Chapter 22 Mahesh M. Mansukhani; Peter L. Nagy; Ali Naini; Vimla Aggarwal A. Glutathione reductase (GSR). B. Catalase (CAT). C. Glutathione peroxidase (GPX). D. Manganese-superoxide dismutase (SOD2). E. Copper/zinc superoxide dismutase (SOD1). 2. Which one of the following represents the central dogma of molecular biology? B. DNA → RNA → protein. C. RNA → protein → DNA. D. RNA → DNA → protein. E. Protein → RNA → DNA. 3a. Which one of the following is the best next step to pursue? B. Ask for another blood sample from the patient because the current result “does not make sense.” C. Test the patient’s blood relatives to determine whether the 7T variant is on the same allele as the R117H mutation. D. Schedule the patient for a chorionic villous sampling to ensure that the fetus is not affected. E. Screen the patient’s reproductive partner for CFTR gene mutations. 3b. The patient’s reproductive partner (i.e., the father of the fetus), who is asymptomatic, is screened with the standard panel. Which one of the following statements best describes the risk for this couple having a child affected by cystic fibrosis? B. Lowest if the father is an Ashkenazi Jew because of the high detection rate among Ashkenazi Jews. C. Highest if the father is of Hispanic origin because of the combination of a high carrier rate and a low detection rate among Hispanics. D. Lowest if the father is of East Asian origin because of the low carrier rate among Asians. E. Can be reduced significantly if the father is of East Asian origin by using an extended panel to include East Asian–specific mutations. 4. These results are from the peripheral blood of a child with acute lymphocytic leukemia who underwent an allogeneic hematopoietic stem cell transplantation 1 year previously. Which one of the following statements regarding the results shown in Figure 22-1 is true? A. If the transplantation had been performed a year ago, this is an ideal result. B. The results are concerning because of the high proportion of recipient DNA in the sample. C. The results are concerning because of the low proportion of recipient DNA in the sample. D. The results are concerning because of the high proportion of donor DNA in the sample. E. This is not the appropriate test to perform in this setting. 5. Cystic fibrosis is caused by mutations in the gene encoding for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The ΔF508 deletion is a common mutation. Which one of the following patient populations in the United States has the highest prevalence of the ΔF508 deletion? B. African Americans. C. Asians. D. Ashkenazi Jews. E. Hispanics. 6. The prevalence of cystic fibrosis in the United States varies in different populations. Which one of the following statements regarding prevalence is true? A. Highest in both the Caucasian and Ashkenazi Jewish populations. B. Highest in both the Caucasian and African American populations. C. Highest in both the Caucasian and Hispanic populations. D. Highest in both the Caucasian and Asian populations. E. Highest in both the Caucasian and Native American populations 7. Which one of the following statements best describes the current approach used to screen newborns for cystic fibrosis in the United States? A. Measurement of immunoreactive trypsinogen and DNA analysis. B. Measurement of both immunoreactive trypsinogen and pancreatitis-associated protein. C. Measurement of both immunoreactive trypsinogen and serum amylase. D. Measurement of both immunoreactive trypsinogen and serum chloride. E. Measurement of both immunoreactive trypsinogen and nasal potential difference. 8. A positive immunoreactive trypsinogen result, along with one DNA mutation, is found when screening a newborn for cystic fibrosis. Which one of the following is the appropriate next step in the management of this patient? A. Make the diagnosis of cystic fibrosis, as recommended by the Cystic Fibrosis Foundation. B. Perform additional genetic and sweat chloride testing to exclude or confirm the diagnosis of cystic fibrosis. C. Repeat the immunoreactive trypsinogen and DNA assays to confirm the abnormal results. D. Measure serum and urine chloride to exclude or confirm the diagnosis of cystic fibrosis. E. Measure serum amylase, lipase, and trypsin to exclude or confirm the diagnosis of cystic fibrosis. 9. The two strands of DNA that compose the double helix are held together by which one of the following? B. Hydrogen bonds. C. Phosphodiester bonds. D. Nitrogen bonds. E. Carbon bonds. 10. Which one of the following is correct for the human genome? A. It has a haploid copy number. B. Nucleated cells contain 22 chromosomes. C. Consists of DNA, which has a circular structure. D. Contains 3 × 109 base pairs of DNA. E. Contains 100,000 protein-coding genes. 11. Which one of the following pathologic conditions is caused by mitochondrial DNA (mtDNA) rearrangement? A. Leber’s hereditary optic neuropathy (LHON). B. Mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS). C. Myoclonus epilepsy and ragged red fibers (MERRF). D. Kearns-Sayre syndrome (KSS). E. Neuropathy, ataxia, and retinitis pigmentosa (NARP). 12. Which one of the following is the best method for detecting mtDNA rearrangements? B. Restriction fragment-length polymorphism (RFLP) analysis. C. Amplification refractory mutation system–polymerase chain reaction (ARMS-PCR). D. Real-time PCR. E. Southern blot analysis. 13. Which one of the following methods is best used to detect multiple mtDNA deletions in a very small muscle biopsy taken from a 7-year-old male patient? B. Southern blotting with a radiolabeled probe. C. Long-range PCR and gel electrophoresis. D. Sequencing by capillary electrophoresis. E. Single-stranded conformation polymorphism (SSCP). 14. Mutation in which one of the following genes causes mtDNA depletion? A. Mitochondrial tRNA lysine (MT-TK). B. Mitochondrial tRNA leucine 1 (MT-TL1). C. Mitochondrial tRNA tryptophan (MT-TW). D. Mitochondrial tRNA isoleucine (MT-TI). E. mtDNA polymerase gamma (POLG). 15. Which of the following methods is best used for accurate determination of mtDNA copy number? A. Southern blot analysis with radiolabeled probes. B. Southern blot analysis with fluorescence detection. C. Real-time PCR with TaqMan probes. D. Sanger sequencing of the whole mitochondrial genome. E. Next-generation sequencing of the whole mitochondrial genome. 16. Which one of the following mutations is associated with MELAS, maternally inherited progressive external ophthalmoplegia (MI-PEO), and maternally inherited diabetes and deafness (MIDD)? B. An adenine-to-guanine transition at nucleotide 8344 position (8344A > G) in mitochondrial tRNA Lys (MTTK). C. A thymidine-to-cytosine transition at nucleotide 8993 position (8993T > C) in mitochondrial ATPase 6 (MTATP6). D. A thymidine-to-cytosine transition at nucleotide 9176 position (9176T > C) in mitochondrial ATPase 6 (MTATP6). E. A guanine-to-adenine at nucleotide 1178 position (11778G > A) in mitochondrial subunit ND4 of complex I (MTND4). 17. Mutations in which one of the following genes cause mitochondrial isolated myopathy? A. Complex I subunit ND4 gene. B. Complex I subunit ND1 gene. C. Cytochrome b (cyt b) gene. D. Cytochrome c oxidase subunit 1 (COX1) gene. E. Succinate dehydrogenase (SDHA) gene. 18. Which one of the following statements is correct? A. All respiratory chain proteins are encoded by mtDNA. B. Sperm do not contain mtDNA. C. All mtDNA-encoded proteins are components of the respiratory chain. D. mtDNA is independent of nuclear DNA (nDNA) and does not require nDNA gene products for its function. E. All mtDNA in the zygote is derived from sperm. 19. A 17-year-old boy presents with severe gastrointestinal dysmotility, progressive external ophthalmoplegia (PEO), cachexia, and peripheral neuropathy, a clinical phenotype consistent with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). A muscle biopsy revealed neurogenic changes and cytochrome c oxidase (COX)-deficient fibers reflecting mitochondrial myopathy. He was referred to your molecular pathology laboratory for diagnosis confirmation. Which one of the following tests is best used to confirm the diagnosis of MNGIE? A. Thymidine phosphorylase (TP) activity in peripheral blood leukocytes. B. Mitochondrial respiratory chain complex I activity. C. Mitochondrial respiratory chain complex II activity. D. Mitochondrial respiratory chain complex IV activity. E. ATPase activity in peripheral blood leukocytes. 20. Which one of the following forms of inheritance is associated with mtDNA-related disorders? B. Autosomal dominant. C. Maternal. D. X-linked recessive. E. X-linked dominant. 21. A change in the nucleotide sequence of the DNA is best known as which of the following? B. Polymorphism. C. Genotype. D. Allele. E. Rare variant. The clinical geneticist at your institution requests your help in formulating a diagnostic approach for a family he is following. The parents are healthy cousins and have three children: one with microcephaly, one with deafness, and one with both conditions. The pedigree for this family is shown in Figure 22-2. Use this scenario to answer the following 12 questions. 22a. Which one of the following best describes the scenario that is most consistent with this clinical situation? B. This is an autosomal dominant deafness syndrome with low penetrance and variable expressivity. C. This is a family with two mutations that cause two separate disorders, each inherited in an autosomal recessive manner. D. This is manifestation of a “mutator” syndrome that causes frequent mutations in the germline of the parents; thus, healthy parents have multiple affected children. E. This situation is due to maternal consumption of genetically modified produce during each pregnancy. 22b. Which one of the following is the best next step to take for this case? B. You recommend comparative genomic hybridization microarray studies to identify regions of homozygosity between the siblings affected by the same disease. This will allow you to narrow the list of genes known to cause the conditions observed in this family. C. You propose targeted testing for all mutations known to cause deafness and microcephaly using Sanger sequencing. D. You propose full genome sequencing because you suspect that the changes responsible for the phenotypes observed are in noncoding regions of the human genome. E. You recommend targeted testing for the most common point mutations associated with deafness and microcephaly. 22c. The targeted sequencing fails to diagnose the cause(s) of deafness and microcephaly in this family. The parents decide to have more children. In your discussions with your clinical geneticist colleagues, which one of the following recommendations is the best advice? B. The family should be advised not to consider additional children because the risk for having additional affected children is uncertain. C. The family should be advised that the risk for having an affected child is one in four for microcephaly or deafness, whereas the chances of having a child with both disorders is one in eight, without providing any further recommendations. D. The family should be advised about the possibility of further genetic testing in case the parents are interested in pursuing prenatal diagnosis or in vitro fertilization. E. The family should be advised that there are no further diagnostic options available for them, and they should consider adoption. 22d. You raise the possibility to your clinical colleagues of using Next-Gen sequencing to help establish a molecular diagnosis for the condition(s) affecting this family. They ask about the difference between Next-Gen sequencing and traditional Sanger sequencing. Which one of the following is the best response? A. It is still experimental, whereas Sanger sequencing is currently available. B. It involves sequencing a single molecule, instead of many copies of the same molecule. C. It can provide megabases worth of nucleotide sequence, whereas Sanger sequencing is only practical for sequencing, at maximum, a few kilobases. D. It is based on irreversible termination of extension on a subpopulation of the templates. E. Interpretation of the data obtained is much more straightforward. 22e. During your explanation of Next-Gen sequencing, you use the phrase “sequence capture.” Your clinical colleagues ask you about the meaning of this term. Which one of the following is the best answer to this question? B. The human genome contains a great deal of noncoding DNA, the role of which is not clearly established in disease. To limit the cost of clinical testing by sequencing, it is practical to focus on certain segments of the genome that have previously been shown to be associated with human disease. Upon sequencing the full genome, these segments are computationally identified and analyzed, whereas the data from noncoding regions of the genome are left in a non–human-readable form; this is sequence capture. C. It refers to the laboratory procedure that selects fragments of diagnostic interest from human genomic DNA for sequence analysis. D. It refers to the method of data collection from the sequencer. E. It refers to the fact that sequences slowly leak out of the reaction chamber of the sequencer, and they need to be prevented from contaminating the instrument by capturing them using a secondary container that is replaced regularly. 22f. The parents decide to pursue further genetic testing. You explain to your clinical colleagues that, in this case, full exome sequencing is probably the best next step. You mention that alternatives would include sequencing all genes implicated in deafness and microcephaly, or full genome sequencing. They ask you to explain your rationale. Which one of the following justifications is not correct? B. Full exome sequencing is less expensive than full genome sequencing, and most known pathogenic mutations are within exons or at intron-exon junctions. C. Analysis of a full exome dataset is less complex than that of a full genome sequencing dataset. D. Sequencing only known genes and loci would be effective if the patients in this family had a mutation previously identified in other families, but it would not detect private mutations specific to this family. E. Exome sequencing provides the most even and reliable sampling of the coding regions of the human genome, even if expense is not a consideration. 22g. You and your colleagues decide to proceed with the exome sequencing approach. They ask you which members of the family should be tested. Which one of the following is the most reasonable answer under these circumstances? A. Both parents and all three children should be sequenced. B. Only the parents should be sequenced. C. The parents, one child with deafness, and one child with microcephaly should be sequenced. D. Only the child with both microcephaly and deafness should be sequenced. E. Only the three siblings should be sequenced. There is no need to sequence the parents. 