Chapter 12 Jeffrey S. Jhang; Richard O. Francis 1. A 2-year-old boy is brought by his mother to his pediatrician with a history of easy bruising and nosebleeds. Results of a complete blood cell count (CBC) and iron studies show a hypochromic microcytic anemia. Platelet aggregation studies for this patient are shown in Figure 12-1. Which platelet receptor is defective in this disorder? B. GPIIb/IIIa. C. P2Y12. D. GPIa. E. GPVI. 2. A 45-year-old woman with no prior past medical history presents to the employee health office for routine preemployment screening. Her CBC results are shown in Table 12-1. You review the peripheral blood smear (Figure 12-2) and recommend repeating the CBC. Which is the best tube type for collecting the next sample? A. Potassium ethylenediaminetetraacetic acid (EDTA) tube at 4°C. B. Foil-wrapped potassium EDTA tube. C. 3.2% sodium citrate tube. D. Glass tube with no additives. E. Serum separator tube. 3a. A 58-year-old woman with a history of renal insufficiency underwent surgery for a total knee replacement. She is placed on low-molecular-weight heparin following her surgery for deep vein thrombosis (DVT) prophylaxis. On postoperative day 5 the patient complains of left calf pain and swelling is noted on physical exam. Duplex ultrasonography demonstrates venous thrombosis in the left leg. Laboratory results from the day before surgery and postoperative day 5 are shown in Table 12-2. A peripheral blood smear from postoperative day 5 is notable only for mild thrombocytopenia. What is the most likely diagnosis? A. Immune thrombocytopenic purpura (ITP). B. Heparin-induced thrombocytopenia (HIT). C. Thrombotic thrombocytopenic purpura (TTP). D. Disseminated intravascular coagulation (DIC). E. Hemodilution. 3b. Based on the clinical suspicion and available laboratory results, which therapeutic intervention should be made for this patient? A. Perform daily plasmapheresis. B. Discontinue heparin. C. Transfuse platelets. D. Discontinue heparin and begin treating with a direct thrombin inhibitor. E. Discontinue heparin and begin warfarin. 4. A 6-year-old Iranian boy is brought to his pediatrician by his mother for frequent nosebleeds and gingival bleeding, which are occasionally severe. There is no history of bleeding disorders in the mother, father, or two older brothers. The CBC shows hemoglobin 11.8 g/dL and platelets 160 × 109/L. Light transmission aggregometry on platelet-rich plasma is shown in Figure 12-3. If this patient is transfused platelets, which isoantibody is most likely going to render the platelet transfusion ineffective? B. Anti-GPIbα. C. Anti-GPIIb/IIIa. D. Anti-GPVI. E. Anti–HPA-1a. 5. Which of the following is found in platelet α-granules? A. Adenosine diphosphate (ADP). B. Magnesium. C. Platelet-derived growth factor (PDGF). D. Pyrophosphate. E. Serotonin. 6. Which inherited platelet disorder is associated with a defect in a platelet membrane surface glycoprotein? B. Chediak-Higashi syndrome. C. Wiskott-Aldrich syndrome. D. Bernard-Soulier (BS) syndrome. E. Gray platelet syndrome. 7. Which platelet function test is considered to be an in vivo assay? A. Turbidometric aggregometry. B. Bleeding time (BT). C. PFA-100. D. Vasodilator-stimulated phosphoprotein (VASP) phosphorylation assay. E. Cone and plate analyzer. 8. Which of the following prolongs the BT? B. Hematocrit of 25%. C. Platelet count of 200 × 109/L. D. Pregnancy. E. Repeating the BT within 4 hours. 9. Which inherited platelet disorder has the classical finding of “large peroxidase-positive granules” in neutrophils? B. Chediak-Higashi syndrome. C. BS syndrome. D. Hermansky-Pudlak syndrome. E. Gray platelet syndrome. 10. A 32-year-old Japanese woman with systemic lupus erythematosus presents with frequent epistaxis. The platelet count is normal. The BT is 15 minutes (reference range, < 7 minutes). Light transmission aggregometry is performed as shown in Figure 12-4. The platelet function studies suggest the presence of an autoantibody against which platelet receptor? B. GPIIb/IIIa. C. GPVI. D. P2Y1. E. P2Y12. 11. Which assay assesses platelet function under shear conditions? B. VerifyNow. C. Impedance aggregometry. D. Assays that measure thromboxane (Tx) A2 metabolites. E. Turbidometric aggregometry. 12. Which inherited platelet disorder is associated with a mutation in the gene for the thrombopoietin (TPO) receptor (MPL)? B. Amegakaryocytic thrombocytopenia with radioulnar synostosis. C. Scott syndrome. D. Wiskott-Aldrich syndrome. E. Congenital amegakaryocytic thrombocytopenia. 13. Which of the following is found in platelet-dense bodies? B. Calcium. C. Fibrinogen. D. P-selectin. E. von Willebrand factor (vWF). 14. Which type of sample is used for flow cytometry–based detection of activated GPIIb/IIIa? A. Fresh nonanticoagulated blood. B. Whole blood. C. Serum. D. Platelet-rich plasma (PRP). E. Platelet-poor plasma (PPP). 15. The decreased presence of which marker, detected by flow cytometry, is used for a definitive diagnosis of Glanzmann thrombasthenia? B. CD41. C. CD154. D. CD49b. E. CD29. 16. Which component of platelet α-granules can be reliably detected in platelets from patients with gray platelet syndrome? B. β-Thromboglobulin. C. Fibrinogen. D. P-selectin. E. Platelet factor 4. 17. Which inherited platelet disorder is associated with oculocutaneous albinism? B. Wiskott-Aldrich syndrome. C. Hermansky-Pudlak syndrome. D. Paris-Trousseau/Jacobsen syndrome. E. BS syndrome. 18. A 19-year-old woman with a history of bruising and thrombocytopenia her entire life presents to a hematologist. Her review of systems is otherwise negative. Results of her CBC are shown in Table 12-3, and examination of the peripheral smear demonstrates platelets the size of the surrounding red blood cells (RBCs) and Döhle-like bodies in neutrophils. This presentation is most consistent with which platelet disorder? B. May-Hegglin anomaly. C. Epstein syndrome. D. Fechtner syndrome. E. Glanzmann thrombasthenia. 19. Which inherited platelet disorder is due to a defect in α-granules? A. Paris-Trousseau/Jacobsen syndrome. B. Chediak-Higashi syndrome. C. BS syndrome. D. Hermansky-Pudlak syndrome. E. Glanzmann thrombasthenia. 20. Which platelet function test uses conditions of high shear blood flow to test platelet function? B. Thromboelastography. C. VerifyNow. D. Light transmission aggregometry. E. PFA-100. 21. A surgeon has completed coronary artery bypass surgery on a 72-year-old man and would like to close the chest, but there is still significant oozing. The CBC is shown in Table 12-4. The surgeon orders thromboelastography and the results with the addition of heparinase are shown in Figure 12-5. Given the results of the thromboelastogram, what is the most appropriate blood product/derivative to administer? B. Fresh frozen plasma. C. Platelets. D. Prothrombin complex concentrates. E. RBCs. 22. Which platelet function test uses detection of platelet aggregation as its end point? B. PFA-100. C. Cone and plate analyzer. D. VASP. E. Thromboelastography. 23. A 27-year-old pregnant woman in her tenth week of pregnancy presents to her physician with a complaint of new-onset bruising, epistaxis, and gum bleeding. Pertinent laboratory values are listed in Table 12-5. Examination of the smear demonstrated thrombocytopenia and normal RBC morphology. What is the most likely cause of this patient’s presentation? B. ITP. C. DIC. D. Syndrome of hemolysis, elevated liver enzymes, and low platelet count (HELLP). E. Hemolytic uremic syndrome (HUS). 24. Which platelet receptor is blocked by the antiplatelet agent abciximab? B. GPIbα. C. GPIIb/IIIa. D. P2X1. E. P2Y12. 25. Which receptor is blocked by the pharmaceutical agent clopidogrel? B. GPIIb/IIIa. C. GPVI. D. P2X1. E. P2Y12. 26. Which platelet disorder is associated with inadequate exposure of negatively charged phospholipids on the platelet surface? B. Fechter syndrome. C. Scott syndrome. D. Epstein syndrome. E. Chediak-Higashi syndrome. 27. What is the basis for using thromboelastography as a platelet function test? A. Activation-dependent changes in platelet surface marker expression. B. In vivo cessation of blood flow by a newly formed platelet plug. C. Shear-induced platelet adhesion. D. Rate and quality/strength of clot formation. E. Platelet-to-platelet aggregation. 28. Which platelet function test is immunoassay based? B. PFA-100. C. Measurement of serum TxB2. D. Measurement of activated GPIIb/IIIa. E. Plateletworks. 29. Which inherited platelet disorder is associated with a defect in platelet signal transduction? A. Arthrogryposis–renal dysfunction–cholestasis. B. Paris-Trousseau/Jacobsen syndrome. C. Chediak-Higashi syndrome. D. Tx synthase deficiency. E. Hermansky-Pudlak syndrome. 30. Which assay is considered the historical gold standard for platelet function testing? B. Impedance aggregometry. C. Turbidometric aggregometry. D. Measurement of TxA2 metabolites. E. Measurement of activated GPIIb/IIIa by flow cytometry. 31. Which test is considered the gold standard for assessing the P2Y12 antagonist effect? A. Turbidometric aggregometry. B. Thromboelastography. C. Plateletworks. D. VASP phosphorylation. E. VerifyNow. 32. Which inherited platelet disorder is associated with immunodeficiency? B. Scott syndrome. C. Glanzmann thrombasthenia. D. Quebec platelet syndrome. E. Wiskott-Aldrich syndrome. 33. Which analyte measured in urine best reflects systemic TxA2 formation? B. 11-dehydro-TxB2. C. Prostaglandin G2. D. TxA2. E. TxB2. 34. Which disorder most likely explains the findings on the platelet aggregation studies shown in Figure 12-6? B. BS syndrome. C. Clopidogrel therapy. D. Glanzmann thrombasthenia. E. von Willebrand disease, type III. 35. Which medication can lead to thrombocytosis? B. Furosemide. C. Piperacillin/tazobactam. D. Romiplostim. E. Tacrolimus. 36. Which mutation is most commonly present in essential thrombocythemia? B. EPO mutation. C. JAK2 V617F. D. MPL mutation. E. PML-RAR fusion gene. 37. Which of the following could best explain a falsely elevated platelet count enumerated on an impedance-based automated hematology analyzer? B. Microclots. C. Platelet satellitism. D. Pseudothrombocytopenia (PTCP). E. Schistocytes. 38. Which condition is most associated with the object indicated by the arrow in Figure 12-7? B. Gray platelet syndrome. C. Myelodysplastic syndrome. D. Pseudothrombocytopenia. E. Wiskott-Aldrich syndrome. 39. Which source of platelets would be the best choice for transfusion in a neonate born with an intracranial hemorrhage to a woman with a history of a pregnancy loss due to neonatal alloimmune thrombocytopenia (NAIT)? B. Whole blood–derived random donor platelets. C. Paternal platelets. D. Single-donor platelets. E. Washed single-donor platelets. 40. Which of the following is an acceptable sample for performing platelet function studies by light transmission aggregometry on PRP? A. 3.2% sodium citrate, light-blue top tube. B. K2-ethylenediaminetetraacetic acid (K2EDTA), purple top tube. C. Lithium heparin, green top tube. D. No additive, red top tube. E. Serum separator, gold top tube. 41. An 8-year-old boy is admitted with bacterial meningitis. His CBC shows a platelet count of 721 × 109/L. Which of the following represents the best advice for the primary physician regarding this patient’s platelet count? A. Perform apheresis immediately. B. Perform apheresis followed by hydroxyurea treatment. C. Treat with hydroxyurea. D. No additional therapy is necessary. E. Perform a splenectomy. 42. A 42-year-old man with end-stage renal disease underwent a deceased donor renal transplant 6 months ago. He was discharged home on tacrolimus and prednisone. Several weeks later he is seen in his physician’s office complaining of fatigue, shortness of breath, and bruising on the soles of his feet. His CBC and relevant laboratory studies are shown in Table 12-6. His peripheral blood smear is shown in Figure 12-8. Which of the following best accounts for the laboratory and peripheral blood smear findings? B. Drug-induced thrombocytopenia. C. ITP. D. TTP. E. Warm autoimmune hemolytic anemia (WAIHA). 43. A 63-year-old man with diabetes, hypertension, and hyperlipidemia is discharged from the hospital on clopidogrel after being treated for an acute myocardial infarction. Several weeks later he is admitted with a second acute myocardial infarction. Which gene is associated with insufficient clopidogrel-induced platelet dysfunction? B. CYP1A1. C. CYP2C9*1. D. UGT1A6 T181A A > G. E. VKORC1 GG. 44. Your laboratory would like to perform “spot” checks of platelet counts reported from a new hematology analyzer using the Fonio method. The laboratory uses a 10 × ocular lens and a 100 × objective lens. The analyzer reports a platelet count of 200 × 109/L. Which of the following represents the approximate number of platelets per high-power field (HPF) that the technologist should count to correlate well with the analyzer? B. 2. C. 5. D. 10. E. 20. 45. A 79-year-old man with diabetes, hypertension, and hyperlipidemia underwent a single-vessel coronary artery bypass graft 4 years ago. His hospitalization was complicated by HIT without thrombotic complications, and his platelet count returned to normal after discontinuation of heparin. He is now admitted for a reoperation to perform a three-vessel coronary artery bypass. The anti-heparin/platelet factor-4 immunoglobulin G (IgG) enzyme-linked immunosorbent assay (ELISA) result is negative and the serotonin release assay result is negative. What is the most appropriate intraoperative management for anticoagulation of the bypass circuit? A. Anticoagulation with unfractionated heparin. B. Anticoagulation with bivalirudin. C. Anticoagulation with argatroban. D. Cancel surgery indefinitely. E. Delay surgery until anti-heparin/platelet factor-4 IgG, IgA, and IgM ELISA is performed. 46. A 22-year-old woman has a history of ITP as a child. She was successfully treated, but she does not know what the treatment consisted of and she has not had any recurrences. She now presents to her obstetrician at 12 weeks of pregnancy for her first prenatal visit. She denies bleeding, including epistaxis, vaginal bleeding, petechiae, and bruising. Laboratory studies are shown in Table 12-7. What is the best choice for treating this patient? B. Cyclophosphamide. C. Corticosteroids. D. Rituximab. E. Splenectomy. 47. A 51-year-old man undergoes presurgical testing prior to a right hernia repair. A CBC, blood type, and red cell antibody screen are performed as shown in Table 12-8. There is no active bleeding. A review of the peripheral blood smear does not show pseudothrombocytopenia. A diagnosis of ITP is made. What would be a first-line treatment for this patient? B. Dapsone. C. Corticosteroids. D. Rituximab. E. Splenectomy. 48. A platelet count is performed on a normal adult volunteer using an impedance-based analyzer. Which of the following best describes the platelet histogram (y-axis: frequency; x-axis size 2 to 20 fL) reported by the analyzer? B. Fourier transform of t-distribution. C. Gaussian (normal) distribution. D. Negatively (left) skewed distribution. E. Positively (right) skewed distribution. 