules contain C2 axes at equal angles to each other and perpendicular to the main axis. This is true, for example of the benzene molecule. This combination is common enough that axes of this type are called dihedral axes.
Rotation about an axis transforms a point with initial coordinates (x, y, z) to the point (x′, y′, z′). The new coordinates are obtained from a linear combination of the original coordinates according to the set of equations



This set of equations can be represented by a matrix equation in which the points are given as column vectors and the coefficients form a (3 × 3) matrix,

A C2 rotation about the z-axis leaves z unchanged but rotates x to –x and y to –y. The transformation matrix for this C2 operation along z is thus,

Inspection of Figure 26.6(a) reveals that we can represent a rotation about z by an angle 2π/n by the matrix

If we were to change the rotation to be about the x-axis, the zeros and ones would move to keep x unchanged and the block of sine and cosine terms would move down an over to transform y and z appropriately.
26.3.1.1 Directed practice
The methane molecule CH4 contain a C3 axis directed along a C–H bond. Looking down this axis (in other words making this axis the z-axis in a molecule-fixed frame) you will see the three other H atoms, which sit at the vertices of an equilateral triangle with the z-axis passing through its center. Starting at the top of the triangle and proceeding clockwise we label the H atoms H1, H2 and H3. Write out the matrix equation that corresponds to performing the C3 operation.
26.3.2 Reflection – plane of symmetry, σ
If reflection of all the nuclei in a molecule through a plane to an equal distance on the opposite side of the molecule produces a configuration indistinguishable from the initial configuration, the molecule is said to possess a plane of symmetry, σ; this is also called a mirror plane. A molecule may possess more than one plane of symmetry, just as it can have multiple axes of symmetry. A plane that contains the z-axis, which is defined to contain the highest-fold axis, is called a vertical plane of symmetry, σv. The H2O molecule is planar with a C2 axis through the central O atom. It also contains two vertical planes of symmetry containing this axis. They are named, according to convention, σv(xz) and σv(yz). The yz-plane is the plane of the molecule. Any planar molecule must contain at least one plane of symmetry. Any molecule with a plane of symmetry cannot be chiral.
The benzene molecule shown in Fig. 26.7 is planar with a C6 axis perpendicular to this plane. The plane of the molecule is a horizontal plane, σh. However, if we look at the vertical planes of symmetry of the molecule – the ones in which the C6 axis lies – we see that there are two ways that we can draw them. The most obvious is that we can draw a vertical axis through the center of opposing C–H bonds. We label these (there are three of them) the σv axes. However, we can also draw three vertical axes through the middle of opposing C–C bonds in the benzene ring. To distinguish these three axes, we label them σd, for dihedral plane of symmetry. Dihedral planes bisect the angles between dihedral axes.

Figure 26.7 The highest-fold axis, a C6 axis for this example of benzene, defines the z-axis and the vertical plane. In benzene the vertical plane is a plane of symmetry, denoted σv. The horizontal plane in benzene is also a plane of symmetry, denoted σh.
Reflection in the xy-plane leaves the coordinates of x and y unchanged but transforms z to –z, (x, y, z) → (x, y, −z). Similarly, reflection in the yz plane leaves y and z unchanged while changing x to –x. The transformation matrix for the operation σxy is thus,

with appropriately similar matrices for σyz and σxz.
26.3.2.1 Directed practice
Describe why H2O has two vertical planes of symmetry but HOD only has one.
26.3.3 Center of inversion, i
Inversion symmetry, i, involves reflection of atoms through the center of the molecule to a point an equal distance on the opposite side of the molecule, (x, y, z) → ( − x, −y, −z). The transformation matrix is thus,

Any molecule possessing inversion symmetry cannot be chiral.
26.3.3.1 Directed practice
Benzene C6H6 contains a center of inversion. Draw isotopologues of benzene formed by the substitution of either one D or two D atoms for H atoms that either do or do not have a center of inversion.
26.3.4 Rotation–reflection axis of symmetry, Sn
A distinct symmetry operation can be generated by performing first a rotation of 2π/n radians, followed by reflection through a plane. The plane is perpendicular to the axis passing through the center of the molecule. The combined symmetry operation is known as n-fold rotation–reflection symmetry, denoted by Sn,

This is also known as an improper rotation. The order of operation is important for matrix multiplication. In other words, matrices do not in general commute and M × N ≠ N × M. Therefore, to obtain the transformation matrix to perform an Sn operation about the z-axis, we must form the cross-product


Any molecule possessing an Sn axis cannot be chiral. In this context it is to be noted that S1 ≡ σ and S2 ≡ i. Benzene contains both S3 and S6 rotation–reflection axes.
26.3.5 Identity element of symmetry, E
The symmetry operation E corresponds to leaving the molecule unchanged. It is sometimes also denoted I in order to avoid confusion between E (for the German Einheit formatted in italics) the identity symmetry element, and E (in Roman type), which is the name of a doubly degenerate irreducible representation. This may seem trivial but it is required by the mathematics of group theory as we shall see below. Performing any n-fold rotation n times corresponds to rotation by 2π and reproduces the initial configuration, for example,

All molecules possess this element of symmetry. Equations (26.41) and (26.44) also demonstrate that we can generate a full range of other symmetry operations by performing cross-products of symmetry elements. The transformation matrix for the identity operation, also called the unit matrix,

is unusual in that it commutes with any other matrix.
26.4 Point groups
Any given molecule possesses a set of symmetry elements. These symmetry elements are not unique to the molecule; instead, they are unique to the shape of the molecule and the juxtapositions of similar and dissimilar atoms within the molecule. Water has the symmetry elements I, C2, σv(xz) and σv(yz). This is true for all bent triatomic molecules of the formula AB2. Similarly, both C6H6 and C6F6 have the symmetry elements I, C6, C3, C2, i, S3, S6, σh, σd and σv. What these sets have in common is that when all of the operations in a set are performed on the molecule, there is a point (more accurately at least one point) in the molecule that does not move. In the case of H2O, every point along the C2 axis is unaffected by the operations in the set. In the benzene molecule, the center of inversion does not move under any operation. For this reason, we call the set of operations a point group.
The members of a point group follow a number of rules. The product of any two operations is defined and is also a member of the group. The inverse of each operation is in the group. The identity operator is a member of every group. All operations of the group are associative,

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