22h. The decision is made to sequence the three siblings. Which one of the following best describes the requirements that need to be satisfied before testing can commence? A. The patients must be older than 18 years. B. Both parents must sign an informed consent form, and the children must sign an assent form. C. At least one parent must sign an informed consent form. The children with microcephaly are exempt from signing the assent form; assent only needs to be obtained from the deaf child. D. You must confirm that the parents’ insurance will cover the cost of testing. E. You have to obtain U.S. Food and Drug Administration (FDA) approval for the method to be used. 22i. All administrative hurdles are now cleared. The sequencing is performed. The results indicate the presence of approximately 40,000 single-nucleotide polymorphisms (SNPs) in each sample. The data analysis software allows for filtering the identified mutations based on various criteria. Which one of the following filtering approaches is not correct? B. Mutations that do not cause amino acid changes, frame shifts, transcriptional stops, or splicing defects should be removed in the initial analysis. C. Emphasis should be placed on mutations for which any one patient is homozygous. D. Mutations that are present in a homozygous form in all three siblings should be excluded as the cause of both deafness and microcephaly. E. The children with deafness should be homozygous for the same mutation; similarly, the children with microcephaly should be homozygous for the same mutation. 22j. Several mutations were found in homozygous form in each sibling. Which one of the following web-based resources is not useful in identifying the mutations responsible for the patients’ conditions? A. Online Mendelian Inheritance in Man (OMIM): http://www.ncbi.nlm.nih.gov/omim B. University of Santa Cruz web browser: http://genome.ucsc.edu C. dbSNP database: http://www.ncbi.nlm.nih.gov/SNP/ D. 1000 genome database: http://www.1000genomes.org E. Leiden database: http://www.dmd.nl 22k. Analysis of the three exome-sequencing datasets is completed, and clearly disruptive homozygous mutations in genes previously implicated in microcephaly and deafness are identified, which are in agreement with the expected phenotypes seen in each sibling. Which one of the following is required before the results can be reported? A. The exome sequencing must be repeated. B. The sequencing data must be reanalyzed using another analysis software program. C. The identity of the samples and the correctness of the mutations must be confirmed using an alternative method. D. The mutations must be confirmed to be absent in 400 healthy control samples. E. You must confirm that the mutations have never been seen previously in any population sampled. 22l. The parents return several months later to request preimplantation testing for the mutations identified in the affected siblings as part of an in vitro fertilization effort. Which one of the following is the best strategy to pursue for this testing? B. Following whole genome amplification, the DNA from each embryo has to be genotyped for the mutations identified in the three siblings with deafness and/or microcephaly. C. Following whole genome amplification, all embryos have to be tested for aneuploidy using a microarray methodology. D. The embryos with the pathogenic mutations need to be preserved for future diagnostic test development. E. Embryos that are carriers for either the deafness or microcephaly mutations can be implanted with their normal counterparts because they would also result in normal children and would improve the chances of successful implantation. 23. Which one of the following answers best describes what a PCR includes? A. Denaturation, digestion, detection. B. Denaturation, annealing, amplification. C. Digestion, annealing, amplification. D. Denaturation, annealing, labeling. E. Denaturation, annealing, digestion. 24. When designing primers for PCR, which one of the following is the best approach? A. Choose primers that show significant complementarity to each other, especially at the 3′ ends. B. Ensure that the forward and reverse primers have a melting temperature (Tm) that differs by at least 5°C. C. Choose primers that are complementary to the same DNA strand. D. Choose primers that have a GC content of at least 90% to ensure strong binding. E. Check that the primers used to amplify genomic human DNA are validated against variant databases (e.g., dbSNP) to avoid primer–binding-site polymorphisms. 25. Which one of the following statements about controls for PCR or reverse-transcriptase PCR (RT-PCR) is true? B. When setting up a PCR, one should add a template to the test samples first, followed by the no-DNA control, and then add the normal and abnormal controls. C. When setting up an RT-PCR, in addition to a no-template (i.e., no RNA) control, one should set up a no-RT control for each RNA sample being tested to test for directly amplifiable RNA. D. When setting up a PCR following an RT reaction, one should set up a separate no-template control in addition to the product of the no-RNA RT reaction. E. The use of deoxyuridine triphosphate (dUTP) in PCR with uracil-N-glycosylase to prevent PCR contamination is of no value for RT-PCR because RNA contains uracil. 26a. A DNA test was performed on a child with severe developmental delay, especially language delay, and an apparently “happy” disposition with inappropriate laughter. PCR of bisulfite-treated DNA with melting curve analysis of an SNRPN exon 1 amplicon showed only an unmethylated allele. Which one of the following statements is correct? A. Only a maternal allele is present; this is consistent with a diagnosis of Angelman syndrome. B. Only the paternal allele is present; this is consistent with a diagnosis of Angelman syndrome. C. Only the paternal allele is present; this is consistent with Prader-Willi syndrome. D. Only the maternal allele is present; this is consistent with Prader-Willi syndrome. E. Only one allele is present. Parental testing to determine the parent of origin of the missing allele will distinguish between Prader-Willi and Angelman syndromes. 26b. Which one of the following statements is true about the inheritance of Prader-Willi and Angelman syndromes? A. Prader-Willi syndrome and Angelman syndrome are caused by different mutations in the same gene. B. Inactivating mutations in UBE3A will cause Prader-Willi syndrome only if present on the paternal allele. C. Inactivating mutations in UBE3A will cause Prader-Willi syndrome only if present on the maternal allele. D. Inactivating mutations of UBE3A cause Angelman syndrome when present on the maternal allele. E. Inactivating mutations of UBE3A cause Angelman syndrome only when present on the paternal allele. 26c. Which one of the following statements is true about genomic imprinting? B. All imprinted loci in placental mammals show expression from only the maternal allele. C. All imprinted loci in placental mammals show expression from only the paternal allele. D. Most cases of Angelman syndrome and Prader-Willi syndrome are caused by imprinting defects. E. Beckwith-Wiedemann syndrome, Russell-Silver syndrome, and transient neonatal diabetes mellitus type 1 are linked to imprinted loci. 27. Which one of the following statements regarding RNA transcription and processing is true? B. Promoters, which are recognized by RNA polymerase, include the TATA box, usually at the -25 position; the GC box that can function in either orientation; and the CAAT box that is usually located at the -80 position. C. Enhancers, which are always in close proximity to promoters, increase transcription activity of the associated gene. D. Noncoding genes are always transcribed by RNA polymerase II. E. The primary transcript of polypeptide-encoding RNAs often contains introns and exons. Introns are characterized by 5′AG and a 3′GU. 28. Which one of the following statements is true regarding sequencing for genetic variants? B. The smaller a deletion, the more likely it is to be missed by Sanger sequencing after DNA PCR. C. Nonsense mutations in the first exon may be missed when performing cDNA sequencing. D. Sequencing of the entire coding region of a gene will identify virtually all significant mutations in a gene. E. When a missense mutation in a gene known to be associated with a condition is seen in a symptomatic individual, the mutation is most likely to be significant. 29. Which one of the following neurologic diseases is caused by a triplet repeat expansion in the coding region of the involved gene? B. Friedreich ataxia. C. Spinocerebellar ataxia type 8 (SCA8). D. Myotonic dystrophy. E. Huntington disease. 30a. Figure 22-3 shows the results of PCR (i.e., capillary electrophoresis) and Southern blot tests for the FMR1 CGG repeat expansion associated with fragile X syndrome. Which one of the following statements regarding the PCR result is true? B. The pretest risk for an expansion is higher in a woman with an affected nephew (brother’s son). C. In a male with mental retardation, the capillary electrophoresis result shown provides strong evidence against fragile X syndrome. D. In a girl with developmental delay, the capillary electrophoresis alone excludes FMR1-associated mental retardation. E. In a mother of a boy with developmental delay, the PCR result rules out an FMR1 expansion as the basis for the child’s condition. 30b. Which of the following statements is true about Southern blot tests and/or the results shown in Figure 22-3? B. Southern blot texting is more reliable than PCR in excluding fragile X tremor ataxia syndrome (FXTAS). C. Southern blot testing is more reliable then PCR in excluding FMR1-associated premature ovarian failure. D. The Southern blot test result adds little to the PCR, which is more sensitive and reliable. E. In a woman undergoing carrier testing, this result is strong evidence against her carrying a premutation or full mutation. 31. A 60-year-old man is being evaluated for placement of a coronary artery stent. His physician plans to start him on regimen of a clopidogrel after the procedure. Pharmacogenomic testing of which one of the following genes would be most useful for predicting whether he is a poor metabolizer? B. CYP2D6. C. CYP2C9. D. VKORC1. E. TPMT. 32. A 65-year-old man with chronic myeloid leukemia is being treated with imatinib. His BCR- ABL1 mRNA transcript levels are being monitored by a laboratory that uses an RT-PCR. Figure 22-4 shows the patient’s BCR-ABL1 transcript levels as measured in the laboratory from time point 1 (i.e., at diagnosis) to time point 6 (i.e., the current sample). Which one of the following is the best next step? A. Test for BCR- ABL1 kinase domain mutations. B. Perform a bone marrow biopsy for pathology and cytogenetics. C. Switch the patient to a second-generation tyrosine kinase inhibitor (TKI). D. Repeat this quantitative PCR assay in 3 to 6 months. E. Consider sending the patient for an urgent allogeneic stem cell transplantation. Major points of discussion ■ The disease starts in adult life, typically between 50 and 60 years of age. ■ The ensuing progressive paralysis is typically fatal within 3 to 5 years of clinical onset, usually as the result of failure of the respiratory system. ■ ALS is one of the more prevalent adult-onset neurodegenerative diseases, with an incidence of 1 to 2 per 100,000 in most populations. The incidence is much higher in the Pacific island of Guam and the Kii Peninsula of Japan, possibly because of consumption of an environmental toxin, β-methylamino-l-alanine. ■ The cause of ALS is unknown in most cases, and these cases are referred to as sporadic ALS (SALS). ■ Approximately 10% of ALS cases are inherited in a dominant manner and are referred to as familial ALS (FALS). ■ To date, mutations in 12 genes have been reported to cause FALS. Among these, SOD1 mutations, which are transmitted in a dominant fashion, account for approximately 20% of all familial ALS forms and about 2% of all ALS cases.3,5,6 2. A. DNA → protein → RNA. Major points of discussion ■ The information between DNA, RNA, and protein is related. ■ The genetic information coded by the DNA determines the sequences of the amino acids in the encoded proteins. ■ First RNA is synthesized from the DNA template through a process called transcription. ■ This RNA, carrying the coded information in a form called messenger RNA, is translated to determine the sequence of the amino acids in the protein. mRNA is decoded by transfer RNA (tRNA) on cellular ribosomes to produce amino acid sequences or proteins in the process called translation. ■ This flow of information is referred to as the central dogma of molecular biology. 3a. A. Perform DNA sequencing with long-range polymerase chain reaction (PCR) to determine whether the 7T allele variant is on the same allele as the R117H mutation. 3b. A. Highest if the father is a non-Jewish Caucasian because the condition is most common among Caucasians. Major points of discussion ■ The disease is characterized by recurrent pulmonary infections resulting from failure to eliminate viscous secretions, with or without pancreatic insufficiency; increased sweat chloride as the result of excessive salt loss; neonatal meconium ileus in 10% to 20% of infants; chronic sinusitis; congenital bilateral absence of vas deferens; and liver disease. The average overall survival of individuals with cystic fibrosis is 30 years. ■ More than 1000 mutations are known in the CFTR gene, with most representing infrequent (< 0.1%) or private mutations. The most common mutation, delF508, accounts for 30% to 70% of mutations, depending on race. When performing carrier screening, the American College of Medical Genetics recommends testing for the 23 most common variants, each of which has a frequency of more than 0.1% among cystic fibrosis patients; this panel has a detection rate of 94.04% among Ashkenazi Jews; 88.29% among non-Hispanic Caucasians; 64.46% among African Americans; 71.72% among Hispanic Americans; and 48% among Asian Americans. ■ When a test result for these mutations is negative, it reduces the risk of being a carrier, but does not eliminate it. The residual risk is 1 in 384 for Ashkenazi Jews; 1 in 206 for non-Hispanic Caucasians; 1 in 171 for African Americans; 1 in 203 for Hispanic Caucasians; and 1 in 183 for Asian Americans. Residual risks should be reported. ■ Clinical tests include genotyping tests that check for specific mutations; mutation scanning assays; direct DNA sequencing assays; and tests for large deletions or rearrangements. Together they may still miss some mutations, and tests such as sweat chloride analysis may be needed to confirm the diagnosis. ■ The R117H mutation is a variant that acts as a severe disease-causing mutation only when it is on the same allele as a 5T intron 8 polymorphism that results in increased alternate splicing, excluding exon 9, with reduced full-length mature mRNA. The longer alleles (7T and 9T) are associated with increased full-length transcript and a milder reduction in function due to R117H. Individuals with a 7T on the same allele as the R117H mutation, along with a severe mutation on the opposite allele, may develop lung disease later in life (in the sixth, and even seventh, decade). The 5T allele itself may be associated with congenital absence of the vas deferens. ■ When the R117H mutation is seen with a 5T variant in an individual, testing of family members may be needed to determine on which allele the 5T variant is located. The 5T allele by itself may be associated with congenital absence of vas deferens, either when it occurs as a homozygous variant or when it occurs with other CFTR mutations. Because of this association, many tests report the 5T/7T and 9T results only when there is an R117H mutation.24,35 4. A. If the transplantation had been performed a year ago, this is an ideal result. Major points of discussion ■ Testing is most commonly performed using multiplex marker kits that use up to 16 STR loci as identity markers. ■ For each informative locus (at least one allele differing between donor and recipient), it is possible to determine the proportions of donor and recipient DNA using peak heights or peak areas corresponding to recipient and donor alleles. ■ After hematopoietic stem cell transplantations for malignancies such as acute lymphocytic leukemia, the recipient’s bone marrow should be repopulated primarily by donor cells. ■ A drop in donor DNA percentage or an increase in recipient DNA percentage may indicate relapse or impending relapse. ■ The results shown here for this patient demonstrate an almost equal proportion of donor and recipient cells, indicating either graft failure or relapse of the acute leukemia.4 5. A. Caucasians. Major points of discussion ■ There are more than 1800 known mutations of the CFTR gene. ■ The American College of Medical Genetics and Genomics recommends that at least 23 different mutations be evaluated when testing for cystic fibrosis. Some testing programs detect more than 40 mutations. ■ Different ethnic populations have different CFTR gene mutations. In locales with a diverse patient population, the selection of the CFTR mutations to be tested will affect the number of cystic fibrosis cases that are detected. ■ Some CFTR mutations that are detected may not be clinically significant.2,13 6. A. Highest in both the Caucasian and Ashkenazi Jewish populations. Major points of discussion ■ The prevalence of cystic fibrosis in the Caucasian population is about 1:3000. ■ The prevalence of cystic fibrosis in the African American population is about 1:15,000. ■ The prevalence of cystic fibrosis in the Hispanic population is about 1:9000. ■ The prevalence of cystic fibrosis in the Asian population is about 1:35,000. ■ The prevalence of cystic fibrosis is higher in the Caucasian than in the African American, Hispanic, and Asian populations. ■ The prevalence of cystic fibrosis in the Ashkenazi Jewish population is about 1:2300 and is similar to the prevalence in the Caucasian population. ■ The prevalence of cystic fibrosis in the Native American population is believed to be about 1:10,000. 