49. Which platelet antigen is most commonly implicated in NAIT? B. HPA-1b. C. HPA-2a. D. HPA-3b. E. HPA-4a. 50. Which platelet receptor is activated by thrombin? B. GPVI. C. P2X1. D. P2Y12. E. Protease-activated receptor 1 (PAR-1). 51. Which of the following is most consistent with the results shown in Table 12-9 for the PFA-100 platelet function analyzer? B. BS syndrome. C. Glanzmann thrombasthenia. D. Gray platelet syndrome. E. Scott syndrome. Table 12-9 52. How many platelets are produced in 1 day by the average adult under normal physiologic circumstances? B. 109 C. 1011 D. 1012 E. 1015 53. Which clinical manifestation is most characteristic of patients with abnormal platelet function? A. Minimal bleeding from superficial cuts. B. Deep muscle hematomas. C. Delayed bleeding. D. Hemarthrosis. E. Petechiae. 54. Which of the following is the ligand for CD110 (the c-mpl gene product)? B. Granulocyte colony-stimulating factor (G-CSF). C. PDGF. D. Signal transducer and activator of transcription 1 (STAT1). E. TPO. 55. A 17-year-old woman with chronic ITP receives a TPO mimetic after her platelet count fails to increase after trials of corticosteroids, Rh immune globulin, and splenectomy. Which of the following is a long-term complication of TPO mimetics that her physician should advise her about? A. Bone marrow reticulin deposition. B. Berry aneurysms with intracranial hemorrhage. C. Pancreatitis. D. Portal vein aneurysms. E. Retinal detachment. 56. Which of the following is stored in the Weibel-Palade bodies of endothelial cells? B. Factor VIII. C. Fibronectin. D. Tx A2. E. vWF. Major points of discussion ■ Glanzmann thrombasthenia is caused by decreased production or dysfunctional platelet glycoprotein GPIIb/IIIa complex. Absence of binding to fibrinogen and/or vWF leads to this bleeding disorder. ■ Platelet aggregation studies show an absence of aggregation with ADP, epinephrine, and collagen. Aggregation with ristocetin is normal because GPIIb/IIIa is not involved with the GPIbα–vWF interaction. ■ Platelet receptors mediate adhesion, activation, and aggregation of platelets. Defects in these receptors can be due to mutations or exogenous substances that inhibit their function. ■ Defects in platelet function can be characterized based on the type of the dysfunction (e.g., adhesion, activation, aggregation). For example, BS syndrome is characterized by a defect in platelet adhesion. ■ Inherited defects in collagen-binding receptors (e.g., glycoprotein VI) are rare.15 2. A. Potassium ethylenediaminetetraacetic acid (EDTA) tube at 4°C. Major points of discussion ■ PTCP is most commonly caused by autoantibodies that recognize platelet antigens (neoantigens) when combined with EDTA. It probably involves the GPIIb/IIIa complex. ■ A way to avoid PTCP is to use a different anticoagulant (e.g., sodium citrate, sodium oxalate, and heparin) when collecting samples. In a subset of patients, it still may not be possible to obtain an accurate platelet count using an alternative anticoagulant. ■ The peripheral blood smear should always be manually reviewed when the automated platelet count is low. This precaution prevents the inappropriate treatment of a falsely low platelet count with steroids, cyclophosphamide, splenectomy, and/or rituximab, which can have devastating consequences. ■ Platelet satellitosis is also associated with EDTA autoantibodies that cause falsely low platelet counts on automated cell counters. Platelets adhere to the surface of neutrophils or monocytes and are not counted by the instrument.3 3a. A. Immune thrombocytopenic purpura (ITP). 3b. A. Perform daily plasmapheresis. Major points of discussion ■ HIT is an immune-mediated response to treatment with heparins. ■ HIT antibodies are directed toward platelet factor 4 (PF4) in complex with heparin. ■ HIT antibodies can develop in response to both unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH). Due to its smaller molecular size, LMWH has a decreased capacity to bind to PF4 tetramers and generate HIT antibodies. Patients treated with LMWH are 2 to 3 times less likely to develop HIT antibodies than are patients treated with UFH. ■ HIT antibodies are necessary, but not sufficient, to cause clinical symptoms of HIT (thrombocytopenia and thrombosis). ■ The hallmark of HIT is an otherwise unexplained thrombocytopenia beginning 4 to 14 days after heparin administration. Thrombocytopenia can present with a platelet count less than 100 to 150 × 103/μL or can present as a 30% to 50% decrease in platelet count from the preheparin baseline. ■ Patients with HIT have an increased risk of thrombotic complications, such as venous or arterial thrombosis and skin lesions (necrosis or erythematous lesions), Thrombosis is estimated to occur in 30% of HIT patients with thrombocytopenia. ■ Patients who initially present without thrombosis have up to a 50% risk of a thrombotic complication within the following 30 days if not treated with a non-heparin anticoagulant. ■ Laboratory assays for HIT include immunological and functional assays. Immunological assays include ELISA to detect the presence of HIT antibodies. Functional assays, such as the serotonin release assay (SRA) and heparin-induced platelet aggregation (HIPA) assay, detect the presence of HIT antibodies that cause platelet aggregation. The SRA is considered the gold standard assay for HIT antibodies. ■ When HIT is suspected, heparin should be discontinued and an alternate, rapidly acting anticoagulant should be started immediately.21 4. A. Anti-GPIa/IIa. Major points of discussion ■ BS syndrome shows thrombocytopenia with giant platelets, a lack of ristocetin-induced platelet aggregation, and a decreased response to thrombin-induced aggregation, ■ GPIbα contains vWF and thrombin binding sites. ■ Bleeding in BS syndrome can be mild to severe, with vascular factors contributing to the severity. Almost all patients with BS syndrome require platelet transfusion at some time in their life. The level of residual glycoprotein remaining on the platelets does not correlate with bleeding tendency. Isoantibodies against missing antigens can block the function of transfused platelets. ■ Thrombocytopenia with normal bone marrow megakaryocytes can lead to a misdiagnosis of ITP, rather than BS syndrome, with resulting incorrect treatment. ■ Giant platelets may be counted as red cells on automated cell counters, which can lead to artifactually lower platelet counts.12 5. A. Adenosine disphosphate (ADP). Major points of discussion ■ α-Granules contain PDGF, β-thromboglobulin, PF4, vWF, fibrinogen, factor V, and other proteins. ■ Dense bodies contain adenosine triphosphate (ATP), ADP, serotonin, pyrophosphate, magnesium, and calcium. ■ Abnormal or deficient α-granules can lead to platelet dysfunction, such as in gray platelet syndrome. ■ In gray platelet syndrome, α-granules are normally formed, but their contents leak from the organelle. Therefore, the α-granules are empty and the platelet does not have its usual granular appearance. 6. A. Quebec platelet syndrome. Major points of discussion • Glanzmann thrombasthenia • BS syndrome ■ BS syndrome is characterized by deficiency of the GPIb/V/IX complex and manifests with bleeding, thrombocytopenia, and large platelets. ■ There are fewer than 1000 estimated cases of BS syndrome worldwide. It is inherited as an autosomal recessive disorder. The genetic defects are in the GPIBA, GPIBB, and GP9 genes (genes encoding GPIb and GPIX, respectively; no defects in the gene encoding GPV have been described) ■ Bleeding often starts in childhood with frequent nosebleeds, gum bleeding, and easy bruising. ■ Diagnosis: Platelets fail to aggregate with ristocetin and the diagnosis can be confirmed using flow cytometry to analyze GPIb-α surface density.4,9 7. A. Turbidometric aggregometry. Major points of discussion ■ The basis of the BT is the in vivo cessation of blood flow by a newly formed platelet plug. ■ Procedure: A blood pressure cuff is applied at a standardized pressure (40 mm Hg) over the upper arm. A standardized cut is made on the volar surface of the forearm. A piece of filter paper is used to absorb the blood every 30 seconds (care must be taken not to dislodge the clot). The time when blood is no longer absorbed is the BT and has a reference range of less than 7 minutes. ■ Major disadvantages of the BT are that it is invasive, can lead to scarring, and has low reproducibility. ■ Qualitative platelet disorders and von Willebrand disease can prolong the BT. ■ Nonetheless, there are many causes of a prolonged or shortened BT. The BT is inversely proportional to the hematocrit and the platelet count. Therefore, the test should not be performed in the presence of anemia or thrombocytopenia.10,18,22 8. A. Cool skin. Major points of discussion ■ It has high variability due to physiologic variation, operator dependence, and technical variation. ■ The BT is performed by applying a blood pressure cuff at a standard pressure (e.g., 40 mm Hg), making a cut(s) on the volar surface of the forearm using a spring-loaded device, absorbing the blood away from the site of injury every 30 seconds using absorbent paper, and then determining the time it takes from the making the cut to cessation of bleeding (no blood on the paper). ■ The BT is highly operator dependent due to variability in how the cuts are made (e.g., pressure applied, direction of the cut), how the blood is blotted away because the clot can be inadvertently dislodged with the filter paper, location of cuts made on the arm, and so on. ■ Lower hematocrit and platelet count are independently associated with an increased BT. Therefore, the clinician should be cautious when the test is performed for anemic or thrombocytopenic patients. In addition, high cuff pressure, deep or long cuts, location, and residual alcohol from cleaning the site can all prolong the BT. ■ Physiologic and other variables can also decrease the BT. Repeating the BT within 4 hours, cool skin, age, gender, ethnicity (e.g., Eskimo), pregnancy, and acute-phase reactions can shorten the BT. ■ Although traditionally used, the BT is not thought to predict bleeding. A prolonged or normal BT does not mean that the patient will or will not bleed during surgery, respectively. 9. A. May-Hegglin anomaly. Major points of discussion ■ The dense-granule disorders include Hermansky Pudlak syndrome; Chediak-Higashi syndrome; and idiopathic dense-granule deficiency. ■ Dense granules are lysosome-related organelles in platelets and megakaryocytes and are members of a family of cell-type-specific organelles that also includes melanosomes, and lytic granules of cytotoxic T lymphocytes and natural killer cells. As such, dense granule disorders are usually part of a more complex congenital disorder with defects in several other lysosome-related organelles. ■ Dense-granule disorders typically present with mild to moderate bleeding, with easy bruising, epistaxis, menorrhagia, and the potential for significant bleeding in the setting of trauma or surgery. ■ Dense-granule disorders may result in defects in platelet aggregation ranging from abnormal responses to all agonists to more subtle changes, such as abnormal responses to low concentrations of agonists. ■ Characteristic aggregometry features include an absence of the secondary wave of aggregation in response to epinephrine; a delayed and reduced response to collagen; impaired aggregation to low concentrations of agonists, such as arachidonic acid and thrombin receptor agonist peptide (TRAP); and high concentrations of ADP eliciting full, irreversible aggregation. ■ A reduction in both the content and ratio of ADP to ATP, or absence of release of ATP, is diagnostic of a platelet dense-granule disorder. ■ A reduction in the number, or absence, of dense granules can be confirmed by electron microscopy. ■ Some characteristics of Chediak-Higashi syndrome include the following: estimated number of cases worldwide: less than 1000; autosomal recessive; genetic defect: LYST gene; oculocutaneous albinism, infections, and a lymphoproliferative accelerated phase; death usually occurs before age 10. ■ Diagnosis: The presence of very large peroxidase-positive cytoplasmic granules in hematopoietic (e.g., neutrophils) and nonhematopoietic cells is a classical finding in Chediak-Higashi syndrome and helps distinguish it from the Hermansky-Pudlak syndrome.4,9 10. A. GPIbα. Major points of discussion ■ Autoantibodies against the collagen receptor have been described. ■ Blockade of collagen receptor(s) by autoantibodies would decrease collagen-induced platelet aggregation. ■ GPVI-deficient platelets have also been described; this is more commonly found among the Japanese. ■ Under high shear stress, collagen receptors are unlikely to result in platelet adhesion. The platelet GPIbα-vWF interaction is most important for platelet adhesion under high shear stress.23 11. A. Cone and plate analyzer. Major points of discussion ■ The number of aggregates, the average size of the aggregates, and the percentage of the total area covered by aggregates are measured by an image analyzer. ■ The aggregation of platelets in this system is dependent on vWF, fibrinogen, platelet GPIbα and GPIIb/IIIa, and the platelet release reaction. ■ CPA instruments are commercially available and can be used with a small sample volume, do not require any manual sample preparation, and produce rapid results. They have a small footprint and can be used at the point of care for near patient testing. Small sample volumes are required; therefore, it is an ideal platelet analyzer for pediatric samples. ■ The disadvantage of the CPA instrument is that it is not widely used and clinical experience is limited. Similar to the BT and PFA-100, the CPA is dependent on the hematocrit and platelet count.10,18,22 12. A. May-Hegglin anomaly. Major points of discussion ■ Inherited platelet disorders are rare causes of bleeding. ■ Inherited platelet disorders characterized by abnormalities of platelet number include: • MYH9 disorders (May-Hegglin anomaly, Sebastian syndrome, Fechter syndrome, Epstein syndrome) • Congenital amegakaryocytic thrombocytopenia • Amegakaryocytic thrombocytopenia with radioulnar synostosis • Thrombocytopenia absent radius syndrome • X-linked thrombocytopenia with dyserythropoiesis ■ Congenital amegakaryocytic thrombocytopenia is characterized by thrombocytopenia with virtually complete absence of megakaryocytes from the bone marrow. ■ There are fewer than 100 cases estimated worldwide. ■ It is inherited in an autosomal recessive inheritance pattern. ■ The genetic defect is in the MPL gene (thrombopoietin receptor gene) ■ Congenital amegakaryocytic thrombocytopenia usually presents during the neonatal period, or soon after, with bleeding and severe thrombocytopenia. ■ Definitive confirmation requires molecular analysis of the MPL gene.4,9 13. A. β-Thromboglobulin. Major points of discussion ■ They have a distinct appearance on electron microscopy referred to as a bull’s-eye. ■ Dense bodies contain ADP, ATP, serotonin, calcium, magnesium, and pyrophosphate. ■ The other type of organelles found in platelets are the α-granules, which are less dense and more numerous by electron microscopy. ■ α-Granules contain PDGF, β-thromboglobulin, factor V, P-selectin, fibrinogen, vWF, PF4, and other proteins. ■ Absent or abnormal α- and dense granules can lead to abnormal platelet function, such as in gray platelet syndrome and Hermansky-Pudlak syndrome, respectively. ■ It should be noted that connections between α-and dense granules may occur in the Golgi apparatus, in which case they may have shared contents. 