7. A. Measurement of immunoreactive trypsinogen and DNA analysis. Major points of discussion ■ There are a considerable number of false-positive immunoreactive trypsinogen results. A positive immunoreactive trypsinogen result triggers the performance of a DNA mutation analysis. ■ A negative immunoreactive trypsinogen result in a patient with cystic fibrosis, although rare, can be obtained and does not routinely trigger a DNA mutation analysis. If cystic fibrosis is suspected, the patient should undergo genetic testing or have a sweat chloride measurement. ■ In the nasal potential difference method, the active transport of ions, mainly sodium and chloride, across the respiratory epithelium generates a potential difference that can be measured. The abnormal sodium and chloride transport in the respiratory epithelia of patients with cystic fibrosis can be measured and used to diagnose patients with this disease. ■ Sweat conductivity and sweat osmolality are screening tests for cystic fibrosis. A positive result must be confirmed by sweat chloride measurements.2,13 8. A. Make the diagnosis of cystic fibrosis, as recommended by the Cystic Fibrosis Foundation. Major points of discussion ■ A positive immunoreactive trypsinogen result followed by detection of two disease-causing mutations is strongly suggestive of cystic fibrosis. In this case, an elevated sweat chloride result will confirm the diagnosis of cystic fibrosis. ■ A positive immunoreactive trypsinogen result followed by a negative DNA mutation result requires another blood sample to be taken 3 weeks later and the immunoreactive trypsinogen and DNA mutation assays repeated. ■ A negative immunoreactive trypsinogen result with a negative DNA mutation result in a second blood sample suggests that the positive immunoreactive trypsinogen result obtained in the first sample was a false-positive result. ■ Serum amylase, lipase, trypsin, chloride, and urine chloride testing are not recommended in the evaluation of cystic fibrosis.2,13 9. A. Sulfhydryl bonds. Major points of discussion ■ The sugar residues are linked by a 3′,5′-phosphodiester bond, in which a phosphate group links the 3′ carbon atom of one sugar to the 5′ carbon atom of the next sugar in the sugar-phosphate backbone. ■ The two strands of DNA are held together by hydrogen bonds to form a duplex. ■ Hydrogen bonding occurs between the laterally opposed complementary base pairs on the two strands of the DNA duplex. ■ The stability of the DNA double helix is maintained by hydrogen bonding between the A-T and G-C base pairs. ■ The individual hydrogen bonds are weak, but the two strands of the double helix are held together by many hydrogen bonds. ■ Hydrogen bonding also permits formation of DNA-RNA duplexes and double-stranded RNA. 10. A. It has a haploid copy number. Major points of discussion ■ The human genome is organized into 46 chromosomes in the nucleus. ■ Each human chromosome consists of a single, continuous DNA double helix. ■ The nuclear genome consists of 46 DNA molecules, totaling approximately 6 billion nucleotides. ■ Each nucleated cell in the body carries its own copy of the genome, which by current estimates contains approximately 20,000 protein-coding genes. ■ The genes are encoded in the DNA of the genome. The DNA is a double helix structure, which was elucidated by James Watson and Francis Crick in 1953. 11. A. Leber’s hereditary optic neuropathy (LHON). Major points of discussion ■ The only novel sequence in the duplicated mtDNA, compared with wild type, is at the boundary of the duplicated region, which is the same as the boundary present in the deleted DNA, indicating that the two mtDNAs (i.e., duplicated and deleted) originated through a common mechanism. ■ Far more patients harbor deleted mtDNA compared with duplicated mtDNA; because of this, and because it is technically difficult to differentiate duplicated mtDNA from deleted mtDNA, the importance of duplicated mtDNA in clinical disease has not been fully recognized. ■ Large-scale partial deletions of mtDNA have been well documented in patients with PEO and KSS. However, the size and location of the deletion, and the proportion of deleted DNA, differ among patients and do not appear to correlate with disease severity. ■ Interestingly, although different patients with KSS harbor different deleted mtDNAs, which cause the loss of different genes, all patients have fundamentally the same clinical presentation.12,25,33 12. A. Sanger cycle sequencing. Major points of discussion ■ Direct sequencing of the whole mitochondrial genome using Sanger sequencing to detect rearrangements is very labor intensive and time consuming. In addition, it cannot determine heteroplasmy level. Next-generation sequencing not only can accurately determine the break points of the deleted segment but it also can determine the heteroplasmy level. This methodology is not yet in routine use but may become the future method of choice for mtDNA analysis. ■ RFLP methodology, which refers to the difference between the sizes of fragments produced by a restriction enzyme, is a very useful technique to detect known mtDNA point mutations. This method has the advantage of being able to determine the heteroplasmy level of the mutation. ■ ARMS-PCR is accurate and convenient for detecting known point mutations in mtDNA. However, it cannot detect new mutations or determine heteroplasmy level. ■ Southern blotting is the method of choice for mtDNA rearrangement analysis. It can easily detect mtDNA duplications or deletions and can also estimate their level of heteroplasmy. The two major inherent disadvantages of this method are that it (1) it requires a relatively large amount of DNA (5 to 10 μg) and (2) has a long turnaround time (2 to 3 days).11,17,25 13. A. RFLP analysis. Major points of discussion ■ The mechanism causing mtDNA deletions is unclear. In muscle morphology, marked accumulation of mitochondria in cytochrome c (COX)-deficient fibers (COX-negative ragged red fibers) is the common finding. It appears that each individual fiber contains a clonal expansion of a single deletion event. ■ It has been reported that mutations in the heart/skeletal muscle isoform of the adenine nucleotide translocator (ANT1), which exchanges adenosine diphosphate and ATP across the mitochondrial inner membrane, causes mtDNA multiple deletions associated with cardiomyopathy. ■ Long-range PCR is commonly used to provide rapid and reliable exclusion of rearrangements in tissue. PCR amplification and electrophoresis of normal mtDNA produces a single large band on the gel. The appearance of additional smaller bands represents multiple mtDNA deletions. ■ Long-range PCR is very sensitive and may detect rearrangements that are not at clinically significant levels. For example, very low levels of deleted mtDNA, which cannot be detected by regular Southern blotting, are detected by long-range PCR. Therefore, it is crucial that the age of the patient is taken into account when interpreting the results. Further investigation is always recommended before issuing a final report on positive results.11,17,25 14. A. Mitochondrial tRNA lysine (MT-TK). Major points of discussion ■ Muscle and liver were originally reported to be affected by MDS, but now it is divided into three clinical categories: myopathic MDS (OMIM #609560), encephalomyopathic MDS (OMIM #612073 and OMIM #612075), and hepatocerebral MDS (OMIM #251880). ■ To date, mutations in at least nine genes have been associated with MDS. Four genes are involved in deoxyribonucleotide triphosphate (dNTP) metabolism: thymidine kinase 2 (TK2), associated with myopathic MDS, deoxyguanosine kinase (DGUOK), associated with hepatocerebral MDS, polymerase gamma (POLG) also with hepatocerebral MDS, and, more specifically, Alper syndrome; and succinate-CoA ligase, adenosine diphosphate (ADP)-forming β-subunit (SUCLA2), associated with encephalomyopathic MDS. ■ Human DNA polymerase is composed of two subunits, a 140-kDa catalytic subunit encoded by POLG on chromosome 15q25, and a 55-kDa accessory subunit encoded by POLG2 on chromosome 17q23-24. The catalytic subunit of mtDNA polymerase gamma, encoded by POLG1, is the only DNA polymerase found in mitochondria, which is responsible for DNA replication and repair. Mutations in POLG can cause mtDNA depletion or multiple mtDNA deletions. To date, more than 120 variants have been associated with different pathologic phenotypes (http://tools.niehs.nih.gov/polg/). ■ Because of the heterogeneous nature of its clinical phenotype, definitive diagnosis of MDS is a daunting task. Based on clinical presentation of the patient, one or more genes can be selected for sequencing. For example, if the presentation is predominantly myopathic, then TK2 sequencing may be considered first, whereas if the presentation is mainly hepatocerebral, sequencing DGOUK should be considered first. 15. A. Southern blot analysis with radiolabeled probes. Major points of discussion ■ mtDNA can also be quantified by PCR. Competitive PCR is one approach, which involves introduction of a serially diluted competitive template into the tissue extract. mtDNA quantitation is achieved by radiometric comparison of the relative amounts of the two products. ■ In recent years, the development of quantitative real-time PCR (qPCR) technology has significantly improved the estimation of mtDNA copy number in clinical specimens, such as blood and tissue. qPCR assays are extremely sensitive and can detect only a few copies, or even a single copy, of target DNA in a clinical specimen. ■ Two chemistries, TaqMan and SYBR green, are most commonly used. Assays based on TaqMan chemistry have the advantage of being capable of simultaneously quantifying more than one target in a sample. SYBR-based qPCR assays, although more convenient than TaqMan-based assays, are less accurate because SYBR green can bind to DNA molecules other than the target. Therefore, highly optimized PCR conditions are a prerequisite for successful application of this approach. ■ Two approaches are used to calculate mtDNA in qPCR assays. (1) The first calculation uses a standard curve, in which known amounts of DNA are used to construct the standard curve from which the copy number of the unknown sample is delineated. (2) Another approach uses relative quantification, in which the relative copy number of target DNA to a reference DNA, amplified simultaneously, is calculated. The advantage of the latter method is that it obviates the need for a standard curve. However, when the latter method is used, it is imperative to ensure that the efficiencies of target and reference amplification are very similar. 16. A. An adenine-to-guanine transition at nucleotide 3243 position (3243A> G) in mitochondrial tRNA Leu(UUR) (MTTL1). Major points of discussion ■ Morphologically, most ragged red fibers are COX positive, which is in contrast to other mitochondrial syndromes involving tRNA mutations that are devoid of COX activity. Moreover, ragged red fibers are most visible in the vasculature, which shows an intense histochemical reactivity for succinate dehydrogenase. ■ MI-PEO, which is phenotypically similar to the sporadic form of the disease, is also associated with the 3243A > G mutation. It has also been associated with mutations in other mitochondrial tRNAs, including tRNA asparagine, tRNA lysine, and tRNA tyrosine. Unlike in MELAS, the ragged red fibers found in MI-PEO are COX negative. ■ MERRF is characterized by myoclonus, generalized seizures, ataxia, and myopathy with ragged red fibers. Again, unlike in MELAS, these ragged red fibers are devoid of COX activity. The most common mutation associated with MERRF is an A > G transition at nucleotide 8344 (8344A > G) in tRNA lysine, which is found in most cases. Another mutation in the same gene (8356T > C) is also a rare cause of the disease. The 8344A > G mutation can be reliably detected by analyzing blood samples. ■ Mutations in the MTATP6 gene at position 8993, predominantly a T > G transversion, account for more than 50% of NARP cases. Another mutation at the same position associated with NARP is a T > C transition, converting leucine to proline at position 156. A high load of these two mutations (8993T > G/C) can also cause maternally inherited Leigh disease.10,25,34 17. A. Complex I subunit ND4 gene. Major points of discussion ■ Mitochondrial disorders, such as NARP, maternally inherited LS, or LHON, are caused by mutations in different subunits of five complexes. ■ Although mutations in mitochondrial tRNA genes affect the synthesis of all mitochondrially encoded proteins, mutations in different tRNA genes produce different clinical phenotypes. For example, mutations in tRNA lysine cause MERRF, whereas mutations in tRNA isoleucine cause cardiomyopathy. ■ Mutations in rRNA genes, which also affect general protein synthesis, cause deafness or cardiomyopathy. The A > G transition mutation in the 12S rRNA gene at nt-1555 causes maternally inherited deafness but usually through interaction with aminoglycoside drugs, such as gentamycin, kanamycin, and streptomycin. ■ Although a relatively large number of mtDNA mutations have been associated with LHON, only four mutations, all located in the complex I gene, are considered primary, pathogenic mutations.9,11 18. A. All respiratory chain proteins are encoded by mtDNA. Major points of discussion ■ Human mtDNA is a 16-kb circular, double-stranded molecule that contains 37 genes: 2 rRNA, 22 tRNA, and 13 structural genes encoding subunits of the mitochondrial respiratory chain. ■ All eukaryotic cells contain many copies of mtDNA, which, at division, distribute randomly among daughter cells. In normal tissues, all copies of mtDNA are identical (homoplasmy). Disease-causing mutations usually affect some, but not all, mtDNA. Therefore, in this case, cells and tissues harbor both wild-type and mutant mtDNA, a condition known as heteroplasmy. A minimal amount of mutant mtDNA must be present before there is a manifestation of respiratory dysfunction and appearance of clinical symptoms; this is known as the threshold effect. ■ The threshold in tissues such as brain, muscle, heart, and endocrine glands with high oxidative energy demand is low compared with other tissues. Therefore, these tissues are especially vulnerable to respiratory chain dysfunction. ■ Because mitochondria are randomly distributed at cell division, the proportion of mutant and wild-type mtDNA in daughter cells may change, and the phenotype may change accordingly. This phenomenon, called mitotic segregation, explains the age-related variability of clinical features frequently observed in mtDNA-related disorders.9,11,18 19. A. Thymidine phosphorylase (TP) activity in peripheral blood leukocytes. Major points of discussion ■ Neurogenic changes and the presence of ragged red fibers and occasional COX-deficient fibers observed on muscle morphology provide further evidence for neuropathy and mitochondrial myopathy. Abnormalities of intestinal smooth muscle and the enteric nervous system are common findings in histologic studies of the gastrointestinal system. ■ MNGIE, which is mapped to chromosome 22q13.32, is caused by mutations in the TP gene. The disease is transmitted in a recessive pattern, and affected individuals are either homozygotes or compound heterozygotes. ■ Human TP catabolizes the dephosphorylation of thymidine to thymine. Its expression level and activity are significantly increased in certain tumors. TP is widely expressed in human tissues, including the brain, peripheral nerves, spleen, lung, bladder, and gastrointestinal system. Paradoxically, skeletal muscle has no TP activity despite the fact that this tissue is usually affected by MNGIE. ■ Various mitochondrial abnormalities, such as decreased mitochondrial respiratory chain activity, ragged red fibers, and multiple deletions, are common findings. However, these are inconsistent and nonspecific. The definitive diagnosis is made by quantifying TP activity in peripheral blood leukocytes and determining plasma thymidine levels because TP levels in leukocytes decrease by more than 90% in MNGIE and plasma thymidine levels are about 50-fold higher than normal. Molecular studies in TP deficiency include sequencing all 10 exons of the gene to search for known and novel mutations.15,20,25 20. A. Autosomal recessive. Major points of discussion ■ Of more than 1500 different proteins found in mitochondria, only 13 are encoded by mtDNA. The rest are encoded by nDNA and imported from the cytoplasm. ■ Mutations in mtDNA lead to respiratory chain diseases. Many of these disorders involve brain and skeletal muscle and are also known as mitochondrial encephalomyopathies. Because the respiratory chain is under dual genetic control, the proteins that make up the respiratory chain involve both Mendelian and mitochondrial genetics. ■ The prevalence of mitochondrial disease is about 10 to 15 cases per 100,000 individuals, which is similar to other neurologic disorders such as ALS and the muscular dystrophies. ■ Mitochondrial genetics differs from Mendelian genetics in three major aspects: maternal inheritance, heteroplasmy, and mitotic segregation. Although paternal mtDNA enters the oocyte at fertilization, these molecules are destroyed during early development, and only mitochondria contributed by oocytes repopulate the embryo. Therefore, if a mother carries an mtDNA point mutation, she will pass it on to all of her children.10,11,14 21. A. Mutation. Major points of discussion ■ Alleles are alternative forms of the same gene or DNA sequence. ■ The genotype refers to the genetic constitution of an individual or the alleles present at one locus. ■ Mutation is a change in DNA sequence, including base-pair substitution, insertion, and deletion. Mutations originate by one of two basic mechanisms: errors introduced during DNA replication or failure to repair after DNA damage. ■ A mutation may lead to complete loss of expression of the gene or to formation of a variant protein with altered properties. ■ Some mutations are not deleterious and have no phenotypic effect, either because the change does not alter the amino acid sequence of the polypeptide or, even if it does, the resulting change in the encoded amino acid sequence does not alter the functional properties of the protein. Therefore, not all mutations have clinical consequences. During evolution, the influx of nucleotide variation ensures genetic diversity and individuality. ■ Many deleterious mutations that lead to genetic disease are rare variants; however, there is no simple correlation between allele frequency and the effect of the allele on health. Many rare variants appear to have no deleterious effect, whereas some variants common enough to be polymorphisms are known to predispose to serious diseases. 22a. A. This is a mitochondrial disease. The mother is a healthy carrier, and the disease manifests in the children differentially because of heteroplasmy. 22b. A. You recommend comparative genomic hybridization microarray studies to identify large structural chromosomal defects. 22c. A. The family should be advised that, because they already have three affected children, they have a good chance of producing a healthy child. 22d. A. It is still experimental, whereas Sanger sequencing is currently available. 22e. A. It refers to a process in which sequence changes known to cause specific disease phenotypes are detected. 22f. A. Full exome sequencing is the best approach because the number of genes implicated in both microcephaly and deafness is large, and there are numerous, as yet unidentified, genes that can cause these phenotypes. 22g. A. Both parents and all three children should be sequenced. 22h. A. The patients must be older than 18 years. 22i. A. All known mutations (i.e., previously detected in other “healthy” individuals) should be excluded from the search for the cause of the disease. 22j. A. Online Mendelian Inheritance in Man (OMIM): http://www.ncbi.nlm.nih.gov/omim 22k. A. The exome sequencing must be repeated. 22l. A. Following full genome amplification, the exome sequencing has to be repeated on cells derived from all embryos. Major points of discussion ■ Differences between Sanger sequencing and high-throughput (Next-Gen) sequencing: • The principle of the currently most successful Next-Gen sequencing instrument, from Illumina, is based on reversible termination of synthesis. Fluorescently labeled nucleotides are added in each extension cycle. No unlabeled nucleotides are present. Incorporation of specific nucleotides is detected simultaneously based on the color of the hundreds of thousands of extended products corresponding to different sequencing reactions. The fluorescent tag interfering with extension is then removed, and a new cycle of extension begins. The Roche and Applied Biosystems instruments use incorporation of unlabeled single nucleotides added one at a time in successive cycles. Incorporation is detected by coupled photochemistry in the Roche instrument (pyrosequencing) and by pH measurements in the Applied Biosystems instrument. ■ Methods of sequence capture: • Site-specific amplification-based methods are more suitable for smaller (e.g., < 200 kb) genomic regions, whereas hybridization-based methods are more appropriate for capturing regions in the 100-kb to 100-Mb range. ■ SNP filtering. Full exome sequencing, depending on the exact capture reagent used and the depth of sequencing applied, can result in identifying 40,000 to 100,000 SNPs in a given sample. The data analysis software allows for filtering the mutations found based on various criteria. The easiest approach simply screens the SNP list generated by the analysis software for known disease-causing mutations. Next, one can focus on mutations in genes that have been previously implicated in disease pathogenesis. Among these mutations, one must prioritize mutations in exon-intron junctions that result in altered splicing, nonsense mutations (introduction of a STOP codon), and nonconserved missense mutations. The latter category is the most problematic. Algorithms have been developed to predict the expected severity of a given missense mutation. These rely on the chemical nature of the amino acid changed, its evolutionary conservation, and its position in the 3D structure of the protein (http://www.gen2phen.org/wiki/tools-predicting-overal-functional-consequences-snps). Such predictions, although generally correct, must be applied with caution for any specific mutation. To assign pathogenicity to a previously unseen missense mutation requires either documenting its higher prevalence in an ethnically matched control population or following its segregation with the disease phenotype in the family. For dominant mutations, which result in a phenotype even in the presence of a single mutant allele, this is difficult because it is rare to have a sufficient number of family members that can be tested. Establishing pathogenicity for loss-of-function mutations is easier. In such cases, both alleles must have the mutation to result in a phenotype. The number of disruptive SNPs that are present in homozygous form is much lower. If one removes genes, such as olfactory receptor genes and genes responsible for the immunologic diversity, only a few such mutations remain to evaluate. ■ The World Wide Web is replete with extremely useful websites relating to human genetic diversity and disease-causing mutations. Some of the most important entry points into this web of resources are listed below. ■ Online Mendelian Inheritance in Man (OMIM): http://www.ncbi.nlm.nih.gov/omim • UCSC web browser: http://genome.ucsc.edu • dbSNP database: http://www.ncbi.nlm.nih.gov/projects/SNP • 1000 genome database: http://www.1000genomes.org • Leiden database: http://www.dmd.nl ■ Next-Gen sequencing will have a profound effect on medicine. It is probable that the availability of this technology in the clinic will result in the generation of hundreds of thousands of datasets. Although screening these datasets for known pathogenic mutations will provide genetic diagnosis for a significant portion of the individuals tested, it will also result in an explosion of disease-causing mutation discovery and, ultimately, better understanding of human biology. This raises important questions about the use of genomic information for improving our analytical abilities by cataloging human genetic diversity while maintaining individual privacy. Additional ethical questions relating to reporting incidental findings must be addressed and continuously reassessed as our understanding of the genetic predictors of human disease improves.19,21,31 23. A. Denaturation, digestion, detection. Major points of discussion ■ PCR is a standard method for analyzing DNA and RNA samples in research and clinical laboratories. ■ This procedure produces millions of copies of a short segment of DNA through repeated cycles of (1) denaturation, (2) annealing, and (3) amplification. ■ It is an alternative to cloning for generating millions of copies of a sequence of DNA. It is extremely sensitive (i.e., allows amplification of minute amounts of target DNA, including DNA from a single cell), rapid, and robust (e.g., can be used with DNA isolated from degraded tissues and formalin-fixed samples). ■ PCR (1) is used to generate a sufficient quantity of DNA to perform a test (e.g., sequence analysis, mutation scanning) or (2) may be a test by itself (e.g., allele-specific amplification, trinucleotide repeat quantification). ■ A wide variety of PCR approaches are used for specific applications, including RT-PCR and real-time PCR (also called quantitative PCR or qPCR). 24. A. Choose primers that show significant complementarity to each other, especially at the 3′ ends. Major points of discussion ■ The design of PCR primers to amplify human genomic targets has been simplified with the completion of the human genome project. Commonly available freeware, such as Primer3, which can design primers according to specifications, and Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/), which is based on this algorithm, can further determine the specificity of the primers. ■ Primers are generally chosen to have a melting temperature of between 52°C and 65°C (ideally between 52°C and 58°C), to be 18 to 22 nucleotides in length, and to have a GC content of approximately 60% to 70%. ■ The primers should not show self-complementarity, or self-priming; they should also not be complementary to each other, especially at their 3′ ends and should, ideally, have a similar Tm. ■ The presence of pseudogenes, gene-family members with high homology to the target, and gene duplications can each complicate efforts to design specific primers and may require testing of multiple primer pairs. ■ Primer–binding-site polymorphisms, especially at the 3′ end of the primer, can result in failure to amplify an allele, with resulting failure to detect any mutation(s) that may be on the same allele. A web-based service to check for this is available from the National Genetics Reference Laboratory (NGRL) at Manchester (www.ngrl.org.uk/Manchester/).29 25. A. When setting up a PCR, one should always add a template to the no-DNA control first to avoid accidentally contaminating it. Major points of discussion ■ Physical controls include separate pre- and post-PCR areas with separate sets of instruments. Procedural approaches to minimizing contamination include ultraviolet irradiation of equipment and surfaces, cleaning of surfaces with bleach or other nucleic acid–damaging agents, autoclaving when possible, the use of single-use aliquots of reagents, and the use of disposable containers when possible. ■ An enzymatic approach to preventing cross-contamination is the use of dUTP in PCR to partially substitute for thymidine. The resulting double-stranded DNA product will contain uracil and can be treated with uracil-N-glycosylase so the PCR product will not be able to act as a template. ■ In addition, the use of proper negative controls is necessary to detect contamination. This includes using a template control (or multiple controls when testing for microorganisms), which must always be set up and capped last to maximize the chances of detecting contamination occurring at any time during reaction setup. ■ When performing RT-PCR, a no-RT control is needed to detect DNA amplification. In addition, a separate negative control is required for the PCR step to detect DNA contamination occurring at this step. The use of single-tube RT-PCR, whenever possible, obviates this issue.29 26a. A. Only a maternal allele is present; this is consistent with a diagnosis of Angelman syndrome. 