14. A. Fresh nonanticoagulated blood. Major points of discussion ■ Platelet function tests are also increasingly used to monitor the therapeutic efficacy of antiplatelet agents used to treat patients at high risk for atherothrombosis. ■ Platelet function studies may be classified based on the testing principle: • In vivo cessation of blood flow by the platelet plug • In vitro cessation of high shear blood flow by the platelet plug • Shear-induced platelet adhesion • Platelet aggregation • Changes in expression of surface markers following activation • Intracellular signaling following activation • Release of substances from platelets following activation • Evaluation of platelet contribution to clot strength ■ Flow cytometry–based assays can measure activation-dependent changes in the expression of platelet surface markers. ■ Platelet surface markers that are measured in these assays include P-selectin, activated GPIIb/IIIa, and leukocyte-platelet aggregates. ■ Characteristics of assays involving activation-dependent changes in platelet surface marker expression: • Advantage: small sample volume • Disadvantage: requires complex sample preparation, a flow cytometer, and a high level of technical experience10,18,22 15. A. CD63. Major points of discussion ■ Glanzmann thrombasthenia is characterized by a normal platelet count, normal platelet morphology, a prolonged BT, and defective platelet aggregation. ■ Glanzmann thrombasthenia characteristics: • Estimated number of cases worldwide: less than 1000 • Autosomal recessive • Genetic defects: ITGA2B, ITGB3 (genes encoding GPIIb and GPIIIa, respectively) • Presentation: Most initial presentations are during the first 5 years of life with purpura or petechiae in the neonatal period or excessive bruising during early childhood. ■ A definitive diagnosis is made by flow cytometry using antibodies against GPIIb (CD41) and GPIIIa (CD61).4,9 16. A. Factor V. Major points of discussion ■ α-Granule disorders include gray platelet syndrome; Paris-Trousseau or Jacobsen syndrome; Quebec platelet syndrome; and arthrogryposis–renal dysfunction–cholestasis. ■ Gray platelet syndrome is characterized by a lack of α-granules when the platelets are analyzed by electron microscopy. The contents of α granules that are both synthesized by the megakaryocyte (PF4, β-thromboglobulin, and PDGF) and taken up from the blood (factor V and fibrinogen) are lacking. P-selectin, an α-granule marker, is retained. ■ There are fewer than 100 cases worldwide. Gray platelet syndrome has both autosomal recessive and autosomal dominant patterns of inheritance. The genetic defects may involve several genes. ■ Platelets appear “gray” (due to lack of α-granules), misshapen, and slightly large (macrothrombocytopenia) on peripheral blood smear. ■ Electron microscopy of platelets shows decreased numbers or absence of α-granules. ■ Clinically, patients have a variable bleeding disorder (mild to severe) associated with both abnormal platelet function and thrombocytopenia.4,9,19 17. A. Sebastian syndrome. Major points of discussion ■ Dense-granule disorders typically present with mild to moderate bleeding with easy bruising, epistaxis, menorrhagia, and the potential for significant bleeding in the setting of trauma or surgery. ■ Dense-granule disorders may result in defects in platelet aggregation, ranging from abnormal responses to all agonists to more subtle changes, such as abnormal responses to low concentrations of agonists. ■ Hermansky-Pudlak syndrome characteristics: • Estimated number of cases worldwide: more than 1000 • Genetically, clinically, and biologically diverse • Most common genetic disorder in Puerto Rico • Autosomal recessive • Genetic defects: HPS1, AP3B1/HPS2, HPS3, HPS4, HPS5, HPS6, DTNBP1, HPS8 genes (protein products may affect trafficking of proteins to new organelles) • Affects platelet dense granules and melanosomes • Other important features: oculocutaneous albinism (common to all forms); pulmonary fibrosis, granulomatous colitis, neutropenia, and mild immunodeficiency (characteristic of certain forms)4,9 18. A. BS syndrome. Major points of discussion ■ May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are MYH9-related diseases that are characterized by mutations in the myosin heavy chain 9 (MYH9) gene. ■ MYH9-related disorders all have giant platelets (i.e., platelets at least the size of RBCs) and are distinguished by the presence or absence of Döhle-like inclusions, hearing loss, nephritis, and cataracts. ■ The majority of the MYH9 mutations show autosomal dominant inheritance. ■ Most patients with MYH9-related disorders do not have clinically significant bleeding problems.4 19. A. Paris-Trousseau/Jacobsen syndrome. Major points of discussion ■ α-Granule disorders include the following: • Paris-Trousseau or Jacobsen syndrome • Quebec platelet syndrome • Arthrogryposis–renal dysfunction–cholestasis ■ Paris-Trousseau/Jacobsen syndrome is characterized by the presence of giant α-granules in a low percentage of platelets. ■ Associated congenital abnormalities include mental retardation, cardiac abnormalities, and craniofacial abnormalities. ■ Paris-Trousseau/Jacobsen syndrome characteristics: • Estimated number of cases worldwide: less than 100 • Autosomal recessive • Genetic defect: 11q23 deletion (includes the FLI1 gene, which is important for megakaryopoiesis) • Presentation: mild bleeding diathesis4,9 20. A. Plateletworks. Major points of discussion ■ The PFA-100 is a whole blood assay where blood flows at physiologic shear stress through an aperture that is coated with a choice of physiological agonists (collagen/ADP, collagen/epinephrine). The interval from the start of the test until the aperture becomes occluded is the closure time. ■ Characteristics of the PFA-100 assay: • Advantages: small sample volume; no sample preparation • Disadvantages: dependent on vWF, hematocrit, and platelet count. ■ Interpretation: • ADP normal and EPI normal = normal • ADP normal and EPI abnormal = aspirin, nonsteroidal antiinflammatory drugs (NSAIDs) • ADP abnormal and EPI abnormal = BS syndrome, Glanzmann thrombasthenia, von Willebrand disease, and so on. ■ If the PFA-100 test result is normal, then further testing is usually not performed because of its excellent negative predictive value. ■ However, if the test result is positive, confirmatory testing with light transmission or whole blood aggregometry is necessary.10,18,22 21. A. Recombinant factor VIIa. Major points of discussion ■ The pattern can be interpreted to provide information on the hemostatic status of the patient, which can guide component therapy, especially during cardiothoracic surgery. ■ The R value is the time from the initiation of the test to the initial formation of the clot. It is reflective of coagulation factor activation. A prolonged R value would suggest the presence of a coagulation factor deficiency and fresh frozen plasma would be indicated. ■ Maximal aggregation (MA) is a measure of the clot strength and is the amplitude of the curve. It is reflective of platelet number or function. A decreased MA would suggest that platelets are indicated. ■ If the MA decreases at an interval shorter than normal, then clot lysis is accelerated, as seen in excessive fibrinolysis. A antifibrinolytic agent may be indicated in this circumstance. 22. A. Plateletworks. Major points of discussion ■ The basis of the Plateletworks assay is platelet aggregation and is performed by counting platelets from a sample before and after activation using agonists. ■ The main use of this assay is the measurement of residual platelet reactivity after the administration of an antiplatelet agent following an acute cardiovascular or neurovascular event. If the residual reactivity remains high, the patient may be at risk of a secondary myocardial infarction or stroke, respectively. ■ Steps in performing the assay: 2. Measure platelet count from a second sample that is collected in the presence of a platelet agonist (e.g., collagen, ADP, or arachidonic acid). 3. Uninhibited platelets in the presence of the agonists will become activated, aggregate, and will not be counted as individual platelets in this second sample. 4. The difference in platelet counts between the two samples provides a measure of aggregation and a ratio of the two counts yields a percentage inhibition. ■ The advantage of this test is that it requires minimal sample preparation. The disadvantage is that it is an indirect test. It assumes that the decrease in platelet count is proportional to amount of platelet aggregation.10,18,22 23. A. TTP. Major points of discussion ■ ITP is the most common autoimmune disorder noted during pregnancy. ■ It is important to understand the normal physiologic changes that occur during pregnancy. For example, the platelet count is expected to decrease by approximately 10% during a normal pregnancy and the greatest decrease is during the third trimester. ■ The differential diagnosis of thrombocytopenia during pregnancy includes ITP, gestational thrombocytopenia (GTP), TTP, HUS, DIC, and the HELLP syndrome. ■ Antiplatelet antibodies in ITP are usually directed against the GPIIb/IIIa or GPIb-IX-V platelet proteins. ■ The majority of antibodies are IgG, are able to cross the placenta and can cause fetal or neonatal ITP.5,11 24. A. α2β1. Major points of discussion ■ GPIIb/IIIa is required for platelet aggregation. Platelets aggregate by GPIIb/IIIa to fibrinogen or vWF. These interactions bridge platelets via fibrinogen and vWF. ■ Other GPIIb/IIIa inhibitors include tirofiban (a nonpeptide agent) and eptifibatide (a heptapeptide agent). All three GPIIb/IIIa inhibitors have different platelet GPIIb/IIIa binding sites. ■ Abciximab is used in percutaneous coronary interventions and for acute coronary syndromes. ■ Glanzmann thrombasthenia is an inherited bleeding disorder caused by a GPIIb/IIIa deficiency. 25. A. GPIbα. Major points of discussion ■ Clopidogrel is a pharmaceutical agent that specifically blocks the P2Y12 receptor and is a strong antiplatelet medication. ■ Other medications that block the P2Y12 receptor and are used as antiplatelet agents include ticlopidine, prasugrel, and cangrelor. ■ Clopidogrel and other P2Y12 receptor blockers are used for prophylaxis against secondary acute coronary and neurovascular thrombosis and in patients who cannot tolerate aspirin. ■ Clopidogrel “resistance”—that is, the lack of platelet inhibition with standard doses of clopidogrel—can be caused by variable hepatic transformation of clopidogrel into its active form by polymorphisms of the CYP2C19 gene. Genetic testing may be useful to determine the appropriate dose or if a change in medication is indicated. 26. A. Sebastian syndrome. Major points of discussion ■ This lack of phospholipid exposure leads to a significantly reduced ability to promote tenase and prothrombinase activity on platelet surfaces. ■ There are fewer than 10 cases of this syndrome reported worldwide. It has an autosomal recessive inheritance pattern. The exact gene defect for this disorder has not been definitively identified. At least one case is due to a mutation in transmembrane protein 16 F (TMEM16F). ■ Patients present with bleeding following invasive procedures. There are also reports of postpartum bleeding. ■ The diagnosis can be made using flow cytometry (by annexin V labeling) to assess whether phosphatidyl serine becomes exposed following platelet activation. ■ The only known treatment is transfusion of normal donor platelets.4,9 27. A. Activation-dependent changes in platelet surface marker expression. Major points of discussion ■ Platelet function tests are also increasingly used to monitor the therapeutic efficacy of antiplatelet agents used to treat patients at high risk for atherothrombosis. ■ The basis of using thromboelastography to evaluate platelet function is to evaluate the contribution of platelets to clot strength in a whole blood sample. ■ Advantages of thromboelastography: It measures global coagulation (i.e., clotting factors, platelets, fibrinolysis) and can potentially be used at the point of care (e.g., in the operating room). ■ Disadvantages of thromboelastography: To test the platelet-specific contribution to clot formation and strength, additional platelet activators (arachidonic acid or ADP) must be used, thereby increasing the complexity of the test.10,18,22 28. A. VerifyNow. Major points of discussion ■ The substances measured in assays based on platelet secretion following activation include platelet-derived microparticles (by flow cytometry), serum TxB2, urinary 11-dehydrothromboxane B2, plasma sCD40L, plasma GPV, and α-granule constituents in plasma (e.g., PF4, β-thromboglobulin, and soluble P-selectin). ■ Serum TxB2, urinary 11-dehydrothromboxane B2, plasma sCD40L, plasma GPV, and α-granule constituents are measured using immunoassays. ■ Serum TxB2 and urinary 11-dehydrothromboxane B2 are indirect measures and are not platelet specific. ■ Serum TxB2 measures the capacity of platelets to synthesize TxA2 and is a measure of the antiplatelet effect of aspirin.10,18,22 29. A. Arthrogryposis–renal dysfunction–cholestasis. Major points of discussion • Platelet cyclooxygenase deficiency • Tx synthase deficiency • Tx A2 receptor defect • ADP receptor defect (P2Y12) ■ In general, patients with this class of defects present with abnormal primary hemostasis manifested by mild bleeding. Platelet number and morphology are usually normal. ■ Tx synthase deficiency or a defect in the TxA2 receptor presents with an aspirin-like defect, but a thorough medication history will not show any ingestion of aspirin or aspirin-containing medications. ■ There is marked impairment of platelet aggregation in response to arachidonic acid; aggregation with ADP and collagen are also reduced. Aggregation with Tx analogs and ristocetin are normal. ■ These cases are diagnosed when there is an abnormal secondary wave of aggregation, but there are normal dense granules on electron microscopy and an absence of aspirin ingestion.4,9 30. A. PFA-100. Major points of discussion ■ The basis of platelet aggregometry is platelet-to-platelet aggregation in response