26b. A. Prader-Willi syndrome and Angelman syndrome are caused by different mutations in the same gene. 26c. A. Contrary to classic Mendelian genetics, the parent of origin affects the expression of most genes in higher mammals. Major points of discussion ■ Maternally imprinted genes are paternally expressed, and paternally imprinted genes are maternally expressed. ■ The clinically distinct Prader-Willi and Angelman syndromes involve the same locus on 15q11-13; a deletion of this locus on the paternal chromosome causes Prader-Willi syndrome and on the maternal chromosome causes Angelman syndrome. ■ The imprinted domain in this locus contains a number of centromeric genes that are paternally expressed and maternally imprinted, and two telomeric genes that are maternally expressed, including UBE3A. ■ Approximately 70% of cases of Prader-Willi syndrome and Angelman syndrome are caused by a de novo large deletion in this locus: Prader-Willi syndrome in the paternal and Angelman syndrome in the maternal allele. About 25% to 30% of cases of Prader-Willi syndrome are caused by maternal uniparental disomy and 1% to 2% of cases of Angelman syndrome by paternal uniparental disomy. These have a recurrence rate of less than 1%. ■ Approximately 1% of cases of Prader-Willi syndrome and 2% to 4% of cases of Angelman syndrome are caused by an imprinting defect. When these are the result of an inherited imprinting center deletion (rare), the recurrence risk in siblings is 50%. ■ Approximately 2% to 5% of cases of Angelman syndrome are caused by inactivating mutations of UBE3A in a maternal allele; if this mutation is present in the mother, the recurrence risk in siblings is 50%. ■ The SNRPN promoter is methylated and inactive in the maternal allele and unmethylated and active in the paternal allele; this is the basis for Southern blot testing with a methylation-sensitive enzyme and an SNRPN probe, or methylation-specific/-sensitive PCR testing. Additional tests include testing for UBE3A mutations and testing for small interstitial deletions of the imprinting center. The latter two are important because of the high recurrence risk in siblings. Less than 1% of cases of Prader-Willi syndrome, but almost 15% to 20% of cases of Angelman’s syndrome, have no identifiable molecular cause. ■ Other genetic conditions that involve imprinted loci include Beckwith-Wiedemann syndrome, an overgrowth syndrome involving imprinted loci on 11p15.5; Russell-Silver syndrome with growth retardation, relative macrocephaly, and a “triangular face,” associated with maternal uniparental disomy of chromosome 7 or hypomethylation of 11p15.5 and/or other imprinted loci; and transient neonatal diabetes mellitus type 1, involving imprinted genes on chromosome 6q24. ■ Imprinted loci have been identified on human chromosomes 6, 7, 11, 14, and 15; about 60 imprinted human genes are known.7,35 27. A. RNA polymerase I, which synthesizes most messenger RNAs (mRNAs), is recruited to promoters by bound transcription factors. Major points of discussion ■ RNA polymerase II recognizes three promoter sites: • The GC box, usually for housekeeping genes. • The CAAT box, usually in the -80 position. ■ Primary transcripts of most RNAs have multiple introns. In DNA, introns are characterized by a 5′GT and a 3′AG, which are splice donor and acceptor sites, respectively (GU and AG in RNA, respectively). Adjacent sequences are also highly conserved. In addition, there is a conserved branch site sequence toward the 3′ end of the intron, which is situated less than 20 nucleotides from a polypyrimidine tract separated by two bases from the splice acceptor AG. In a minority of cases, the splice donor may be an AU and the acceptor an AC. ■ RNA transcripts undergo 5′ capping with 7-methylguanine and polyadenylation at the 3′ end. The 3′ polyadenylation site is usually separated from the stop codon by a sequence of variable length, called the 3′-untranslated region (3′UTR). There may be a sequence upstream of the first translated codon, which is called the 5′UTR. The 3′UTR may contain sequences complementary to one or more miRNAs, which may regulate mRNA translation. ■ Polypeptides are encoded by mRNA, with each amino acid encoded by a triplet codon. The first translated codon is invariably an ATG and encodes a methionine. The last coding triplet is followed by one of three stop codons, which are followed by the 3′UTR. This translation takes place on ribosomes.30 28. A. Genomic DNA is a better template to evaluate the presence and effect of deep intronic variants, as these are not present in mRNA or complementary DNA (cDNA). Major points of discussion ■ cDNA sequencing, performed after reverse transcription of RNA, will detect the effect of intronic mutations on the final transcript. However, in addition to the general lability of RNA, cDNA sequencing can only be performed on cells that express the gene. In addition, the phenomenon of “nonsense-mediated decay” should be guarded against. RNAs that have nonsense mutations within the first exon are released early from the ribosome and are degraded, resulting in sequencing of only the normal allele. ■ With modern methods, DNA base calling is performed by software. Sequences should be of high quality, and to ensure both sensitivity and specificity, sequencing both strands of each segment is recommended. ■ When a missense mutation is found, interpretive reports should indicate the certainty that the mutation is or is not deleterious. The American College of Medical Genetics and Genomics recommends a set of interpretive categories. ■ The American College of Medical Genetics and Genomics categories include the following: 1. Known sequence variation that is a recognized cause of the disorder. 2. A previously unreported sequence variation that is expected to cause the disorder (e.g., mutations of the initiation codon; splice junctions; frame-shift mutations). 3. A previously unreported sequence variant that may or may not cause the condition (e.g., nonconservative substitution of an evolutionarily conserved residue). 4. A previously unreported variant that is probably not causative of the disease. 5. A previously reported variation that is known not to be causative of the disease. 6. A variation that is not expected to be causative of a disease but is associated with a clinical syndrome in population studies. ■ The sequence variation should be related to a reference sequence in a public database such as GenBank. If available, the reference sequence should be obtained from the National Center for Biotechnology Information Reference Sequence Database. ■ When two variants are found in a gene, it cannot be assumed that they are on separate alleles. Family testing should be recommended. When performing family testing, the risk for detection of unsuspected nonpaternity should be considered.23 29. A. Fragile X. Major points of discussion ■ A rapid progressive variant of HD, which presents with rigidity and intellectual decline before the age of 20 years, occurs in about 5% of affected patients. ■ The HD gene is large (11 kb), contains 67 exons, and is on the short arm of chromosome 4 (4p16.3). The encoded protein, huntingtin (htt), consists of 3136 amino acid residues with a molecular weight of 350 kDa. The mutation associated with clinical manifestations of the disease is expansion of an unstable CAG repeat encoding a polyglutamine tract in the first exon of the htt gene. ■ The range of repeat length in the unaffected population varies from 6 to 35 repeats. ■ Normal alleles are defined as alleles with no more than 26 CAG repeats. ■ Alleles with 27 to 35 repeats are defined as mutable normal. These alleles have not been convincingly associated with the HD phenotype, but they can be meiotically unstable in sperm, and pathologic expansion of the paternally derived allele in this size range has been described. ■ HD alleles with reduced penetrance are defined as alleles with 36 to 39 CAG repeats. ■ HD alleles with full penetrance are defined as alleles with at least 40 CAG repeats. ■ CAG-repeat expansion mutations account for more than 99% of cases of HD. Therefore, tests that effectively detect and measure the CAG repeat in the htt gene have more than 99% sensitivity. ■ With regard to specificity, the absence of HD pathology has not been documented in any individual with an HD allele size of at least 40 repeats who died, disease-free, after living up to or past the normal life expectancy. Therefore, positive results are 100% specific.1,16,27 30a. A. Only one allele is seen on the capillary electrophoresis image. Therefore, this must be from a male. 30b. A. The results of the capillary electrophoresis and Southern blot test, taken together, are consistent with a normal male. Major points of discussion ■ Definitions: normal alleles (~ 5 to 44, most commonly 29 or 30, repeats); intermediate or “gray-zone” alleles (~ 45 to 54 repeats) carry no risk for expansion to full mutations in the next generation, but future generations or distant relatives may have expansions; premutations (~ 55 to ~ 200 to 230 repeats) carry an increased risk for expansion to full mutations with maternal transmission; full mutations (> 200 or > 230 repeats) are usually fully hypermethylated. ■ PCR with primers flanking the repeat can measure the number of repeats; very large repeats may fail to amplify. Southern blot analysis following restriction enzyme digestion with two enzymes (one methylation-sensitive) identifies expansions and the methylation status of alleles; this is the gold standard. ■ In approximately 40% of women who show a single band by PCR with flanking primers, a “repeat-primed” PCR that binds to the CGG repeats and can detect the presence of expanded alleles that fail to amplify with flanking primers will distinguish between true homozygotes and the rare woman who carries a very large second allele. ■ “Size mosaicism,” or the presence of alleles of different sizes, occurs most commonly in males with (unmethylated) premutation and (methylated) full-mutation alleles; this can complicate analysis in rare cases. Therefore, a very clean background on the Southern blot test is critically important. ■ Women with premutation alleles have an increased risk for premature ovarian failure or early menopause. This does not change the protein sequence and requires transcription of the allele (full mutations are not associated with this risk). This implies RNA toxicity. Similarly, males (and some females) with premutation alleles develop FXTAS during adulthood. ■ Prenatal testing is available for this disorder. When testing chorionic villous samples, methylation analysis of the allele, to distinguish premutations from full mutations, can be misleading because of absent/incomplete methylation of extraembryonic tissue. ■ The FMRP protein regulates the translation of proteins in polyribosomes and at dendrites. Deregulated translation of proteins in neurons is the basis of the phenotype expressed in this disorder. Some of the targets regulated by FMRP are genes known to be associated with autism spectrum disorder.22,26 31. A. CYP2C19. Major points of discussion ■ Clopidogrel is a prodrug and is converted by CYP2C19 to its active metabolite, which then inhibits the platelet P2Y12 (i.e., ADP) receptor. ■ CYP2C19 loss-of-function alleles (e.g., CYP2C19*2) appear to be associated with higher rates of recurrent cardiovascular events in patients receiving clopidogrel. If the patient is a poor metabolizer, an alternate P2Y12 inhibitor may be indicated. ■ Pharmacogenomic testing is not recommended for all patients receiving clopidogrel. ■ Testing is recommended when there is recurrent stent thrombosis despite compliance with taking medications and when dual-antiplatelet therapy is otherwise indicated. 32. A. Test for BCR- ABL1 kinase domain mutations. Major points of discussion ■ In a real-time PCR assay, a positive reaction is detected by accumulation of a fluorescent signal. ■ The cycle threshold (Ct) is defined as the number of cycles needed for the fluorescent signal to cross a defined threshold. Therefore, Ct levels are inversely proportional to the amount of target nucleic acid in the sample (i.e., the lower the Ct, the higher the amount of target nucleic acid in the sample). ■ A major molecular response (MMR) in CML is a 3-log reduction (i.e., 1000 times) in the level of BCR-ABL1 transcript. Because each cycle approximately doubles the transcript, an increase in the Ct by approximately 10 cycles (210 = 1024) would be approximately a 3-log reduction. ■ BCR-ABL1 kinase domain mutation analysis is recommended both in the case of MMR with increasing quantitative PCR on serial PCR and an increasing quantitative PCR that crosses a defined threshold. It should also be considered if MMR is not achieved.28
Molecular Pathology
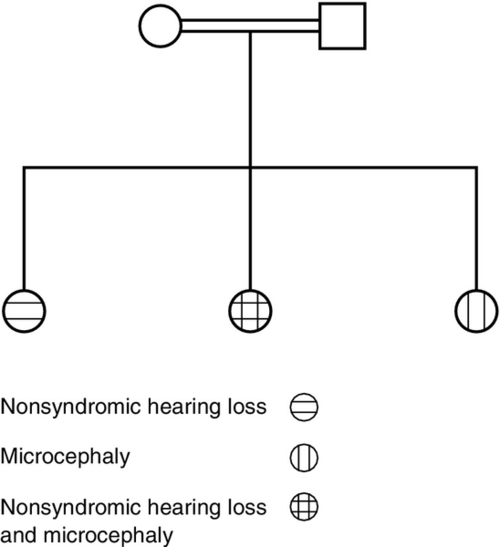
Cystic fibrosis screening results obtained from an otherwise asymptomatic 34-year-old pregnant woman show compound heterozygosity for two mutations in the CFTR gene: delF08 (c.F508del) and R117H; in addition, her two intron 8 alleles are 7T/9T. These results are confirmed by repeat testing on the original blood tube. Use this scenario to answer the following two questions.


B. DNA → RNA → protein.
C. RNA → protein → DNA.
D. RNA → DNA → protein.
E. Protein → RNA → DNA.
Rationale: DNA is transcribed into RNA, and the RNA is translated into protein.
Rationale: In this instance, the 7T allele is almost certainly in linkage with the R117H mutation.
B. Ask for another blood sample from the patient because the current result “does not make sense.”
Rationale: This result can be seen in a 34-year-old. The CFTR protein containing the R117H mutation will have some function.
C. Test the patient’s blood relatives to determine whether the 7T variant is on the same allele as the R117H mutation.
Rationale: This is not necessary because in this instance the 7T allele is almost certainly associated with the R117H mutation.
D. Schedule the patient for a chorionic villous sampling to ensure that the fetus is not affected.
Rationale: This is premature because it would depend on whether the father is a cystic fibrosis carrier.
E. Screen the patient’s reproductive partner for CFTR gene mutations.
Rationale: This patient is at least a cystic fibrosis carrier, and she might develop lung disease later in life. However, the most important step is to ensure that the father is not a carrier.
Rationale: Although Caucasians are likely to have the highest carrier rate, the standard panel has the highest detection rate among Caucasians.
B. Lowest if the father is an Ashkenazi Jew because of the high detection rate among Ashkenazi Jews.
Rationale: The detection rate of the standard American College of Medical Genetics panel is even higher among Ashkenazi Jews.
C. Highest if the father is of Hispanic origin because of the combination of a high carrier rate and a low detection rate among Hispanics.
Rationale: The residual risk is lower among Hispanics than East Asians.
D. Lowest if the father is of East Asian origin because of the low carrier rate among Asians.
Rationale: Asians have the highest residual risk because of the low detection rate.
E. Can be reduced significantly if the father is of East Asian origin by using an extended panel to include East Asian–specific mutations.
Rationale: Extended panels do not help with East Asian populations because of the high percentage of private mutations.
Rationale: This is not an ideal result. The presence of a high proportion of recipient DNA 1 year after transplantation would be indicative of either graft failure or relapse of the acute leukemia.
B. The results are concerning because of the high proportion of recipient DNA in the sample.
Rationale: The presence of a high proportion of recipient DNA is indicative of either graft failure or relapse of the acute leukemia.