to various platelet agonists. ■ Turbidometric (light transmittance) aggregometry measures platelet-to-platelet aggregation in PRP in response to classic agonists. The assay is time consuming because PPP and PRP must be prepared from whole blood. In addition, most of the steps in this assay are manual and are of high complexity. Nonetheless, this test is considered the gold standard for platelet function testing. ■ Impedance aggregometry measures changes in impedance in response to classic agonists. It is a whole blood assay. ■ Classic agonists include collagen, ADP, epinephrine, arachidonic acid, thrombin, TRAP, phorbol 12-myristate 13-acetate (PMA), ristocetin, and U44619 (a TxA2 analog). ■ Both impedance and turbidometric aggregometry are used to diagnose a variety of inherited and acquired platelet defects.10,18,22 31. A. Turbidometric aggregometry. Major points of discussion ■ The basis of the VASP phosphorylation assay is measurement of activation-dependent signaling by flow cytometry. ■ It is a whole blood assay considered the gold standard for assessing the P2Y12 antagonist effect. ■ Its major disadvantage is that it requires complex sample preparation, a flow cytometer (i.e., high capital investment), and a high level of technical experience. ■ P2Y12 inhibitors used as antiplatelet agents include clopidogrel, prasugrel, cangrelor, and ticagrelor.10,18,22 32. A. BS syndrome. Major points of discussion ■ Estimated number of cases worldwide: less than 1000. ■ X-linked inheritance; genetic defect: WAS gene.4,9 ■ Bleeding manifestations: bruising and purpura in the neonatal period with an increased risk of intracranial hemorrhage and bleeding after circumcision. ■ Eczema develops during first year of life. ■ Immunodeficiency is manifested by infections (most commonly bacterial) that occur in the first 6 months of life. ■ Confirmatory molecular diagnosis is obtained by analyzing the WAS gene. 33. A. 2,3-dinor TxB2. Major points of discussion ■ Cyclooxygenase is required to convert arachidonic acid to prostaglandin G2, which is then quickly converted to PGH2. PGH2 is then converted to TxA2 by Tx synthase. ■ TxA2 is rapidly hydrolyzed to form TxB2, which is also rapidly metabolized and cleared from the circulation. ■ Clinically, measuring TxA2 metabolites has been used to assess the effectiveness of aspirin therapy. If aspirin therapy is effective in inhibiting cyclooxygenase, then TxA2 levels should be significantly reduced. ■ TxB2 is mainly metabolized into 11-dehydro-TxB2 and 2,3-dinor-TxB2. Small amounts of both are excreted in the urine unchanged. ■ TxB2 is also formed during blood storage, so it is not a useful marker for TxA2 formation. ■ 11-dehydro- TxB2 and 2,3-dinor-TxB2 are found in urine and can be measured as markers of TxA2 formation. 11-dehydro-TxB2 is considered the better marker because of its longer half-life, greater abundance in urine, and less variability.7 34. A. Aspirin therapy. Major points of discussion ■ Cyclooxygenase is required for converting arachidonic acid to prostaglandin G2, which is then quickly converted to PGH2. PGH2 is then converted to Tx (TxA2) by Tx synthase. ■ TxA2 binds to the thromboxane receptor by an autocrine or paracrine mechanism. TxA2 is essential for recruiting additional platelets into the nascent clot. ■ Aspirin irreversibly inhibits cyclooxygenase by acetylation of a serine residue and strongly inhibits platelet function. NSAIDs reversibly inhibit cyclooxygenase and may transiently inhibit platelet function. ■ Aspirin is effective for primary and secondary prophylaxis against myocardial infarction and cerebrovascular ischemic events. 35. A. Amphotericin. Major points of discussion ■ Reactive causes of thrombocytosis include infection, inflammation, splenectomy, hyposplenism, surgery, and iron deficiency anemia. ■ Thrombocytosis can also be caused by myeloproliferative disorders such as essential thrombocytosis, polycythemia vera, and chronic myelogenous leukemia. ■ TPO agonists such as eltrombopag and romiplostim, which are used to treat chronic refractory ITP, can lead to thrombocytosis. ■ Reactive thrombocytosis is generally not treated because it does not increase the risk of thrombosis. ■ Thrombocytosis due to a myeloproliferative disorder increases the risk of thrombosis at very high platelet counts (e.g., > 1 million/μL). It is treated with aspirin, hydroxyurea, or anagrelide or with plateletpheresis as single or combination therapy. 36. A. BCR-ABL fusion gene. Major points of discussion ■ There is a high rate of thrombosis seen in patients with ET. ■ According to the 2008 World Health Organization (WHO) guidelines, the diagnosis of ET requires the following: 1. A platelet count greater than 450 × 109/L 2. Megakaryocyte proliferation on bone marrow biopsy and/or aspirate; megakaryocytes must be enlarged and mature; an absence of granulopoiesis or erythropoiesis 3. Not meeting WHO criteria for polycythemia vera, primary myelofibrosis, chronic myelogenous leukemia, myelodysplastic syndrome, or other myeloid disorder 4. Demonstration of the JAK2 V617F mutation or no evidence of reactive thrombocytosis in the absence of a clonal marker ■ Patients with ET can be asymptomatic or they can present with vasomotor symptoms (e.g., headache, light-headedness, syncope). More serious complications result from thrombosis or hemorrhage. Although the platelet count is increased, the platelets that are produced do not function normally, which increases the risk for bleeding. ■ The normal function of the JH2 (Janus homology 2) domain is to negatively regulate the JH1 (Janus homology 1) catalytic domain. The JAK2 V617F mutation is located in the JH2 autoinhibitory domain, which leads to autonomous JAK2 function. ■ The JAK2 V617F mutation is present in 50% of patients with ET; 4% of patients have MPL mutations (i.e., in the TPO receptor).25 37. A. Macrothrombocytes. Major points of discussion ■ The cell count is determined by creating a histogram of cell frequency (y-axis) versus the cell size (x-axis). In general, platelets range from 2 to 20 fL in size (this is instrument dependent); therefore, the area under the curve from 2 to 20 fL is the platelet count. Red cells can be enumerated by performing the same calculations between roughly 50 and 250 fL in size. ■ The mean platelet volume is the average of the distribution from 2 to 20 fL. ■ Microorganisms, extracellular parasites (e.g., Babesia), cytoplasmic cell fragments (e.g., in acute myeloid leukemia), schistocytes (e.g., in burns, TTP), and severely microcytic red cells can be counted as platelets. These could cause falsely elevated platelet counts. ■ Optical measurement of platelets is an alternative to impedance counting. Optical counting may include a fluorescent dye to help distinguish platelets from other cell types and particles. For example, schistocytes would be the size of a platelet but would not fluorescently stain like a platelet. Therefore, these two populations can be separated.16,26 38. A. Babesiosis. Major points of discussion ■ The cytoplasm can be blue-gray with bluish granules on Wright-Giemsa stain. The periphery may be scalloped or ruffled. ■ Giant platelets can be mistaken for reticulocytes (polychromatophilic red cells), non–megakaryocyte cell fragments (e.g., cytoplasmic fragments of blast cells), neutrophils, megakaryocytes, and agglutinated platelets. ■ Giant platelets are associated with various acquired and inherited diseases. Most of them are premalignant or malignant (e.g., myelodysplastic syndrome, acute leukemia, multiple myeloma). They can also be formed as a physiologic response to splenectomy, hemorrhage, and ITP. ■ Inherited disorders associated with giant platelets include BS syndrome, the May-Hegglin anomaly, and familial macrothrombocytopenia. ■ Small platelets are seen in Wiskott-Aldrich syndrome, an X-linked immune deficiency associated with eczema and thrombocytopenia. 39. A. Maternal platelets. Major points of discussion ■ NAIT is a cause of severe fetal/neonatal thrombocytopenia that can lead to severe bleeding, including intracranial hemorrhage and death. The greatest risk in neonates is during the first 96 hours after birth and generally resolves after 2 weeks. However, most cases of intracranial hemorrhage occur in utero. ■ If the antibody specificity is known, then platelets can be obtained from rare platelet donors who do not express the corresponding antigen. Approximately 95% of cases are caused by maternal alloimmunization to the HPA-1, -2, -3, -5, or -15 antigens. Anti-HPA-1a is the most commonly detected antibody. ■ Laboratory testing that shows maternal serum reacting with paternal platelets in the presence of a negative HLA class I antibody screen supports the presence of an anti-HPA antibody. If the reaction of maternal serum is against a large number of random donor platelets, then an antibody against a high-frequency platelet antigen should be considered. ■ Another cause of severe fetal/neonatal thrombocytopenia that should be considered is the transplacental transfer of maternal antiplatelet autoantibodies in a mother with idiopathic thrombocytopenic purpura. In this case, the maternal platelet count would also be low. ■ Although maternal antigen-negative platelets are an optimal source for transfusing the affected neonate, if they are not available, then random donor platelets (apheresis- or whole blood–derived) would be the next best choice for emergently providing platelets to treat bleeding. Nonetheless, attempts to provide antigen-negative donor platelets should be made.1 40. A. 3.2% sodium citrate, light-blue top tube. Major points of discussion ■ Samples are drawn into 3.2% sodium citrate anticoagulant in a ratio of 9 parts whole blood to 1 part sodium citrate. Sodium citrate is a weak chelator of calcium and inhibits clotting. However, some unbound calcium remains available for platelet aggregation. ■ EDTA cannot be used for this purpose because it binds 10 times more calcium than sodium citrate, making calcium less available, which will blunt platelet aggregation responses. Heparin has physiological levels of calcium and spontaneous platelet aggregation can occur. Acid-citrate-dextrose (ACD) has a low pH, which decreases platelet aggregation. Therefore, these anticoagulants should not be used. ■ Anticoagulant levels should be adjusted if the hematocrit (HCT) is elevated because the decreased plasma volume leads to over-anticoagulation of the sample. Some unbound calcium is required for platelet aggregation to occur. ■ Anticoagulant volume = (1.85 × 10–3)(100 – HCTpercent)(Vblood). ■ Samples should be prepared and analyzed within 4 hours of sample collection, preferably in less than 2 hours, because platelets undergo activation during prolonged storage.8,14 41. A. Perform apheresis immediately. Major points of discussion ■ Secondary thrombocytosis is a reactive response to bacterial and viral infection, surgery, trauma, iron deficiency, and inflammatory states. ■ Other causes of secondary thrombocytosis include rebound after chemotherapy or splenectomy. ■ Secondary thrombocytosis rarely requires intervention because it is not associated with an increased risk of thrombosis. ■ Because they are associated with an increased risk of thrombosis, high platelet counts in patients with primary thrombocytosis are treated with cytoreductive therapy (e.g., hydroxyurea), aspirin, and/or plateletpheresis.6,26 42. A. DIC. Major points of discussion ■ TTP is classically associated with a pentad of signs/symptoms: fever, mental status changes, microangiopathic hemolytic anemia, thrombocytopenia, and renal failure. However, all five findings may not be present in all cases. ■ Congenital TTP is caused by an inherited deficiency of the ADAMTS13 enzyme. ADAMTS13 cleaves ultralarge multimers of vWF. A deficiency in ADAMTS13 leads to an excess of ultralarge VWF multimers, platelet thrombi, and shear stress of red cells that leads to hemolysis. Simple plasma infusion can replace the enzyme in congenital TTP. ■ Idiopathic TTP is caused by an autoantibody against ADAMTS13 that leads to its deficiency. Subsequently, ultralarge VWF multimers and platelet thrombi form, which causes hemolysis by the shearing of red cells. The treatment of choice in antibody-mediated TTP is plasmapheresis using fresh frozen plasma as the replacement fluid. This procedure removes the autoantibody from the circulation and provides ADAMTS13 in the plasma replacement fluid. Immunosuppressants, usually steroids, are administered to inhibit antibody production. ■ Drug-induced TTP is caused by multiple medications; cyclosporine, tacrolimus, clopidogrel, mitomycin C, and gemcitabine are the ones most commonly reported. Treatment requires the cessation of the medication inducing the TTP. Although plasmapheresis is often used, it may not be beneficial in drug-induced TTP.13 43. A. CYP2C19*2. Major points of discussion ■ Clopidogrel is a P2Y12 antagonist that is commonly used as an antiplatelet agent for secondary prophylaxis after an acute ischemic cardiovascular or cerebrovascular event. ■ Clopidogrel is metabolized to an active metabolite that has the antiplatelet activity. It is metabolized to its active form by the P450 enzyme CYP2C19. ■ Polymorphisms in the CYP2C19 gene are associated with “normal” or “slow” metabolism of clopidogrel to its active form. CYP2C19*1 encodes the protein with normal clopidogrel metabolism, whereas CYP2C19*2 or *3 is associated with slower metabolism. ■ Patients receiving clopidogrel for secondary prophylaxis against stroke or myocardial infarction may still have an event. Some of these cases may be due to inadequate clopidogrel-induced platelet inhibition (clopidogrel resistance) due to reduced clopidogrel metabolism. 44. A. 1. Major points of discussion ■ The Fonio method for estimating platelets is based on review of a peripheral blood smear using a light microscope. Using a × 10 ocular and × 100 objective lens, the average number of platelets per HPF can be determined. The platelet estimate is [(20) × (average number of platelets per HPF)]. ■ If there are 10 platelets per HPF, then the platelet estimate using the Fonio method would be 200 × 109/L. ■ Modern hematology analyzers measure the platelet count using impedance-, optical-, fluorescence-, or antibody-based methods. ■ Automated analyzers are more accurate, reproducible, and less labor intensive than manual methods. 