C. The results are concerning because of the low proportion of recipient DNA in the sample.
Rationale: Recipient DNA is present in a high proportion in this sample, and as such, is very concerning. The presence of a high proportion of recipient DNA is indicative of either graft failure or relapse of the acute leukemia.
D. The results are concerning because of the high proportion of donor DNA in the sample.
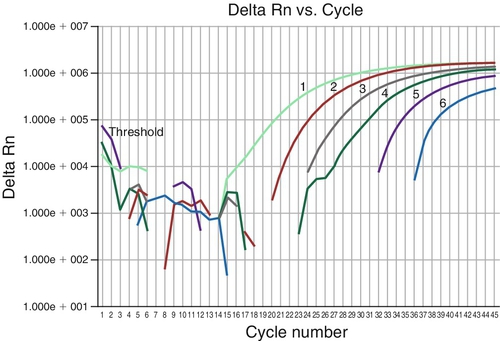
Rationale: A high proportion of donor DNA in the sample would be a reassuring finding.
E. This is not the appropriate test to perform in this setting.
Rationale: This is the standard test performed in this type of post-transplantation setting.
Rationale: The ΔF508 deletion is found in approximately 72% of the Caucasian population in the United States.
B. African Americans.
Rationale: The ΔF508 deletion is found in approximately 44% of the African American population.
C. Asians.
Rationale:The ΔF508 deletion is found in approximately 39% of the Asian American population.
D. Ashkenazi Jews.
Rationale:The ΔF508 deletion is found in approximately 31% of the Ashkenazi Jewish population.
E. Hispanics.
Rationale: The ΔF508 deletion is found in approximately 54% of the Hispanic population.
B. Highest in both the Caucasian and African American populations.
C. Highest in both the Caucasian and Hispanic populations.
D. Highest in both the Caucasian and Asian populations.
E. Highest in both the Caucasian and Native American populations.
Rationale: Caucasian and Ashkenazi Jewish populations have the highest prevalence cystic fibrosis.
Rationale: This is currently the most common procedure used to screen newborns for cystic fibrosis in the United States.
B. Measurement of both immunoreactive trypsinogen and pancreatitis–associated protein.
Rationale: This procedure is used in Europe for screening newborns for cystic fibrosis.
C. Measurement of both immunoreactive trypsinogen and serum amylase.
Rationale: Serum amylase, a marker for pancreatitis, is not used to screen for cystic fibrosis.
D. Measurement of both immunoreactive trypsinogen and serum chloride.
Rationale: Serum chloride is not used to screen for cystic fibrosis.
E. Measurement of both immunoreactive trypsinogen and nasal potential difference.
Rationale: Measurement of nasal potential difference is a manual, time-consuming test that can be used to diagnose cystic fibrosis. However, it is not a screening procedure.
Rationale: This result cannot be used to diagnose cystic fibrosis.
B. Perform additional genetic and sweat chloride testing to exclude or confirm the diagnosis of cystic fibrosis.
Rationale: This is the recommended approach.
C. Repeat the immunoreactive trypsinogen and DNA assays to confirm the abnormal results.
Rationale: Immunoreactive trypsinogen and DNA testing are usually not repeated.
D. Measure serum and urine chloride to exclude or confirm the diagnosis of cystic fibrosis.
Rationale: These tests are not used to assess patients for cystic fibrosis.
E. Measure serum amylase, lipase, and trypsin to exclude or confirm the diagnosis of cystic fibrosis.
Rationale: These tests are not used to assess patients for cystic fibrosis.
Rationale: There are no sulfhydryl (-SH) groups in the DNA double helix. In proteins, the disulfide bonds are formed between the thiol groups of cysteine residues.
B. Hydrogen bonds.
Rationale: Two strands of the DNA are held together by hydrogen bonds to form a duplex.
C. Phosphodiester bonds.
Rationale: The 3′,5′-phosphodiester bonds link the adjacent nucleotides in a DNA polynucleotide chain.
D. Nitrogen bonds.
Rationale: These are not involved in maintaining the DNA double helix.
E. Carbon bonds.
Rationale: Carbon bonds do not hold together the two strands of the double helix.
Rationale: Except for the gametes (which are haploid), all nucleated cells are diploid.
B. Nucleated cells contain 22 chromosomes.
Rationale: The number of chromosomes in a nucleated cell is 46, which is double the number found in the gametes (23 chromosomes).
C. Consists of DNA, which has a circular structure.
Rationale: The human genome consists of DNA, which has a double helix structure.
D. Contains 3 × 109 base pairs of DNA.
Rationale: This is the correct answer.
E. Contains 100,000 protein coding genes.
Rationale: The number of protein-coding genes in the human genome is approximately 20,000.
Rationale: LHON is most often caused by missense point mutations in genes encoding complex I subunits.
B. Mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS).
Rationale: To date, all cases of MELAS have been reported to be caused by point mutations in several mitochondrial tRNA genes or genes encoding subunits of the respiratory chain.
C. Myoclonus epilepsy and ragged red fibers (MERRF).
Rationale: MERRF is most often caused by mutations in mitochondrial lysine tRNA.
D. Kearns-Sayre syndrome (KSS).
Rationale: KSS is caused by a single deletion in mitochondrial DNA.
E. Neuropathy, ataxia, and retinitis pigmentosa (NARP).
Rationale: NARP is associated predominantly with a point mutation in the ATPASE6 gene.
Rationale: Direct sequencing of mtDNA is rarely used for detecting mtDNA rearrangements.
B. Restriction fragment-length polymorphism (RFLP) analysis.
Rationale: RFLP analysis is commonly used to detect mtDNA point mutations.
C. Amplification refractory mutation system–PCR (ARMS-PCR).
Rationale: ARMS-PCR can only be used to detect known mutations.
D. Real-time PCR.
Rationale: In the context of mtDNA analysis, real-time PCR is used to determine mtDNA copy number.
E. Southern blot analysis.
Rationale: Southern blot analysis is regarded as the gold standard for detecting mtDNA rearrangements.
Rationale: RFLP analysis is best used to detect known point mutations in mtDNA.
B. Southern blotting with a radiolabeled probe.
Rationale: Southern blots needs large amounts of DNA (5 μg or more).
C. Long-range PCR and gel electrophoresis.
Rationale: mtDNA multiple deletions can be quickly detected by long-range PCR on small biopsy specimens.
D. Sequencing by capillary electrophoresis.
Rationale: Multiple deletions are extremely difficult to detect by conventional sequencing.
E. Single-stranded conformation polymorphism (SSCP).
Rationale: SSCP can best detect nucleotide variations in small PCR-amplified fragments and should not be used to detect multiple deletions in mtDNA.
Rationale: Mutations in MT-TK are associated with MERRF syndrome.
B. Mitochondrial tRNA leucine 1 (MT-TL1).
Rationale: Mutations in MT-TL1 most commonly cause MELAS.
C. Mitochondrial tRNA tryptophan (MT-TW).
Rationale: Mutations in MT-TW have been associated with dementia and chorea (DEMCHO) but not depletion.
D. Mitochondrial tRNA isoleucine (MT-TI).
Rationale: Mutations in MT-TI are most commonly associated with cardiomyopathy.
E. mtDNA polymerase gamma (POLG).
Rationale: POLG is one of the nine genes with mutations that have been reported to cause mtDNA depletion.8,17,32
Rationale: Southern blot with radiolabeled probe detection is valuable for evaluating mtDNA rearrangements, such as deletions or duplications.
B. Southern blot analysis with fluorescence detection.
Rationale: Southern blot analysis with a fluorescence detection system is very sensitive for qualitative, but not quantitative, evaluation of mtDNA.
C. Real-time PCR with TaqMan probes.
Rationale: Real-time PCR accompanied by TaqMan probe technology is highly sensitive and accurate and is currently available for determining mtDNA copy number.
D. Sanger sequencing of the whole mitochondrial genome.
Rationale: Direct sequencing of mtDNA detects sequence variation but cannot determine mtDNA copy number.
E. Next-generation sequencing of the whole mitochondrial genome.
Rationale: Next-generation sequencing of the mitochondrial genome can determine any base-pair variation with a high level of accuracy but it does not determine copy number.
Rationale: This mutation is the most common cause of MELAS. It is also associated with MI-PEO and MIDD.
B. An adenine-to-guanine transition at nucleotide 8344 position (8344A> G) in mitochondrial tRNA Lys (MTTK).
Rationale: This mutation is the most common cause of MERRF but has not been associated with MELAS, MI-PEO, or MIDD.
C. A thymidine-to-cytosine transition at nucleotide 8993 position (8993T > C) in mitochondrial ATPase 6 (MTATP6).
Rationale: This mutation is the most common cause of NARP but has not been associated with MELAS, PEO, or MIDD.
D. A thymidine-to-cytosine transition at nucleotide 9176 position (9176T > C) in mitochondrial ATPase 6 (MTATP6).
Rationale: This mutation, similar to 8993T > C, is associated with NARP (i.e., maternally inherited Leigh disease) but has not been associated with MELAS, PEO, or MIDD.
E. A guanine-to-adenine at nucleotide 1178 position (11778G > A) in mitochondrial subunit ND4 of complex I (MTND4).
Rationale: This mutation is commonly associated with LHON but has not been associated with MELAS, PEO, or MIDD.
Rationale: Mutations in ND4 are associated with LHON, MELAS, and Leigh syndrome (LS).
B. Complex I subunit ND1 gene.
Rationale: Mutations in ND1 cause MELAS, LHON, and MELAS/LHON overlap but not isolated myopathy.
C. Cytochrome b (cyt b) gene.
Rationale: Mutations in myogenic stem cells, presumably occurring after germ-layer differentiation, cause isolated myopathy. To date, most known mutations in cyt b occur in this category.
D. Cytochrome c oxidase subunit 1 (COX1) gene.
Rationale: COX1 mutations are associated with multisystem disorders, sideroblastic anemia, and sensorineuronal hearing loss.
E. Succinate dehydrogenase (SDHA) gene.
Rationale: Succinate dehydrogenase, encoded by SDHA, is a nuclear gene; its mutations do not cause isolated mitochondrial myopathy.
Rationale: The mitochondrial respiratory chain consists of about 75 protein subunits organized in five separate complexes. Only 13 of these are encoded by mtDNA, the rest by nuclear DNA.
B. Sperm do not contain mtDNA.
Rationale: Sperm contain thousands of mitochondria in each cell, each containing 5 to 10 copies of mtDNA.
C. All mtDNA-encoded proteins are components of the respiratory chain.
Rationale: mtDNA encodes 13 proteins that are incorporated into mitochondrial respiratory chain complexes.
D. mtDNA is independent of nuclear DNA (nDNA) and does not require nDNA gene products for its function.
Rationale: mtDNA functions under the overarching control of nDNA because it requires several nDNA-encoded proteins for its maintenance and proper functioning.
E. All mtDNA in the zygote is derived from sperm.
Rationale: All mitochondria and, therefore, all mtDNA in the zygote are derived from the ovum.
Rationale: MNGIE is caused by TP deficiency, the activity of which is easily measured in peripheral blood leukocytes.
B. Mitochondrial respiratory chain complex I activity.
Rationale: Complex I activity of the respiratory chain remains unaltered in TP deficiency.
C. Mitochondrial respiratory chain complex II activity.
Rationale: Complex II activity of the respiratory chain remains unaltered in TP deficiency.
D. Mitochondrial respiratory chain complex IV activity.
Rationale: Although the mosaic pattern of COX-deficient fibers is a common histologic finding in MNGIE, it is not specific for MNGIE, and the same pattern may be observed in other mitochondrial myopathies.
E. ATPase activity in peripheral blood leukocytes.
Rationale: ATPase activity levels in blood leukocytes are not informative because they are not altered in MNGIE.
B. Autosomal dominant.
Rationale: mtDNA inheritance does not follow Mendelian genetics rules.
C. Maternal.
Rationale: A unique feature of mitochondria (and all mtDNAs) is that they are inherited only from the mother. Therefore, a mother harboring an mtDNA point mutation passes it on to all of her children, but only her daughters transmit it to their progeny.
D. X-linked recessive.
E. X-linked dominant.
Rationale: mtDNA is not a constituent of the X chromosome.
Rationale: See Major Points of Discussion.
B. Polymorphism.
Rationale: When a change in DNA sequence (i.e., a variant) is so common that it is present at more than a 1% frequency in the general population, the variant is known as polymorphism.
C. Genotype.
Rationale: This refers to the genetic constitution of an individual.
D. Allele.
Rationale: This refers to one of the two or more forms of a gene.
E. Rare variant.
Rationale: Alleles with a frequency of less than 1% are called rare variants.
Rationale: Deafness and microcephaly can both be caused by mitochondrial dysfunction. Relatively common and maternally inherited, aminoglycoside-induced nonsyndromic deafness is caused by the A1555G mutation in mtDNA, whereas microcephaly in the Old Order Amish population is autosomal recessive due to a c.530G > C transversion in SLC25A19. There is no mutation in mtDNA known to cause both deafness and microcephaly.
B. This is an autosomal dominant deafness syndrome with low penetrance and variable expressivity.
Rationale: The two deaf children show an equally severe phenotype, as do the two microcephalic children; thus, variable expressivity is not likely. In addition, having three affected children in three births is not consistent with low penetrance.
C. This is a family with two mutations that cause two separate disorders, each inherited in an autosomal recessive manner.