45. A. Anticoagulation with unfractionated heparin. Major points of discussion ■ HIT is characterized by a drop in the platelet count by approximately 50% within 5 to 10 days of the initiation of heparin therapy and can be associated with the development of thrombosis. ■ The anti-heparin/PF4 ELISA assay result is often used to screen for the antibodies implicated in causing HIT. Although this assay is highly sensitive, it has poor specificity. The serotonin release assay is a functional assay that assesses the presence of anti-heparin/PF4 antibodies that can activate platelets. This assay has high sensitivity and specificity (98% and 95%, respectively). ■ If a patient has a remote history of HIT with negative immunologic and functional assay results for HIT, unfractionated heparin can be used for cardiovascular surgery. ■ If a patient has acute HIT with positive immunologic and functional assays for HIT, unfractionated heparin should not be used for cardiovascular surgery. Surgery can be delayed until the assay results are negative and then unfractionated heparin can be used. If surgery cannot be delayed, then an agent such as bivalirudin can be used as an alternate anticoagulant.17 46. A. Azathioprine. Major points of discussion ■ ITP is caused by autoantibodies against platelet antigens. The antibody-coated platelets are cleared by the reticuloendothelial system. Recent evidence suggests that there is a concomitant platelet production defect. ■ First-line treatment of ITP in adults includes corticosteroids, intravenous immunoglobulin (IVIG), and intravenous Rh immune globulin in Rh-positive patients. A longer course of corticosteroids is preferred over a shorter course of corticosteroids or IVIG. IVIG and Rh immunoglobulin are preferred, if steroids are contraindicated (e.g., uncontrolled diabetes mellitus). ■ Pregnant patients should be treated in the same manner as adult patients, so corticosteroids and IVIG are suggested as first-line treatments. However, randomized controlled trials in pregnant patients have not been performed. ■ When pregnant patients with ITP are delivering, the method of delivery is based on the current recommendations for the obstetric indication. ■ Neonates born to women with ITP are at increased risk of thrombocytopenia.20 47. A. Apheresis. Major points of discussion ■ Bone marrow biopsy shows normal erythropoiesis and myelopoiesis and increased megakaryocytes. ■ ITP is caused by autoantibodies against platelet antigens. The antibody-coated platelets are cleared by the reticuloendothelial system. Recent evidence suggests there is a concomitant platelet production defect. ■ First-line treatment of ITP includes corticosteroids, IVIG, and intravenous Rh immune globulin in Rh-positive patients. A longer course of corticosteroids is preferred over a shorter course of corticosteroids or IVIG. IVIG and Rh immunoglobulin are preferred if steroids are contraindicated. ■ Second-line treatments for patients unresponsive to first-line therapy include rituximab, dapsone, and splenectomy. Splenectomy is avoided in pediatric patients because of the long-term risk of sepsis from encapsulated bacteria such as Meningococcus, Pneumococcus, and Haemophilus. ■ Platelet transfusion is generally not useful because the transfused platelets are cleared at the same rate as the patient’s own platelets. However, if there is significant bleeding or bleeding risk, then platelet transfusion may be indicated.20 48. A. Fonio distribution. Major points of discussion ■ The platelet distribution (histogram) provides information beyond just the platelet count. It provides the platelet distribution width (PDW; platelet anisocytosis), mean platelet volume (MPV; average size), and the total platelet count (area under the curve). ■ The platelet distribution is positively (right) skewed. Taller on the left side (small particles) than the right side (larger particles) of the distribution. ■ The mean platelet volume (MPV) can be calculated as: MPV (fL) = Platelet crit (%)/Platelet count (109/L). ■ An increase in larger platelets would increase the right side of the platelet distribution and also increase the MPV. An increase in larger platelets suggests new platelet production after platelet destruction (e.g., immune thrombocytopenia, TTP) or marrow recovery (e.g., after bone marrow transplant). 49. A. HPA-1a. Major points of discussion ■ Antiplatelet antibodies cross the placenta and lead to platelet clearance and fetal/neonatal thrombocytopenia. ■ NAIT increases the risk of intracerebral hemorrhage in utero, perinatally, and postnatally. ■ The platelet antigen HPA-1a (or, equivalently, PLA1) is most commonly implicated in NAIT. ■ Diagnostic workup includes antigen phenotyping and/or genotyping of maternal, paternal, and fetal platelet antigens. ■ Treatment may include fetal monitoring and intrauterine or postdelivery platelet transfusion with antigen-negative platelets (often collected from the mother).1 50. A. GPIIb/IIIa. Major points of discussion ■ Adhesion is mediated by platelet GPIbα binding to subendothelial vWF and platelet GPVI binding to subendothelial collagen. ■ Aggregation is mediated by platelet GPIIb/IIIa binding to fibrinogen and vWF. ■ Platelets are also activated when specific receptors are bound to their ligands—for example: ADP-P2Y12 or P2Y1; ATP-P2X1; serotonin-serotonin receptor; thromboxane-thromboxane receptor; epinephrine–α-adrenergic receptor, and so on. ■ Thrombin is the most potent activator of platelets via the PAR-1 and PAR-4 receptors. Thrombin activates the platelet by irreversibly cleaving the first extracellular loop of the PAR, leading to a tethered ligand. ■ Antiplatelet pharmaceuticals have been used for primary and secondary prophylaxis of acute myocardial infarction and cerebrovascular accidents (e.g., clopidogrel-P2Y12). 51. A. Aspirin, 325 mg per day. Major points of discussion ■ In this test, whole blood flows through an orifice coated with collagen and a platelet agonist (Epi or ADP) under high shear stress. As platelets adhere to the orifice and become activated, a platelet plug is formed and closes the orifice. ■ The time it takes for a sample to close the orifice is called the closure time. A normal closure time with Col/Epi and Col/ADP suggests normal platelet function. ■ However, the PFA-100 test may not be sensitive enough to detect mild von Willebrand disease and mild platelet function disorders. ■ Scott syndrome is a disorder of platelet procoagulant activity caused by defective exposure of platelet membrane phosphatidyl serine upon platelet activation. In this disorder, the PFA-100 closure time would be expected to be normal for both the Col/Epi and Col/ADP cartridges. 52. A. 106 Major points of discussion ■ The lifespan of a platelet is approximately 7 to 10 days. ■ Production of platelets can increase approximately 10-fold under physiologic stress. ■ TPO is a factor that stimulates megakaryocytes to produce additional platelets. ■ TPO analogs, such as eltrombopag and romiplostim, can be used to stimulate platelet production. 53. A. Minimal bleeding from superficial cuts. Major points of discussion ■ Coagulation disorders are characterized by deep hematomas, minimal bleeding from superficial cuts, hemarthroses, and delayed postoperative bleeding. ■ Platelet disorders are more commonly seen in women than men. Coagulation disorders are more commonly seen in men because of the sex-linked inheritance. ■ von Willebrand disease is a quantitative or qualitative disorder of vWF. Platelet function is normal, but bleeding characteristic of a platelet disorder is seen (e.g., mucosal bleeding, epistaxis, petechiae, menorrhagia). This is due to the impaired ability of platelets to adhere to the subendothelium in the absence of vWF. ■ In severe von Willebrand disease, factor VIII levels can be decreased and bleeding characteristic of a coagulation factor deficiency can be seen. This is due to the decreased stability of factor VIII in the absence of vWF. 54. A. Erythropoietin (EPO). Major points of discussion ■ TPO stimulates megakaryocyte proliferation and differentiation, as well as platelet production. ■ TPO is the ligand for CD110, the TPO receptor. The c-mpl gene encodes the CD110 protein. ■ After binding TPO, CD110 forms dimers, leading to phosphorylation and activation of CD110, Janus kinases (JAKs), signal transducers and activators of transcription (STATs), and other downstream pathways, including mitogen-activated protein (MAP) kinases. ■ Liver parenchymal and endothelial cells, as well as bone marrow stromal cells, are the main sites of TPO production. ■ A recombinant form of TPO was used as a pharmaceutical, but trials were stopped when study volunteers developed anti-TPO autoantibodies and subsequent thrombocytopenia. ■ Current TPO mimetics stimulate the TPO receptor but are not structurally related to TPO. Therefore, autoantibodies recognizing TPO are not a long-term complication. ■ However, the TPO mimetics have long-term side effects of thromboembolism, hepatic toxicity, and bone marrow reticulin deposition. ■ TPO mimetics are used as a second-line treatment for chronic immune thrombocytopenia when there is a risk of bleeding. 55. A. Bone marrow reticulin deposition. Major points of discussion ■ TPO mimetics, such as eltrombopag and romiplostim, stimulate megakaryocytes to produce platelets. ■ These agents are used as a second-line treatment of chronic ITP when patients do not respond to treatment with corticosteroids, intravenous globulins, or splenectomy and when there is a risk of bleeding. They should not be used solely to normalize platelet counts. ■ TPO mimetics can also be used to increase the platelet count in patients with hepatitis C so they qualify to receive α-interferon. ■ Long-term complications of therapy include hepatotoxicity, thromboembolism, increased blast counts, and cataracts. ■ TPO mimetics can either initiate or worsen bone marrow deposition of reticulin-type collagen. They should not be used in patients with myelodysplastic syndrome.2 56. A. ADAMTS13. Major points of discussion ■ Weibel-Palade bodies are secretory organelles found in endothelial cells. ■ They appear as oval or elongated organelles with a whorled or fingerprint-like appearance on electron microscopy. ■ Weibel-Palade bodies store vWF and P-selectin. vWF participates in primary hemostasis by binding platelets to the subendothelium and also stabilizes factor VIII in the plasma. P-Selectin participates in leukocyte adhesion. ■ Qualitative or qualitative disorders of vWF can lead to a mild to severe bleeding disorder known as von Willebrand disease. ■ Other proteins contained in Weibel-Palade bodies include interleukin 8, endothelin, and tissue plasminogen activator. ■ Desmopressin-induced increase in VWF is most likely due to the direct release of multimeric vWF from Weibel Palade bodies.
Hematology
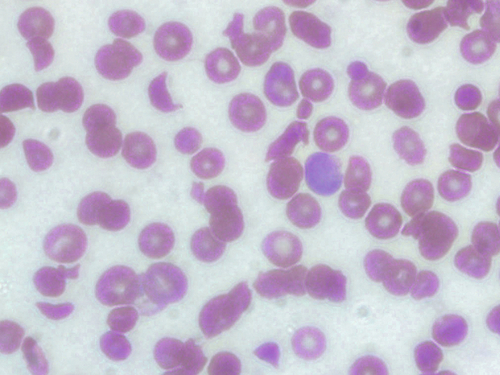
Platelets (Qualitative and Quantitative)
Cartridge
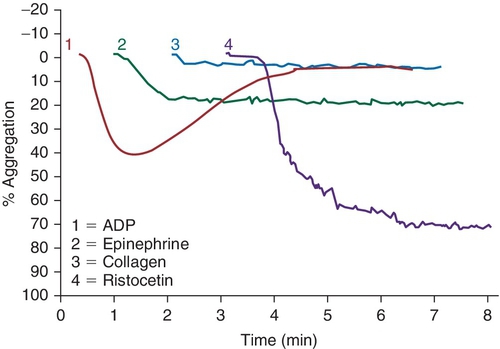
Result
Reference Range
Collagen/epinephrine (Col/Epi)
89 sec
< 120 sec
Collagen/adenosine diphosphate (Col/ADP)
113 sec
< 180 sec
Rationale: PTCP is almost exclusively caused by the presence of EDTA and is thought to be enhanced at colder temperatures.
B. Foil-wrapped potassium EDTA tube.
Rationale: Foil wrapping would not prevent pseudothrombocytopenia. Foil is used for light-sensitive analytes, such as bilirubin, erythrocyte protoporphyrin, and carotene.
C. 3.2% sodium citrate tube.
Rationale: In the absence of EDTA, platelet clumps are less likely to form and an accurate automated platelet count can be determined.
D. Glass tube with no additives.
Rationale: A glass tube with no additives would clot the sample and remove coagulation factors and platelets. However, if analyzed when the sample is very fresh, an accurate platelet count may be obtained with this approach.
E. Serum separator tube.
Rationale: A serum separator tube would clot the sample and remove coagulation factors and platelets.
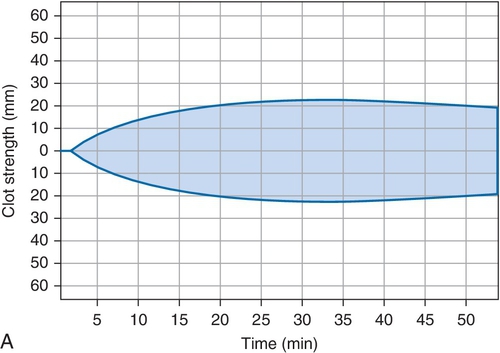
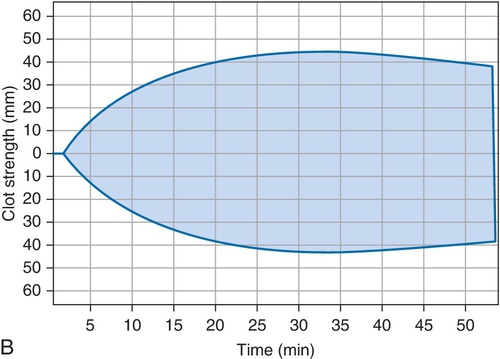
Rationale: The platelet count would be expected to be lower in ITP. Clinical presentation is better for HIT.
B. Heparin-induced thrombocytopenia (HIT).
Rationale: The treatment of the patient with heparin, the timing and degree of thrombocytopenia, and the presence of thrombosis are all consistent with HIT.
C. Thrombotic thrombocytopenic purpura (TTP).
Rationale: No schistocytes are found upon examination of the peripheral smear, the LDH is not elevated, and the hemoglobin is stable, making TTP unlikely.
D. DIC.
Rationale: The coagulation parameters, including the activated partial thromboplastin time (aPTT), prothrombin time (PT), fibrinogen, and d-dimer are all within the reference range, and these would be abnormal in DIC.
E. Hemodilution.
Rationale: The decreased platelet count is most likely not due to hemodilution, because the other parameters in the CBC are stable.
Rationale: Plasmapheresis is not a first-line therapy for HIT.
B. Discontinue heparin.
Rationale: Heparin should be discontinued; however, HIT is suspected, which puts the patient at an increased risk of thrombosis. Therefore an alternate anticoagulant should be given.
C. Transfuse platelets.
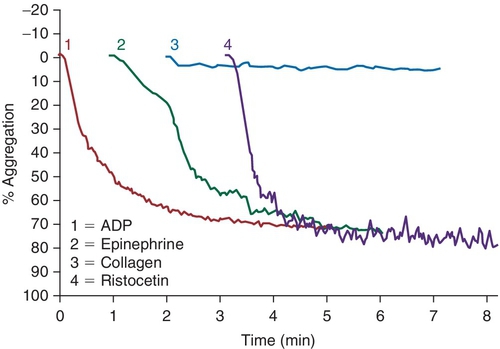
Rationale: There is no evidence of bleeding, and the platelet count is at the lower limit of normal with no evidence of decreased platelet function. Therefore, platelet transfusion would not be appropriate.
D. Discontinue heparin and begin treating with a direct thrombin inhibitor.
Rationale: Heparin should be discontinued and the patient should start an alternate anticoagulant.
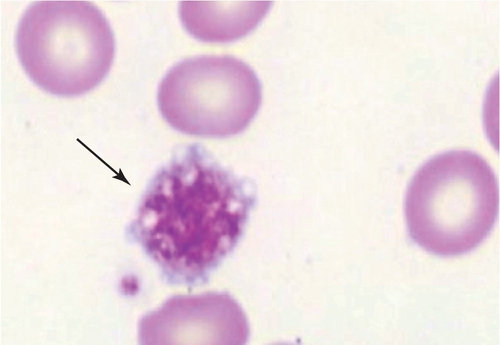
E. Discontinue heparin and begin warfarin.
Rationale: Heparin should be discontinued; however, warfarin is inappropriate in the acute setting.
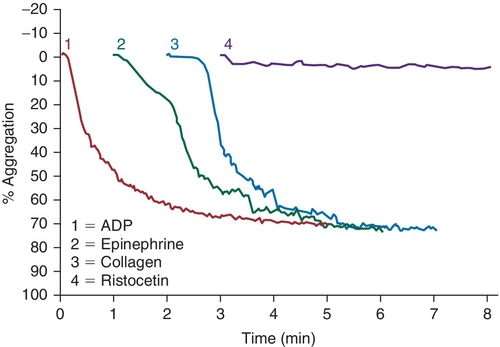
Rationale: Light transmission aggregometry is not consistent with a defect in GPIa/IIa, which is a collagen receptor.
B. Anti-GPIbα.
Rationale: The platelets from patients with BS syndrome aggregate with physiologic agonists, but do not aggregate when induced with ristocetin. In the absence of GPIb/IX/V, isoantibodies against GPIb can form, leading to ineffective responses to platelet transfusions.
C. Anti-GPIIb/IIIa.
Rationale: Patients with Glanzmann thrombasthenia have absent or nonfunctioning platelet glycoprotein GPIIb/IIIa. Thus, platelet binding to fibrinogen and vWF is impaired and platelets do not aggregate with physiologic agonists. The absence of only ristocetin-induced platelet aggregation suggests a defect in the GPIb/V/IX complex.
D. Anti-GPVI.
Rationale: The light transmission aggregometry results in this case are not consistent with a defect in GPVI, which is a collagen receptor.
E. Anti–HPA-1a.
Rationale: HPA-1 is an antigen that is commonly implicated in posttransfusion purpura. The light transmission aggregometry results in the case presented would not be seen in this disorder.
Rationale: ADP is found in platelet-dense bodies.
B. Magnesium.
Rationale: Magnesium is found in platelet-dense bodies.
C. Platelet-derived growth factor (PDGF).
Rationale: PDGF is found in platelet α-granules.
D. Pyrophosphate.
Rationale: Pyrophosphate is found in platelet-dense bodies.
E. Serotonin.
Rationale: Serotonin is found in platelet-dense bodies.
Rationale: Quebec platelet syndrome is associated with a defect of the α-granules.
B. Chediak-Higashi syndrome.
Rationale: Chediak-Higashi syndrome is associated with a defect of the β-granules.
C. Wiskott-Aldrich syndrome.
Rationale: Wiskott-Aldrich syndrome is associated with defects in platelet signal transduction and maintenance of the cytoskeleton.
D. Bernard-Soulier (BS) syndrome.
Rationale: BS syndrome is associated with a defect of the membrane GPIb-IX-V complex.
E. Gray platelet syndrome.
Rationale: Gray platelet syndrome is associated with a defect of the α-granules.
Rationale: Turbidometric aggregometry is considered the gold standard for platelet function testing; however, it is not an in vivo test.
B. Bleeding time (BT).
Rationale: The BT is considered an in vivo platelet function test.
C. PFA-100.
Rationale: The PFA-100 measures the time required for the cessation of blood flow in an in vitro assay.
D. Vasodilator-stimulated phosphoprotein (VASP) phosphorylation assay.
Rationale: The VASP phosphorylation assay is a flow cytometry–based in vitro assay.
E. Cone and plate analyzer.
Rationale: The cone and plate analyzer is an in vitro assay that measures shear-induced platelet adhesion.
B. Hematocrit of 25%.
Rationale: This prolongs the bleeding time (BT).
C. Platelet count of 200 × 109/L.
Rationale: A normal platelet count should not affect the BT.
D. Pregnancy.
E. Repeating the BT within 4 hours.
Rationale for A, D, and E: This shortens the BT.
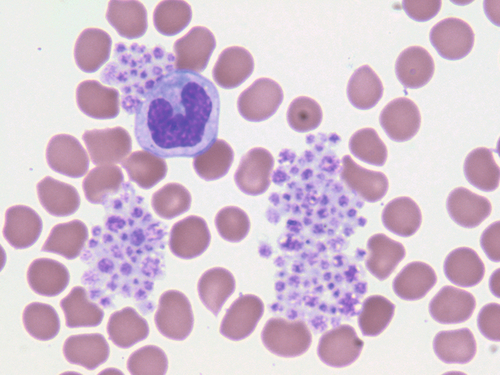
Rationale: Döhle-like bodies in neutrophils on May-Grünwald-Giemsa–stained peripheral blood are suggestive of MYH-9–related syndromes (of which May-Hegglin anomaly is one).
B. Chediak-Higashi syndrome.
Rationale: Large peroxidase-positive granules in both hematopoietic (neutrophils) and nonhematopoietic cells are found.
C. BS syndrome.
Rationale: Absent ristocetin-induced platelet aggregation is a key finding in BS syndrome. Large platelets are also seen.
D. Hermansky-Pudlak syndrome.
Rationale: Large peroxidase-positive granules are not found in the Hermansky-Pudlak syndrome, which, like Chediak-Higashi syndrome, is associated with oculocutaneous albinism.
E. Gray platelet syndrome.
Rationale: A classical finding in gray platelet syndrome is a lack of α-granules when platelets are examined by electron microscopy.
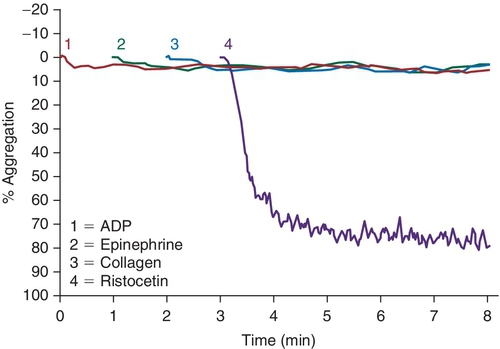
Rationale: In this case, the platelet function study would show an absence of ristocetin-induced platelet aggregation, but normal aggregation with the other agonists.
B. GPIIb/IIIa.
Rationale: In this case, the platelet function study would show an absence of aggregation with ADP, epinephrine, and collagen, but the ristocetin results would be normal.
C. GPVI.
Rationale: GPVI is a collagen binding and signaling receptor. In this case, abnormal aggregation with collagen would be expected.
D. P2Y1.
E. P2Y12.
Rationale for D and E: P2Y12 is an ADP receptor. In this case, the aggregation assay would be expected to show an abnormal ADP response.
Rationale: The cone and plate analyzer evaluates high shear platelet adhesion onto a surface.
B. VerifyNow.
Rationale: The VerifyNow assay does not use shear conditions.
C. Impedance aggregometry.
Rationale: Impedance aggregometry does not use shear conditions.
D. Assays that measure thromboxane (Tx) A2 metabolites.
Rationale: Assays that measure TxA2 metabolites are immunoassays that measure stable Tx metabolites in the serum or urine.
E. Turbidometric aggregometry.
Rationale: Turbidometric aggregometry does not use shear conditions.
Rationale: May-Hegglin anomaly is associated with mutation of MYH9 gene.
B. Amegakaryocytic thrombocytopenia with radioulnar synostosis.
Rationale: Amegakaryocytic thrombocytopenia with radioulnar synostosis is associated with a mutation of the HOXA11 gene.
C. Scott syndrome.
Rationale: Scott syndrome is associated with mutation of the ABCA1 gene.
D. Wiskott-Aldrich syndrome.
Rationale: Wiskott-Aldrich syndrome is associated with mutation of the WAS gene.
E. Congenital amegakaryocytic thrombocytopenia.
Rationale: Congenital amegakaryocytic thrombocytopenia is associated with mutation of the MPL gene.
Rationale: α-Granules contain β-thromboglobulin.
B. Calcium.
Rationale: Dense bodies contain calcium, magnesium, and pyrophosphate.
C. Fibrinogen.
Rationale: α-Granules contain fibrinogen.
D. P-selectin.
Rationale: α-Granules contain P-selectin.
E. von Willebrand factor (vWF).
Rationale: α-Granules contain vWF.
Rationale: Fresh nonanticoagulated blood is required for the Görög thrombosis test, an assay that assesses high shear–dependent platelet function and thrombolysis.
B. Whole blood.
Rationale: Whole blood samples are used for flow cytometry–based platelet function tests.
C. Serum.
Rationale: Serum is used for measuring TxA2 metabolites in the blood.
D. Platelet-rich plasma (PRP).
Rationale: Preparation of PRP is a step in the sample preparation for turbidometric aggregometry.
E. Platelet-poor plasma (PPP).
Rationale: Preparation of PPP is a step in the sample preparation for turbidometric aggregometry.
Rationale: CD63 is decreased/absent in Hermansky-Pudlak syndrome.
B. CD41.
Rationale: CD41 (GPIIb) is decreased/absent in Glanzmann thrombasthenia.
C. CD154.
Rationale: CD154 is the CD40 ligand.
D. CD49b.
Rationale: CD49b (GPIa) is a component of the collagen receptor (GPIa/IIa).
E. CD29.
Rationale: CD29 (GPIIa) is a component of the collagen receptor (GPIa/IIa).
Rationale: Platelet factor V is absent in gray platelet syndrome.
B. β-Thromboglobulin.
Rationale: β-Thromboglobulin is absent in gray platelet syndrome.
C. Fibrinogen.
Rationale: Platelet fibrinogen is absent in gray platelet syndrome.
D. P-selectin.
Rationale: P-selectin is retained in Gray platelet syndrome.
E. Platelet factor 4.
Rationale: Platelet factor 4 is absent in gray platelet syndrome.
Rationale: Sebastian syndrome is not associated with oculocutaneous albinism.
B. Wiskott-Aldrich syndrome.
Rationale: Wiskott-Aldrich syndrome is not associated with oculocutaneous albinism.
C. Hermansky-Pudlak syndrome.
Rationale: Hermansky-Pudlak syndrome is associated with oculocutaneous albinism.
D. Paris-Trousseau/Jacobsen syndrome.
Rationale: Paris-Trousseau/Jacobsen syndrome is not associated with oculocutaneous albinism.
E. BS syndrome.
Rationale: BS syndrome is not associated with oculocutaneous albinism.
Rationale: Neutrophil inclusions would not be present.
B. May-Hegglin anomaly.
Rationale: Presents with thrombocytopenia, giant platelets, and Döhle-like inclusions in neutrophils without hearing loss, nephritis, or cataracts.
C. Epstein syndrome.
Rationale: Characterized by thrombocytopenia, giant platelets, hearing loss, and nephritis. Döhle-like bodies are not present.
D. Fechtner syndrome.
Rationale: Characterized by thrombocytopenia, giant platelets, Döhle-like inclusions in neutrophils, hearing loss, nephritis, and cataracts.
E. Glanzmann thrombasthenia.
Rationale: Does not present with thrombocytopenia.
Rationale: Paris-Trousseau/Jacobsen syndrome is characterized by the presence of giant α-granules.
B. Chediak-Higashi syndrome.
Rationale: Chediak-Higashi syndrome is associated with a defect of the dense granules.
C. BS syndrome.
Rationale: BS syndrome is associated with a defect in the GPIb/V/IX complex.
D. Hermansky-Pudlak syndrome.
Rationale: Hermansky-Pudlak syndrome is associated with a defect of the dense granules.
E. Glanzmann thrombasthenia.
Rationale: Glanzmann thrombasthenia is associated with a defect of GPIIb/IIIa.
Rationale: The Plateletworks assay does not use high shear blood flow conditions to test platelet function.
B. Thromboelastography.
Rationale: Thromboelastography does not use high shear blood flow conditions to test platelet function.
C. VerifyNow.
Rationale: The VerifyNow assay does not use high shear blood flow conditions to test platelet function.
D. Light transmission aggregometry.
Rationale: Light transmission aggregometry does not use high shear blood flow conditions to test platelet function.
E. PFA-100.
Rationale: The PFA-100 uses high shear blood flow conditions to test platelet function.
B. Fresh frozen plasma.
C. Platelets.
Rationale: The decreased maximum amplitude with a sufficiently hemostatic platelet count (> 50 mg/dL) suggests that there is platelet dysfunction. A platelet transfusion may be useful.
D. Prothrombin complex concentrates.
Rationale for A, B, and D: The thromboelastogram does not suggest a coagulation factor deficiency.
E. RBCs.
Rationale: A hemoglobin level higher than 10 g/dL is adequate for a patient with cardiac disease.
Rationale: The Plateletworks assay uses detection of platelet aggregation as its end point.
B. PFA-100.
Rationale: The PFA-100 is based on high shear blood flow.
C. Cone and plate analyzer.
Rationale: The cone and plate analyzer uses shear-induced platelet adhesion as its end point.
D. VASP.
Rationale: The VASP phosphorylation assay does not use detection of platelet aggregation as its end point. It is a flow cytometry–based assay.
E. Thromboelastography.
Rationale: Thromboelastography uses global clot formation as its end point.
Rationale: An elevated lactate dehydrogenase (LDH) in serum and schistocytes on the peripheral smear would be expected.
B. ITP.
Rationale: The only abnormality that would be expected in ITP is a low platelet count.
C. DIC.
Rationale: DIC often presents with a prolonged PT and APTT, increased LDH, and schistocytes on the peripheral smear.
D. Syndrome of hemolysis, elevated liver enzymes, and low platelet count (HELLP).
Rationale: HELLP presents with elevated aspartate aminotransferase (AST), alanine aminotransferase (ALT), LDH, and schistocytes on the peripheral smear.
E. Hemolytic uremic syndrome (HUS).
Rationale: HUS presents with elevated LDH, blood urea nitrogen (BUN), and creatinine, as well as changes in RBC morphology, including schistocytes.
Rationale: α2β1 is a platelet collagen receptor and is not blocked by abciximab.
B. GPIbα.
Rationale: GPIbα is a platelet vWF receptor involved in platelet adhesion and is not blocked by abciximab.
C. GPIIb/IIIa.
Rationale: Abciximab contains antibody Fab fragments that block GPIIb/IIIa (αIIbβ3). It is used to treat acute coronary syndromes and for percutaneous coronary intervention (i.e., cardiac catheterization).
D. P2X1.
Rationale: P2X1 is the platelet ATP receptor and is not blocked by abciximab.
E. P2Y12.
Rationale: P2Y12 is the platelet ADP receptor and is not blocked by abciximab.
Rationale: There are no Food and Drug Administration (FDA)-approved pharmaceuticals that inhibit GPIbα.
B. GPIIb/IIIa.
Rationale: GPIIb/IIIa can be pharmaceutically blocked by abciximab, tirofiban, and eptifibatide.
C. GPVI.
Rationale: Currently, there are no FDA-approved pharmaceuticals that inhibit GPVI.
D. P2X1.
Rationale: P2X1 is an ATP receptor and is not specifically blocked by clopidogrel.
E. P2Y12.
Rationale: Clopidogrel is a strong antiplatelet agent that blocks P2Y12, the platelet ADP receptor.
Rationale: Sebastian syndrome is associated with macrothrombocytopenia.
B. Fechter syndrome.
Rationale: Fechter syndrome is associated with macrothrombocytopenia.
C. Scott syndrome.
Rationale: Scott syndrome is associated with inadequate exposure of negatively charged phospholipids.
D. Epstein syndrome.
Rationale: Epstein syndrome is associated with macrothrombocytopenia.
E. Chediak-Higashi syndrome.
Rationale: Chediak-Higashi syndrome is associated with a dense-granule defect.
Rationale: Flow cytometry to detect various proteins, such as P-selectin and platelet surface activated GPIIb/IIIa, or to identify leukocyte-platelet aggregates, is used to evaluate activation-dependent changes in platelet surface marker expression.
B. in vivo cessation of blood flow by a newly formed platelet plug.
Rationale: The BT measures the time required for in vivo cessation of blood flow by a newly formed platelet plug.
C. Shear-induced platelet adhesion.
Rationale: The cone and plate analyzer evaluates shear-induced platelet adhesion.
D. Rate and quality/strength of clot formation.
Rationale: Thromboelastography evaluates the rate and quality/strength of clot formation.
E. Platelet-to-platelet aggregation.
Rationale: Aggregometry evaluates platelet-to-platelet aggregation.
Rationale: The VerifyNow assay is based on platelet aggregation.
B. PFA-100.
Rationale: The PFA-100 is based on high shear blood flow.
C. Measurement of serum TxB2.
Rationale: Measurement of serum thromboxane B2, a metabolite of TxA2, uses an immunoassay.
D. Measurement of activated GPIIb/IIIa.
Rationale: Measurement of activated GPIIb/IIIa is based on flow cytometry.
E. Plateletworks.
Rationale: The Plateletworks assay is based on platelet aggregation.
Rationale: Arthrogryposis–renal dysfunction–cholestasis is associated with a defect in the α-granules.
B. Paris-Trousseau/Jacobsen syndrome.
Rationale: Paris-Trousseau/Jacobsen syndrome is associated with a defect in the α-granules.
C. Chediak-Higashi syndrome.
Rationale: Chediak-Higashi syndrome is associated with a defect in the α-granules.
D. Tx synthase deficiency.
Rationale: Tx synthase deficiency is associated with a defect in platelet signal transduction.
E. Hermansky-Pudlak syndrome.
Rationale: Hermansky-Pudlak syndrome is associated with a defect in the dense granules.