Rationale: This is the most probable scenario because the parents are both healthy, and the disorders manifest early with identical phenotypes and clearly segregate, as expected with a Mendelian autosomal recessive model.
D. This is manifestation of a “mutator” syndrome that causes frequent mutations in the germline of the parents; thus, healthy parents have multiple affected children.
Rationale: Hereditary nonpolyposis colon cancer (HNPCC) is caused by a defect in DNA mismatch repair (MMR) genes. However, such patients do not have children with microcephaly or deafness.
E. This situation is due to maternal consumption of genetically modified produce during each pregnancy.
Rationale: There is no confirmed scientific evidence that genetically modified produce causes human disease.
Rationale: Although developmental delay and mental retardation are associated with loss of chromosomal segments that can be identified using microarray technologies, most known causes of microcephaly and deafness are due to single-nucleotide changes present at low allele frequencies in the population.
B. You recommend comparative genomic hybridization microarray studies to identify regions of homozygosity between the siblings affected by the same disease. This will allow you to narrow down the list of genes that are known to cause the conditions observed in this family.
Rationale: Although this approach will narrow the regions that harbor the disease-causing genes, the resolution is not high enough based on six observed meiotic events.
C. You propose targeted testing for all mutations known to cause deafness and microcephaly using Sanger sequencing.
Rationale: Sanger sequencing is most cost effective when used to identify mutations in high-probability gene targets. Unfortunately, both microcephaly and deafness can be caused by many low-frequency alleles.
D. You propose full genome sequencing because you suspect that the changes responsible for the phenotypes observed are in noncoding regions of the human genome.
Rationale: Full genome sequencing is still not cost effective for screening for mutations in candidate genes. As new technologies appear, this approach may become more widely used.
E. You recommend targeted testing for the most common point mutations associated with deafness and microcephaly.
Rationale: ASPM (locus name MCPH5; chromosome 1q31) accounts for 37% to 54% of cases of microcephaly. GJB2 (which encodes connexin 26) and GJB6 (which encodes connexin 30) account for about 50% of autosomal recessive nonsyndromic hearing loss. It would be most sensible to first test these three genes for mutations.
Rationale: The chance of having an affected child with either disease is one in four, whereas having a child affected with both diseases is one in eight. Already having children with the disease does not alter the risk for future children.
B. The family should be advised not to consider additional children because the risk for having additional affected children is uncertain.
Rationale: The risk can be calculated with certainty. The chance of having an affected child with either disease is one in four, whereas having a child affected with both diseases is one in eight. Already having children with the disease does not alter the risk for future children.
C. The family should be advised that the risk for having an affected child is one in four for microcephaly or deafness, whereas the chances of having a child with both disorders is one in eight, without providing any further recommendations.
Rationale: The parents should be informed that preimplantation screening for affected embryos can improve their chances of having a healthy child.
D. The family should be advised about the possibility of further genetic testing in case the parents are interested in pursuing prenatal diagnosis or in vitro fertilization.
Rationale: The introduction of high-throughput sequencing into clinical practice allows for the simultaneous screening of a large number of disease-causing genes. After the disease-causing mutation is identified, targeted testing of the embryos for the deleterious mutations would be feasible.
E. The family should be advised that there are no further diagnostic options available for them, and they should consider adoption.
Rationale: High-throughput sequencing provides a powerful tool for identifying rare disease-causing mutations.
Rationale: Next-Gen sequencing is currently available using multiple platforms from companies such as Roche, Illumina, and Applied Biosystems. Multiple academic and commercial entities currently offer Next-Gen sequencing for clinical diagnostic purposes.
B. It involves sequencing a single molecule, instead of many copies of the same molecule.
Rationale: Currently available commercial systems (e.g., from Roche, Illumina, and Applied Biosystems) all sequence multiple copies of the same molecule. Many thousands of independent clones are sequenced simultaneously. Instruments that work by sequencing single molecules, such as those commercially available from Pacific Biosystems, may soon become sufficiently reliable for clinical use.
C. It can provide megabases worth of nucleotide sequence, whereas Sanger sequencing is only practical for sequencing, at maximum, a few kilobases.
Rationale: The output of the HiSeq 2500 from Illumina in a single run is approximately 1 terabase (Tb), which corresponds to more than 100 times the size of the human diploid genome.
D. It is based on irreversible termination of extension on a subpopulation of the templates.
Rationale: Sanger sequencing uses irreversible termination of synthesis. At each cycle, fluorescently labeled dideoxynucleotides are mixed in with normal, unlabeled deoxynucleotides and are incorporated in a small percentage of extension products, which are irreversibly terminated. The length and color of the various terminated DNA molecules are then determined, and the sequence is derived from these data. In contrast, the principle of the Illumina instrument is based on reversible termination of synthesis. Fluorescently labeled nucleotides are added together in each extension cycle. No unlabeled nucleotides are present. Incorporation of specific nucleotides is detected simultaneously based on the color of the hundreds of thousands of extended products corresponding to different sequencing reactions. The fluorescent tag interfering with extension is then removed, and a new cycle of extension begins. The Roche and Applied Biosystems instruments use incorporation of unlabeled single nucleotides added one at a time in successive cycles. Incorporation is detected by coupled photochemistry in the Roche instrument (pyrosequencing) and by pH measurements in the Applied Biosystems instrument.
E. Interpretation of the data obtained is much more straightforward.
Rationale: Interpretation of Next-Gen datasets requires sophisticated computational algorithms and large amounts of computing and data storage capacity.
Rationale: Detection of sequence variations, neutral or pathogenic, is a process that occurs after the region of interest is sequenced and compared with a reference sequence that is considered normal.
B. The human genome contains a great deal of noncoding DNA, the role of which is not clearly established in disease. To limit the cost of clinical testing by sequencing, it is practical to focus on certain segments of the genome that have previously been shown to be associated with human disease. Upon sequencing the full genome, these segments are computationally identified and analyzed, whereas the data from noncoding regions of the genome are left in a non-human-readable form; this is sequence capture.
Rationale: It is a process that takes place before sequencing, not after the sequence is obtained. Regions of interest are either amplified using PCR-based or other locus-specific amplification methods, or captured using hydrogen bonding between complementary RNA or DNA baits; the DNA library created from the sample can then be analyzed.
C. It refers to the laboratory procedure that selects fragments of diagnostic interest from human genomic DNA for sequence analysis.
Rationale: Site-specific amplification-based methods are more suitable for smaller (e.g., up to 200 kb) genomic regions, whereas hybridization-based methods are more appropriate for capturing regions in the 100-kb to 100-Mb range.
D. It refers to the method of data collection from the sequencer.
Rationale: Sequence capture is a biochemical process based on amplification by synthesis or hydrogen bond formation between complementary strands on nucleic acids.
E. It refers to the fact that sequences slowly leak out of the reaction chamber of the sequencer, and they need to be prevented from contaminating the instrument by capturing them using a secondary container that is replaced regularly.
Rationale: Sequences are recorded interpretations of data obtained from the observation of chemical reactions taking place during sequencing. They are not a physical substance.
Rationale: Full exome sequencing will provide a comprehensive list of mutations in annotated protein-coding genes. Because, in this case, we are looking for homozygosity for two nontolerated/disruptive mutations, the chances that the cause of the disease can be determined with certainty are good.
B. Full exome sequencing is less expensive than full genome sequencing, and most known pathogenic mutations are within exons or at intron-exon junctions.
Rationale: With currently available technology, full genome sequencing is approximately 10 times more expensive than full exome sequencing.
C. Analysis of a full exome dataset is less complex than that of a full genome sequencing dataset.
Rationale: The size of the dataset is approximately 50 times larger for a full genome sequencing experiment. Interpretation is also more difficult because there is currently no good method for predicting the pathogenicity of mutations present in non–protein-coding regions of the genome.
D. Sequencing only known genes and loci would be effective if the patients in this family had a mutation previously identified in other families, but it would not detect private mutations specific to this family.
Rationale: Although there are many known cases of deafness and microcephaly, there are also many regions implicated in the pathogenesis of these diseases, where the genes involved are not yet identified.
E. Exome sequencing provides the most even and reliable sampling of the coding regions of the human genome, even if expense is not a consideration.
Rationale: With currently available capture reagents, even at 100-fold medium coverage depth, approximately 10% of the regions targeted are not sufficiently sampled (at least 10-fold coverage). At 200-fold coverage, this is reduced to approximately 5%.
Rationale: The key to finding the cause of the diseases is to look for homozygosity for disruptive mutations in the affected children. Once such differences are found, they can be verified to be heterozygous in the parents using a targeted sequencing approach.
B. Only the parents should be sequenced.
Rationale: Sequencing the parents would not be sufficient to identify the disease-causing allele(s) because the parents share about 25% of their genome.
C. The parents, one child with deafness, and one child with microcephaly should be sequenced.
Rationale: The power of the analysis is provided by comparing the three siblings; that is, the six underlying meiotic events.
D. Only the child with both microcephaly and deafness should be sequenced.
Rationale: Valuable extra information can be gained from the children who are affected only with one disease because they should be homozygous only for one set of disruptive mutations.
E. Only the three siblings should be sequenced. There is no need to sequence the parents.
Rationale: The key to finding the causes of these diseases is to identify homozygosity for disruptive mutations in the affected children. After such differences are found, they can be verified to be heterozygous in the parents using a targeted sequencing approach.
Rationale: Patients younger than 18 years need to sign an assent document, which explains to them, at their level of comprehension, the reasons for testing and the events associated with testing. Consent is signed by the parent or legal guardian. Testing can only be performed for conditions that are either present or will manifest before age 18 years.
B. Both parents must sign an informed consent form, and the children must sign an assent form.
Rationale: The signature of one parent is sufficient, and the children with microcephaly might be so severely affected that the assent document cannot be explained to them and they cannot sign it. There should be a court document assigning the decision-making capacity to the parent or legal guardian.
C. At least one parent must sign an informed consent form. The children with microcephaly are exempt from signing the assent form; assent only needs to be obtained from the deaf child.
Rationale: The signature of one parent is sufficient, and the children with microcephaly might be so severely affected that the assent document cannot be explained to them and they cannot sign it. There should be a court document assigning the decision-making capacity to the parent or legal guardian.
D. You must confirm that the parents’ insurance will cover the cost of testing.
Rationale: Prior insurance clearance is not required for testing to be performed; however, it is important to explain to the patients the amount that they might be billed for, in case insurance does not cover the testing expenses.
E. You have to obtain U.S. Food and Drug Administration (FDA) approval for the method to be used.
Rationale: Most tests in molecular pathology are developed and validated by the laboratories themselves. The College of American Pathologists (CAP) is the agency that supervises the performance characteristics of such tests. In some states, New York for example, the state authorities need to provide written approval for performing the test based on a submitted written validation summary.
Rationale: Disease-causing alleles are often present in carriers who are “healthy” and who participate as healthy controls in genotyping and sequencing projects mapping human genetic diversity. Thus, SNPs that have been observed previously should not be excluded from the analysis.
B. Mutations that do not cause amino acid changes, frame shifts, transcriptional stops, or splicing defects should be removed in the initial analysis.
Rationale: This will decrease the number of SNPs evaluated to approximately 1000 per sample.
C. Emphasis should be placed on mutations for which any one patient is homozygous.
Rationale: Because we are trying to determine the molecular cause of two independently segregating disease phenotypes based on an autosomal recessive mode of inheritance, the affected children have to be homozygous for either or both of the disease-causing mutations.
D. Mutations that are present in a homozygous form in all three siblings should be excluded as the cause of both deafness and microcephaly.
Rationale: Because only two of the three children have deafness and microcephaly, respectively, any mutation that is present in a homozygous form in all three children is, by definition, not the cause of either condition.
E. The children with deafness should be homozygous for the same mutation; similarly, the children with microcephaly should be homozygous for the same mutation.
Rationale: The unaffected child for either condition should be either wild-type or heterozygous for the SNPs that are thought to cause the deafness or the microcephaly.
Rationale: OMIM is a comprehensive compendium of human genes and genetic phenotypes. It contains information on all known Mendelian disorders and on more than 12,000 genes. It focuses on the relationship between phenotype and genotype and is updated daily, and its entries contain links to other resources.
B. University of Santa Cruz web browser: http://genome.ucsc.edu
Rationale: This site contains the reference sequence and working draft assemblies for a large collection of genomes, as well as coding and noncoding RNAs and epigenetic modifications. The site also displays currently curated SNPs and highlights known pathogenic SNPs in red.
C. dbSNP database: http://www.ncbi.nlm.nih.gov/SNP/
Rationale: This database is curated by the National Center for Biotechnology Information (NCBI) and provides tools to search for all previously detected SNPs. It provides data concerning allele frequencies for most common SNPs in various ethnic populations.
D. 1000 genome database: http://www.1000genomes.org
Rationale: Because full genome sequences are compared, genomic deletions and rearrangements are also included in the list of genetic polymorphisms. This is a work in progress, and its usefulness will increase as more genome sequences are added and analyzed.
E. Leiden database: http://www.dmd.nl
Rationale: This database is specifically designed for scientists performing research and/or diagnosis in the setting of Duchenne and Duchenne-like muscular dystrophies (e.g., Duchenne, Becker, limb-girdle). Although some additional databases about various disease groups are linked to this site, it has a somewhat limited disease-specific focus.
Rationale: Verification of mutations identified using high-throughput sequencing is more economically done using a targeted sequencing approach.