Rationale: The PFA-100 assay is not considered the historical gold standard for platelet function testing.
B. Impedance aggregometry.
Rationale: Impedance aggregometry is not considered the historical gold standard for platelet function testing.
C. Turbidometric aggregometry.
Rationale: Turbidometric aggregometry is considered the historical gold standard for platelet function testing.
D. Measurement of TxA2 metabolites.
Rationale: Measurement of TxA2 metabolites is not considered the historical gold standard for platelet function testing. These metabolites are not specific for platelets.
E. Measurement of activated GPIIb/IIIa by flow cytometry.
Rationale: Measurement of activated GPIIb/IIIa by flow cytometry is not considered the historical gold standard for platelet function testing.
Rationale: Turbidometric aggregometry is considered the gold standard overall for platelet function testing, but not specifically for assessing the P2Y12 antagonist effect.
B. Thromboelastography.
Rationale: Thromboelastography is not considered the gold standard for assessing the P2Y12 antagonist effect.
C. Plateletworks.
Rationale: The Plateletworks assay is not considered the gold standard for assessing the P2Y12 antagonist effect.
D. VASP phosphorylation.
Rationale: VASP phosphorylation is considered the gold standard for assessing the P2Y12 antagonist effect.
E. VerifyNow.
Rationale: The VerifyNow assay is not considered the gold standard for assessing the P2Y12 antagonist effect.
Rationale: BS syndrome is not associated with immunodeficiency.
B. Scott syndrome.
Rationale: Scott syndrome is not associated with immunodeficiency.
C. Glanzmann thrombasthenia.
Rationale: Glanzmann thrombasthenia is not associated with immunodeficiency.
D. Quebec platelet syndrome.
Rationale: Quebec platelet syndrome is not associated with immunodeficiency.
E. Wiskott-Aldrich syndrome.
Rationale: Wiskott-Aldrich syndrome is associated with immunodeficiency.
Rationale: 2,3-dinor TxB2 is formed by β-oxidation of TxA2. Compared with 11-dehydro-TxB2, it has a shorter half-life and is present in smaller amounts in urine.
B. 11-dehydro-TxB2.
Rationale: 11-dehydro-TxB2 is formed by dehydrogenation of TxB2. Compared with 2,3-dinor TxB2, it is more abundant in urine and has a longer half-life.
C. Prostaglandin G2.
Rationale: Prostaglandin G2 is rapidly converted to PGH2 by prostaglandin G/H synthase.
D. TxA2.
Rationale: TxA2 is rapidly hydrolyzed to TxB2.
E. TxB2.
Rationale: TxB2 is rapidly metabolized and only a small fraction is excreted into urine. It is also produced during storage of blood samples.
Rationale: Aspirin irreversibly inhibits cyclooxygenase, which leads to impaired TxA2 production, and platelet aggregation is inhibited. The secondary wave of aggregation is usually missing after ADP or epinephrine agonists. Collagen aggregation is impaired. Ristocetin aggregation is unaffected.
B. BS syndrome.
Rationale: BS syndrome is the absence of the GPIb/V/IX complex, which leads to defective binding of vWF. Ristocetin aggregation is absent, but ADP, epinephrine, and collagen all induce aggregation.
C. Clopidogrel therapy.
Rationale: Clopidogrel inhibits the P2Y12 receptor, which is the main receptor for ADP. Aggregation with ADP should not be present.
D. Glanzmann thrombasthenia.
Rationale: Glanzmann thrombasthenia is a disorder of GPIIb/IIIa. Aggregation with ADP, epinephrine, and collagen are absent, and ristocetin-induced aggregation is normal.
E. von Willebrand disease, type III.
Rationale: von Willebrand disease, type III shows the absence of vWF multimers. Ristocetin would not induce aggregation.
Rationale: Amphotericin is a common cause of drug-induced thrombocytopenia.
B. Furosemide.
Rationale: Furosemide has been implicated as a cause of drug-induced thrombocytopenia.
C. Piperacillin/tazobactam.
Rationale: Piperacillin/tazobactam is a common cause of drug-induced thrombocytopenia.
D. Romiplostim.
Rationale: Romiplostim is a TPO analog that stimulates platelet production.
E. Tacrolimus.
Rationale: Tacrolimus is a cause of drug-induced thrombocytopenia, including TTP.
Rationale: The Philadelphia chromosome results in the BCR-ABL fusion gene; it is associated with chronic myelogenous leukemia.
B. EPO mutation.
Rationale: Mutation of the EPO gene is not involved in the pathogenesis of essential thrombocythemia (ET).
C. JAK2 V617F.
Rationale: JAK2 V617F is found in 50% of patients with ET.
D. MPL mutation.
Rationale: MPL mutations are found in 4% of patients with ET.
E. PML-RAR fusion gene.
Rationale: The PML-RAR fusion gene is found in acute promyelocytic leukemia.
Rationale: Large platelets can be counted as red cells, causing a falsely low platelet count.
B. Microclots.
Rationale: Platelets can become part of microclots, causing a falsely low platelet count.
C. Platelet satellitism.
Rationale: Platelets can adhere to the periphery of white cells, causing a falsely low platelet count.
D. Pseudothrombocytopenia (PTCP).
Rationale: PTCP, caused by anti-EDTA/platelet antibodies, results in the formation of platelet clumps. This causes a falsely low platelet count.
E. Schistocytes.
Rationale: Schistocytes, which are red cell fragments created by mechanical shear stress, can be small enough to be counted as platelets by impedance-based analyzers. This causes a falsely elevated platelet count.
Rationale: Extracellular Babesia can be mistaken for normal platelets. These organisms usually measure 1 to 2 μm, whereas giant platelets are larger than 10 μm.
B. Gray platelet syndrome.
Rationale: Platelets in gray platelet syndrome are usually normal in size but can be enlarged. However, the platelets lack the basophilic granules, which gives them a hypogranular, gray, amorphous appearance.
C. Myelodysplastic syndrome.
Rationale: Giant platelets are associated with various conditions, including myelodysplastic syndrome and leukemias.
D. Pseudothrombocytopenia.
Rationale: In pseudothrombocytopenia, many agglutinated platelets are seen; these agglutinates should not be mistaken for giant platelets.
E. Wiskott-Aldrich syndrome.
Rationale: Wiskott-Aldrich syndrome is associated with thrombocytopenia and small platelets.
Rationale: Maternal platelets will lack the antigen to which the maternal antibody is directed against. However, the plasma will contain maternal antibodies that would be transfused along with the platelets. Some transfusion medicine physicians recommend volume reducing or washing the maternal platelets before transfusion.
B. Whole blood–derived random donor platelets.
Rationale: Random donor platelets would most likely carry the antigen implicated in NAIT.
C. Paternal platelets.
Rationale: Paternal platelets will express the antigen that immunized the mother of the newborn. Therefore, they are a poor choice for transfusion.
D. Single-donor platelets.
E. Washed single-donor platelets.
Rationale: Single-donor platelets will most likely carry the antigen implicated in NAIT. The plasma from such a donor is unlikely to contain the antiplatelet antibodies implicated in NAIT; therefore, washing would not be useful. There is also some concern that washing platelets decreases their number and functionality.
Rationale: Sodium citrate is a weak calcium chelator and is the anticoagulant of choice for platelet function studies.
B. K2-ethylenediaminetetraacetic acid (K2EDTA), purple top tube.
Rationale: K2EDTA is a strong calcium chelator and is not used in platelet function studies.
C. Lithium heparin, green top tube.
Rationale: Heparin affects platelets by decreasing the response to epinephrine and collagen agonists. In addition, spontaneous aggregation can occur due to the presence of physiological concentrations of calcium in the anticoagulant.
D. No additive, red top tube.
E. Serum separator, gold top tube.
Rationale: Clotting in these tubes will consume platelets.
B. Perform apheresis followed by hydroxyurea treatment.
C. Treat with hydroxyurea.
Rationale: Apheresis and hydroxyurea do not a have a role in treating reactive thrombocytosis.
D. No additional therapy is necessary.
Rationale: Reactive thrombocytosis is not associated with an increased risk of thrombosis. Thrombocytosis caused by myeloproliferative disorders does require treatment when platelet counts are in the range of 1 million/μL or higher.
E. Perform a splenectomy.
Rationale: Splenectomy does not a have a role in reactive thrombocytosis.
Rationale: The absence of coagulation abnormalities is less consistent with DIC than with TTP. Although the PT, aPTT, and fibrinogen can be normal in DIC, more often the consumption of coagulation factors leads to a prolonged PT and aPTT, as well as to low fibrinogen levels.
B. Drug-induced thrombocytopenia.
Rationale: Medications are a common cause of thrombocytopenia and should always be considered as an explanation. However, in this case, medications alone do not explain the microangiopathic hemolytic anemia along with the thrombocytopenia.
C. ITP.
Rationale: ITP presents with thrombocytopenia in the absence of other hematologic abnormalities (e.g., anemia, leukopenia). Therefore, the presence of microangiopathic hemolytic anemia in this case is not consistent with ITP.
D. TTP.
Rationale: TTP is a microangiopathic hemolytic anemia that presents with hemolytic anemia with schistocytes on the peripheral blood smear and thrombocytopenia. Classically, TTP is accompanied by mental status changes, fever, and renal failure, but these three findings may not always be present. In this case, the TTP is drug induced (i.e., by tacrolimus).
E. Warm autoimmune hemolytic anemia (WAIHA).
Rationale: WAIHA alone does not present with low platelets or schistocytes.
Rationale: Clopidogrel metabolism to its active metabolite is slower with the *2 or *3 polymorphisms of the p450 enzyme gene CYP2C19.
B. CYP1A1.
Rationale: This gene encodes the enzyme that metabolizes the R isomer of warfarin into its inactive metabolites.
C. CYP2C9*1.
Rationale: CYP2C9 metabolizes S-warfarin to its inactive metabolites.
D. UGT1A6 T181A A > G.
Rationale: UGT1A6 encodes for UDP-glucuronosyltransferase 1A6, which metabolizes aspirin.
E. VKORC1 GG.
Rationale: The VKORC1 gene encodes the vitamin K epoxide reductase complex involved in vitamin K recycling and warfarin anticoagulation.
Rationale: This is equivalent to approximately 20 × 109/L.
B. 2.
Rationale: This is equivalent to approximately 40 × 109/L.
C. 5.
Rationale: This is equivalent to approximately 100 × 109/L.
D. 10.
Rationale: This is equivalent to approximately 200 × 109/L.
E. 20.
Rationale: This is equivalent to approximately 400 × 109/L.
Rationale: Anticoagulation with unfractionated heparin for a remote history of HIT with negative immunologic and functional assays for HIT is recommended for patients undergoing cardiovascular surgery.
B. Anticoagulation with bivalirudin.
Rationale: In patients with acute HIT and positive immunologic and functional assay results for HIT, delay of cardiovascular surgery until assay results are negative is recommended. However, bivalirudin can be administered if surgery cannot be delayed.
C. Anticoagulation with argatroban.
Rationale: In patients with remote HIT undergoing percutaneous coronary intervention and who have negative immunologic and functional assay results for HIT, a nonheparin alternate anticoagulant can be used (e.g., bivalirudin, argatroban, danaparoid).
D. Cancel surgery indefinitely.
Rationale: Cardiovascular surgery can be performed with remote HIT and negative immunologic and functional assay results for HIT. Alternatively, bivalirudin can be used if surgery cannot be delayed.
E. Delay surgery until anti-heparin/platelet factor-4 IgG, IgA, and IgM ELISA is performed.
Rationale: Delaying surgery until anti-heparin/platelet factor-4 IgG, IgA, IgM ELISA is negative when the IgG assay result is already negative will not change anticoagulation management for this patient.
Rationale: Azathioprine is not recommended as a first-line treatment in a pregnant patient with ITP.
B. Cyclophosphamide.
Rationale: Cyclophosphamide is cytotoxic and teratogenic and should not be used in pregnancy.
C. Corticosteroids.
Rationale: Corticosteroids or intravenous immunoglobulin are the first-line treatments of ITP in pregnancy.
D. Rituximab.
Rationale: Rituximab is a second-line treatment for ITP.
E. Splenectomy.
Rationale: Splenectomy is a second-line treatment and surgery can be difficult because the gravid uterus presents a large obstacle.
Rationale: Plasmapheresis is not used in the treatment of ITP.
B. Dapsone.
Rationale: Dapsone is a second-line treatment for ITP.
C. Corticosteroids.
Rationale: A long course of corticosteroids is the first choice for the treatment of ITP in adults with platelet count less than 30/μL.
D. Rituximab.
Rationale: Rituximab is a second-line treatment for ITP.
E. Splenectomy.
Rationale: Splenectomy is used for patients with ITP in whom corticosteroid therapy has failed. Aside from the morbidity and mortality from the surgical procedure, there is also the long-term risk of infection and sepsis from encapsulated bacteria such as Meningococcus, Pneumococcus, and Haemophilus.
Rationale: Fonio is a method for counting platelets, not a distribution of platelets.
B. Fourier transform of t-distribution.
Rationale: This is irrelevant for platelet counting.
C. Gaussian (normal) distribution.
Rationale: Red cells have a normal distribution when presented as a histogram.
D. Negatively (left) skewed distribution.
Rationale: Platelets do not have a left skewed distribution; it is right skewed.
E. Positively (right) skewed distribution.
Rationale: The platelet histogram is right skewed.
Rationale: HPA-1a (or, equivalently, PLA1) is a common platelet antigen that is most often implicated in NAIT. Antibodies recognizing it account for up to 80% of serologically confirmed cases of NAIT.
B. HPA-1b.
Rationale: HPA-1b (or, equivalently, PLA2) is rarely implicated in NAIT.
C. HPA-2a.
Rationale: HPA-2a is implicated in fewer than 1% of NAIT cases.
D. HPA-3b.
Rationale: HPA-3b is rarely implicated in NAIT.
E. HPA-4a.
Rationale: HPA-4a is commonly implicated in NAIT in Asian populations, but is not the most common among all ethnic groups.
Rationale: GPIIb/IIIa on platelets binds to vWF and fibrinogen to mediate platelet aggregation.
B. GPVI.
Rationale: GPVI on platelets binds to collagen and mediates platelet adhesion under low shear stress.
C. P2X1.
Rationale: ATP binds to the P2X1 receptor and increases intracellular calcium.
D. P2Y12.
Rationale: ADP binds to the platelet P2Y12 and the P2Y1 receptors to mediate platelet activation.
E. Protease-activated receptor 1 (PAR-1).
Rationale: The most potent activator of platelets is mediated by the thrombin–PAR-1 interaction.
Rationale: Therapeutic aspirin effect would show a prolonged closure time with collagen (Col) and epinephrine (Epi; Col/Epi) and a normal closure time with Col/ADP.
B. BS syndrome.
Rationale: Both Col/Epi and Col/ADP would be prolonged in BS syndrome.
C. Glanzmann thrombasthenia.
Rationale: Both Col/Epi and Col/ADP would be prolonged in Glanzmann thrombasthenia.
D. Gray platelet syndrome.
Rationale: Both Col/Epi and Col/ADP would be prolonged in gray platelet syndrome
E. Scott syndrome.
Rationale: Scott syndrome is a disorder of platelet procoagulant activity caused by defective exposure of platelet membrane phosphatidyl serine upon platelet activation.
B. 109
C. 1011
D. 1012
E. 1015
Rationale: Approximately 1011 platelets are produced by an adult on a daily basis under normal, physiologic conditions.
Rationale: Persistent bleeding from superficial cuts is characteristic of platelet disorders.
B. Deep muscle hematomas.
Rationale: Deep hematomas are characteristic of coagulation factor deficiencies.
C. Delayed bleeding.
Rationale: Delayed postoperative bleeding is characteristic of coagulation factor deficiencies.
D. Hemarthrosis.
Rationale: Joint bleeding is characteristic of coagulation factor deficiencies.
E. Petechiae.
Rationale: Petechiae are seen in platelet-type bleeding.
Rationale: EPO is an essential factor for red cell production and initiates its action by binding to EpoR (the EPO receptor).
B. Granulocyte colony-stimulating factor (G-CSF).
Rationale: G-CSF stimulates the proliferation and maturation of cells of the neutrophil lineage. It binds to the G-CSF receptor.
C. PDGF.
Rationale: PDGF stimulates mesenchymal cell proliferation and signals through the PDGF receptor α and β subunits.
D. Signal transducer and activator of transcription-1 (STAT-1).
Rationale: STAT-1 is a downstream target involved in intracellular signaling. It is not an extracellular ligand.
E. TPO.
Rationale: TPO is the ligand for the c-mpl gene product, the TPO receptor. It stimulates platelet production by acting on megakaryocytes and platelets.
Rationale: TPO mimetics may increase the risk or progression of bone marrow reticulin deposition.
B. Berry aneurysms with intracranial hemorrhage.
Rationale: TPO mimetics are not associated with aneurysms or bleeding. However, they can be associated with thromboembolic complications.
C. Pancreatitis.
Rationale: TPO mimetics can cause hepatotoxicity but are not known to be a risk for pancreatitis.
D. Portal vein aneurysms.
Rationale: Portal vein thrombosis, but not aneurysms, can be seen with TPO mimetics.
E. Retinal detachment.
Rationale: Cataracts may increase with TPO mimetics, but retinal detachment is not a complication.
B. Factor VIII.
C. Fibronectin.
D. Tx A2.
Rationale: ADAMTS13, factor VIII, fibronectin, and TxA2 are not stored in endothelial Weibel-Palade bodies.
E. vWF.
Rationale: vWF and P-selectin are stored in Weibel-Palade bodies.24
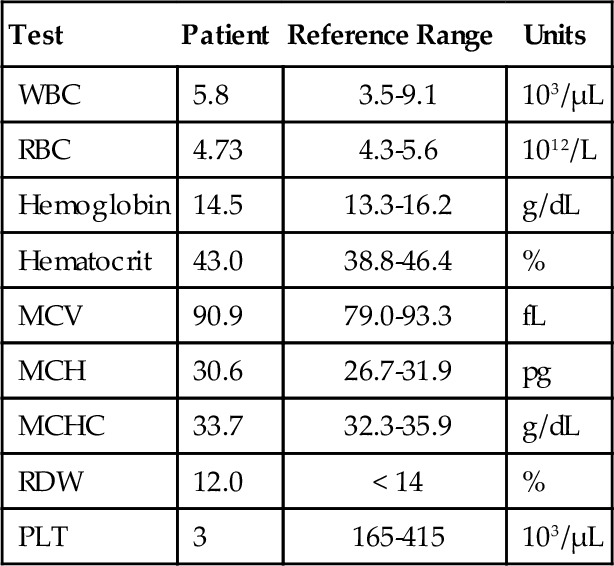
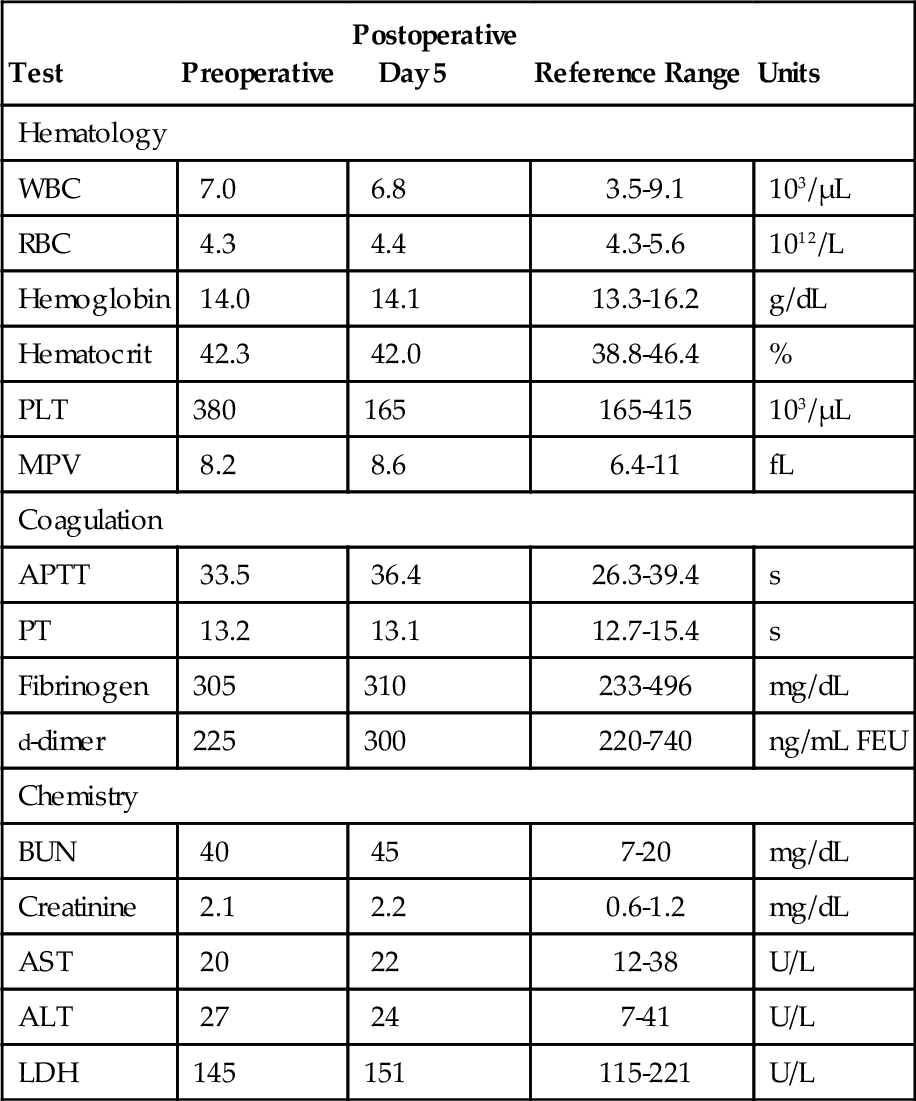
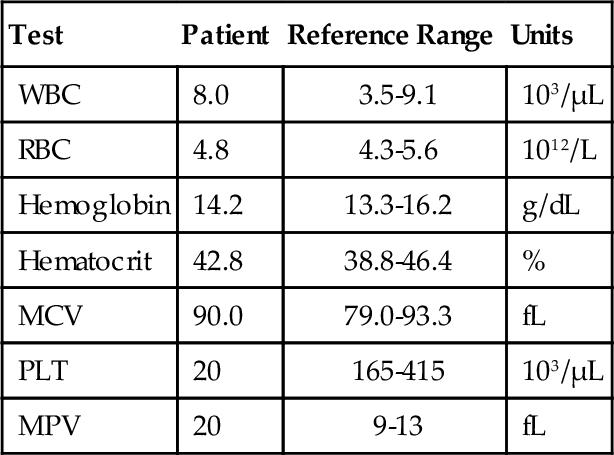
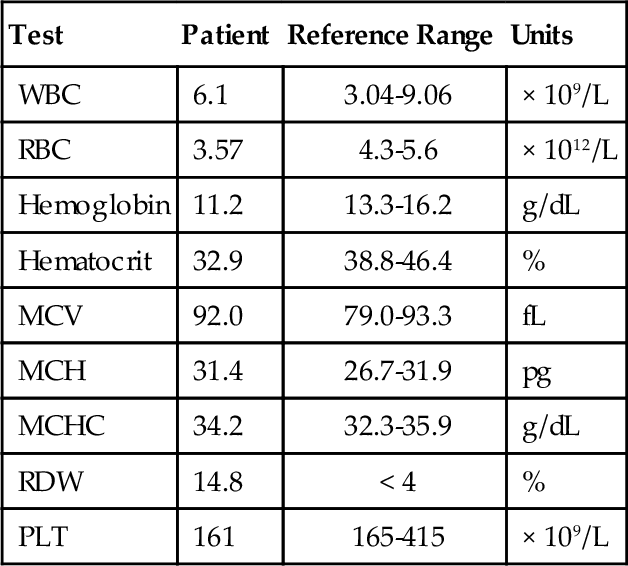
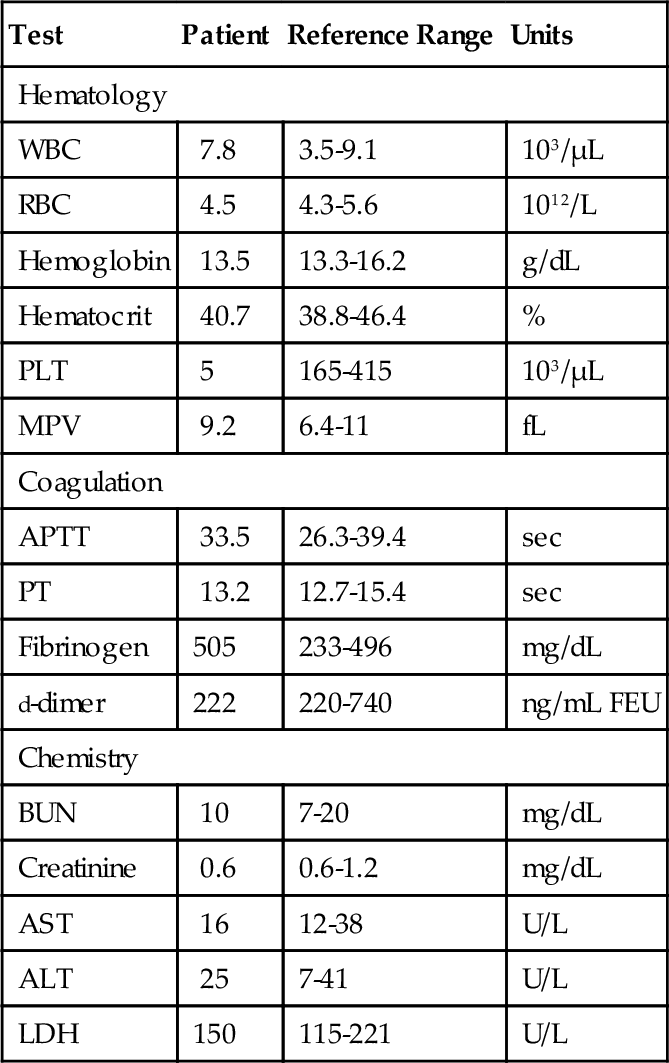
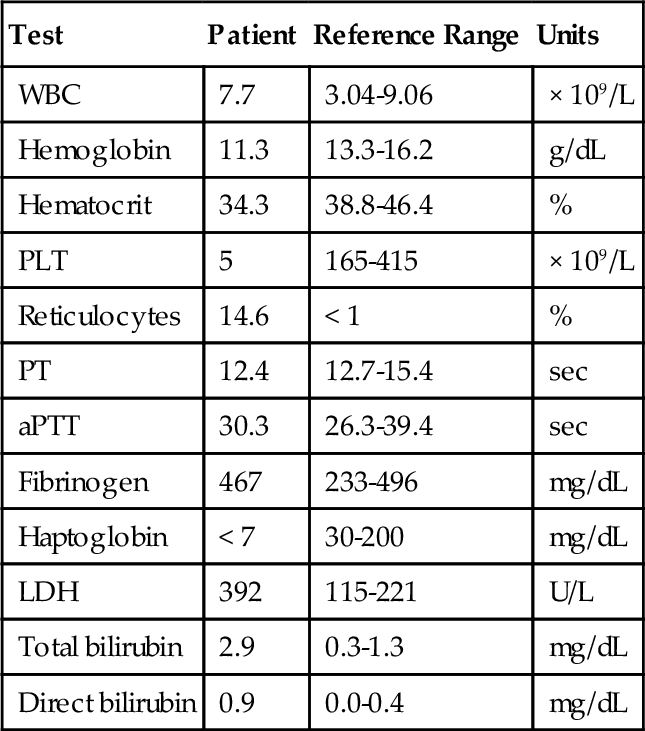
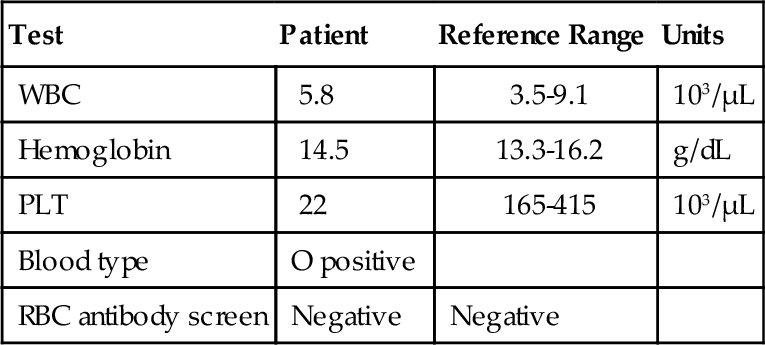
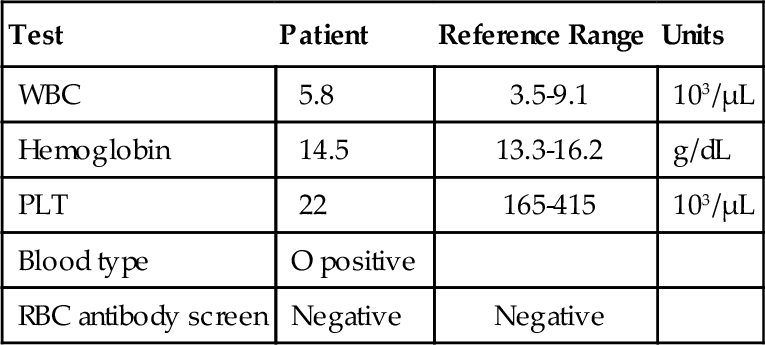
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