B. The sequencing data must be reanalyzed using another analysis software program.
Rationale: All mutations identified by the software of choice are visually verified. Observing the coverage depth, the quality of the alignments, and the individual reads allows one to judge the level of certainty of a specific mutation call. Using an alternative alignment method would be appropriate, if one wanted to evaluate the overall accuracy of the mutation-calling algorithm during validation of the test.
C. The identity of the samples and the correctness of the mutations must be confirmed using an alternative method.
Rationale: It is vital that results are confirmed to belong to the relevant patient. This is ensured by genotyping DNA from the original sample using an alternative method, such as SNP microarray studies, and comparing the genotyping and sequencing results. The presence of the pathogenic mutation also needs to be confirmed using targeted sequencing.
D. The mutations must be confirmed to be absent in 400 healthy control samples.
Rationale: Disease-causing mutations are often family or clan specific and might not be present in controls from even ethnically matched populations.
E. You must confirm that the mutations have never been seen previously in any population sampled.
Rationale: Disease-causing alleles are often present in carriers who are “healthy” and participate as healthy controls in genotyping and sequencing projects mapping human genetic diversity. Thus, SNPs that have been observed previously should not be excluded from the analysis.
Rationale: After a disease-causing mutation has been identified with certainty, a targeted sequencing approach is more appropriate.
B. Following whole genome amplification, the DNA from each embryo has to be genotyped for the mutations identified in the three siblings with deafness and/or microcephaly.
Rationale: Whole genome amplification is required because the DNA content of the cell consists of two copies only and the chances that the site-specific PCR will not work at such a low template level is possible. In addition, there is a potential risk of cross-contamination from other sources. Whole genome amplification using random primers decreases the chance of false-negative, as well as false-positive, results.
C. Following whole genome amplification, all embryos have to be tested for aneuploidy using a microarray methodology.
Rationale: Because we are looking for specific changes, genome-wide microarray testing is not required. Microarray testing for aneuploidy has not been shown to improve the chances of children being born with such conditions. This is because most aneuploid fetuses are spontaneously aborted early during pregnancy.
D. The embryos with the pathogenic mutations need to be preserved for future diagnostic test development.
Rationale: There is no medical need to preserve the embryos. DNA from such embryos could be preserved for potential use as positive controls in future studies.
E. Embryos that are carriers for either the deafness or microcephaly mutations can be implanted with their normal counterparts because they would also result in normal children and would improve the chances of successful implantation.
Rationale: Because the number of embryos generated is typically not limiting, there is no pressure to use carrier embryos.
Rationale: Digestion and detection are not the basic steps in PCR.
B. Denaturation, annealing, amplification.
Rationale: The three steps of PCR include denaturation of DNA to convert it from double-stranded DNA to single-stranded DNA by heating; binding (annealing) of oligonucleotide primers that are complementary to the DNA sequence; and synthesis of the new DNA strands that are complementary to the target DNA strand (amplification).
C. Digestion, annealing, amplification.
Rationale: The PCR reaction does not include DNA digestion.
D. Denaturation, annealing, labeling
Rationale: A standard PCR reaction does not include labeling.
E. Denaturation, annealing, digestion.
Rationale: The PCR reaction does not include DNA digestion.
Rationale: Primers should not show significant complementarity, especially at their 3′ ends, to avoid primer-dimer formation.
B. Ensure that the forward and reverse primers have a melting temperature (Tm) that differs by at least 5° C.
Rationale: Primers should, ideally, have a Tm that differs by less than 5°C.
C. Choose primers that are complementary to the same DNA strand.
Rationale: Primers should be complementary to opposite strands. (Note that because DNA sequences are always written in the 5′-to-3′ direction, and because DNA strands are antiparallel, each PCR primer, as written, is the “reverse complement” of the strand that it binds.
D. Choose primers that have a GC content of at least 90% to ensure strong binding.
Rationale: PCR primers should ideally have a GC content in the 60% to 70% range. However, this may vary depending on the target and application.
E. Check that the primers used to amplify genomic human DNA are validated against variant databases (e.g., dbSNP) to avoid primer–binding-site polymorphisms.
Rationale: This should always be done to avoid problems with allelic dropout; in particular, polymorphisms should especially be avoided toward the 3′ ends of the primers.
B. When setting up a PCR, one should add a template to the test samples first, followed by the no-DNA control, and then add the normal and abnormal controls.
Rationale: The template should be added last.
C. When setting up an RT-PCR, in addition to a no-template (i.e., no RNA) control, one should set up a no-RT control for each RNA sample being tested to test for directly amplifiable RNA.
Rationale: This control is performed to check for directly amplifiable DNA.
D. When setting up a PCR following an RT reaction, one should set up a separate no-template control, in addition to the product of the no-RNA RT reaction.
Rationale: The additional no-template control will distinguish between contamination of the RT step (the additional control will show no contamination) and of the PCR step; both controls will show contamination.
E. The use of deoxyuridine triphosphate (dUTP) in PCR with uracil-N-glycosylase to prevent PCR contamination is of no value for RT-PCR because RNA contains uracil.
Rationale: The dUTP with uracil-N-glycosylase works well with both DNA-PCR and RT-PCR.
Rationale: The maternal SNRPN allele is methylated.
B. Only the paternal allele is present; this is consistent with a diagnosis of Angelman syndrome.
C. Only the paternal allele is present; this is consistent with Prader-Willi syndrome.
Rationale: Absence of the maternal allele at this locus is the most common cause of Angelman syndrome.
D. Only the maternal allele is present; this is consistent with Prader-Willi syndrome.
Rationale: The maternal SNRPN allele is methylated.
E. Only one allele is present. Parental testing to determine the parent of origin of the missing allele will distinguish between Prader-Willi and Angelman syndromes.
Rationale: The absence of a methylated allele at this locus is consistent with Angelman syndrome.
Rationale: These two clinically distinct conditions are not caused by the same gene; however, the same deletion on chromosome 15 will cause Prader-Willi syndrome when on the paternal allele and Angelman syndrome when on the maternal allele.
B. Inactivating mutations in UBE3A will cause Prader-Willi syndrome only if present on the paternal allele.
C. Inactivating mutations in UBE3A will cause Prader-Willi syndrome only if present on the maternal allele.
Rationale: Inactivating mutations of UBE3A cause Angelman syndrome when present on the maternal allele.
D. Inactivating mutations of UBE3A cause Angelman syndrome when present on the maternal allele.
Rationale: UBE3A is mainly expressed from the maternal allele. Therefore, inactivating mutations of UBE3A causes Angelman syndrome.
E. Inactivating mutations of UBE3A cause Angelman syndrome only when present on the paternal allele.
Rationale: The paternal UBE3A allele is methylated, and only the maternal allele is expressed. Therefore, only mutations in the maternal allele cause Angelman syndrome.
Rationale: There are only a limited number of imprinted loci in the genome.
B. All imprinted loci in placental mammals show expression from only the maternal allele.
C. All imprinted loci in placental mammals show expression from only the paternal allele.
Rationale: Some imprinted loci are paternally imprinted and maternally expressed, whereas others are maternally imprinted and paternally expressed.
D. Most cases of Angelman syndrome and Prader-Willi syndrome are caused by imprinting defects.
Rationale: Although these diseases involve an imprinted locus, most cases result from deletions or uniparental disomy. Primary imprinting defects are rare.
E. Beckwith-Wiedemann syndrome, Russell-Silver syndrome, and transient neonatal diabetes mellitus type 1 are linked to imprinted loci.
Rationale: In addition to Prader-Willi syndrome and Angelman syndrome, these are some conditions that involve imprinted loci.
Rationale: This describes RNA polymerase II.
B. Promoters, which are recognized by RNA polymerase, include the TATA box, usually at the -25 position; the GC box that can function in either orientation; and the CAAT box that is usually located at the -80 position.
Rationale: See Major points of discussion.
C. Enhancers, which are always in close proximity to promoters, increase transcription activity of the associated gene.
Rationale: Enhancers can be anywhere in relation to the transcription start site.
D. Noncoding genes are always transcribed by RNA polymerase II.
Rationale: Noncoding genes may be transcribed by any of the three RNA polymerases.
E. The primary transcript of polypeptide-encoding RNAs often contains introns and exons. Introns are characterized by 5′AG and a 3′GU.
Rationale: They are characterized by 5′GU and a 3′AG.
Rationale: Most DNA sequencing tests will miss deep intronic variants, and their effects cannot be easily evaluated. Nonetheless, if deep intronic variants affect splicing, they will show with cDNA sequencing.
B. The smaller a deletion, the more likely it is to be missed by Sanger sequencing after DNA PCR.
Rationale: Large deletions are more likely to be missed by DNA PCR sequencing.
C. Nonsense mutations in the first exon may be missed when performing cDNA sequencing.
Rationale: Nonsense-mediated decay can lead to loss of the mutant transcript.
D. Sequencing of the entire coding region of a gene will identify virtually all significant mutations in a gene.
Rationale: This is not always the case, and deep intronic mutations and large deletions and rearrangements can account for a large percentage of mutations in some genes (e.g., NF1).
E. When a missense mutation in a gene known to be associated with a condition is seen in a symptomatic individual, the mutation is most likely to be significant.
Rationale: This is not necessarily true; the identified mutation could be a neutral nonsynonymous variant.
Rationale: Fragile X is caused by a CGG expansion in the 5′-UTR of the FMR gene.
B. Friedreich ataxia.
Rationale: Friedreich ataxia is caused by the expansion of GAA repeats in intron 1 of the FA gene.
C. Spinocerebellar ataxia type 8 (SCA8).
Rationale: SCA8 is caused by the expansion of CTG repeats in the 5′-UTR of the SCA8 gene.
D. Myotonic dystrophy.
Rationale: Myotonic dystrophy is caused by the expansion of CTG repeats in the 3′-UTR of the DM gene.
E. Huntington disease.
Rationale: Huntington disease is caused by the expansion of CAG repeats in the first exon of the huntingtin (htt) gene, leading to the expansion of a polyglutamine tract in the huntingtin protein.
Rationale: Whereas a normal male will show a single normal allele, this result may also be seen when a woman has two identical alleles or one normal allele and one expanded allele that fails to amplify.
B. The pretest risk for an expansion is higher in a woman with an affected nephew (brother’s son).
Rationale: The affected nephew must have inherited the expanded allele from his mother; therefore, this woman will have a risk equal to that of the general population.
C. In a male with mental retardation, the capillary electrophoresis result shown provides strong evidence against fragile X syndrome.
Rationale: More than 99% of fragile X mental retardation is caused by FMR1 CGG expansions. Although rare cases of mosaicism or other inactivating FMR1 mutations may be missed by this test, the result shown excludes most cases of fragile X mental retardation in males.
D. In a girl with developmental delay, the capillary electrophoresis alone excludes FMR1-associated mental retardation.
Rationale: The result shows only one allele; a full expansion of the second allele, which can be associated with mental retardation in girls, may be missed by this test.
E. In a mother of a boy with developmental delay, the PCR result rules out an FMR1 expansion as the basis for the child’s condition.
Rationale: The woman may have an expansion that is not detected by PCR.
Rationale: Both methylated and unmethylated, nonexpanded alleles are seen on the Southern blot analysis, consistent with two X chromosomes (XX female, or XXY male).
B. Southern blot texting is more reliable than PCR in excluding fragile X tremor ataxia syndrome (FXTAS).
Rationale: FXTAS is associated with premutations, which are better assessed by PCR.
C. Southern blot testing is more reliable then PCR in excluding FMR1-associated premature ovarian failure.
Rationale: Premature ovarian failure is associated with premutations.
D. The Southern blot test result adds little to the PCR, which is more sensitive and reliable.
Rationale: Large expansions may be missed by PCR. Therefore, the Southern blot test confirms that the woman is not a carrier.
E. In a woman undergoing carrier testing, this result is strong evidence against her carrying a premutation or full mutation.
Rationale: Both the capillary electrophoresis and Southern blot test show normal results. Together, they have a high negative predictive value for FMR1 repeat expansions.
Rationale: CYP2C19 loss-of-function alleles appear to be associated with higher rates of recurrent cardiovascular events in patients receiving clopidogrel.
B. CYP2D6.
Rationale: Tamoxifen is metabolized by CYP2D6 to the more potent endoxifen.
C. CYP2C9.
D. VKORC1.
Rationale: The clinical pharmacogenetic tests relevant for warfarin use are CYP2C9 and VKORC1.
E. TPMT.
Rationale: TPMT testing is recommended before starting treatment with azathioprine.
Rationale: BCR-ABL1 kinase domain mutation analysis is recommended when quantitative PCR rises and/or when there is suboptimal response to imatinib.
B. Perform a bone marrow biopsy for pathology and cytogenetics.
Rationale: Bone marrow analysis by pathology and cytogenetics is performed at the time of diagnosis of chronic myelogenous leukemia (CML).
C. Switch the patient to a second-generation tyrosine kinase inhibitor (TKI).
Rationale: The sample at time point 1 has the highest amount of BCR- ABL1 mRNA, whereas the sample at time point 6 has the least. Therefore, the transcript level has decreased progressively. A second-generation TKI (e.g., dasatinib and nilotinib) is approved for the treatment of imatinib-intolerant or -resistant patients.
D. Repeat this quantitative PCR assay in 3 to 6 months.
Rationale: A major molecular response is defined as a 3-log reduction in transcript level. Monitoring should be repeated in 3 to 6 months using this type of quantitative PCR assay.
E. Consider sending the patient for an urgent allogeneic stem cell transplantation.
Rationale: Allogeneic stem cell transplantation should be considered for patients with imatinib or second-generation TKI resistance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


