In adults, a variety of histologic subtypes of non-Hodgkin lymphoma have been encountered. Most are diffuse large B-cell lymphomas, but some are follicular, and rarely, they are of T-cell lineage. In children, ovarian lymphomas are almost always diffuse, high-grade B-cell lymphomas that are often Burkitt lymphomas. Ovarian involvement has been described in Hodgkin disease (36) and plasmacytoma (37), but each of these is exceptionally rare. A primary ovarian diffuse large B-cell lymphoma arising in a mature cystic teratoma has also been reported (38).
Differential Diagnosis
Lymphoma in the ovary may be confused with several primary ovarian tumors. In such cases, it is helpful that diffuse infiltration of the adjacent fallopian tube and broad ligament is much more common in lymphomas than in most of the tumors in the differential diagnosis.
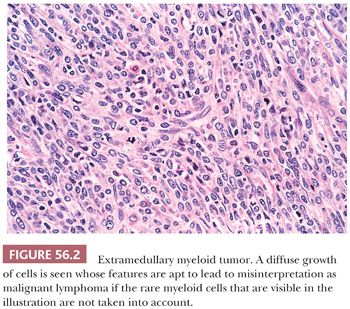
Lymphomas may be indistinguishable on gross examination from dysgerminomas. The former, however, are bilateral in 50% of cases in contrast to 10% of dysgerminomas. Although these tumors may resemble each other superficially on microscopic examination, their nuclei and immunohistochemical features are strikingly different. The single-file arrangement of lymphoma and leukemic cells may simulate metastatic carcinoma, particularly one of breast origin. Diffuse lymphoma can resemble undifferentiated carcinoma, granulosa cell tumor, or small cell carcinoma of the hypercalcemic type on low magnification. Numerous immunohistochemical markers are available to identify tumors as lymphomas. Extramedullary myeloid tumors (Fig. 56.2) can be distinguished with immunohistochemistry for myeloperoxidase (33) or chloracetate esterase staining. The possibility of extramedullary myeloid tumor should be considered when one is entertaining a diagnosis of ovarian lymphoma because many cases of extramedullary myeloid tumor have been initially misinterpreted as such. However, extramedullary myeloid tumor on routine stains is often composed of cells with more finely dispersed nuclear chromatin and may have more abundant cytoplasm, which may be deeply eosinophilic, compared to lymphoma cells with nuclei of the same size. Identification of eosinophilic myelocytes with confirmatory immunostaining may be crucial in making the diagnosis.
SMALL CELL CARCINOMA, HYPERCALCEMIC TYPE
This tumor (39) has occurred in females 7 months to 44 years of age, but the great majority occur between 15 and 30 years, with a peak in the early 20s, when it is the most common form of undifferentiated ovarian carcinoma. Approximately two-thirds of the tumors are associated with paraendocrine hypercalcemia, accounting for approximately 60% of all reported ovarian tumors associated with that disorder. Rare familial cases have been reported, including a remarkable occurrence in three sisters (39). Small cell carcinomas are almost always unilateral, although involvement of the opposite ovary may be seen as part of the abdominal spread encountered at laparotomy in approximately a third of the cases. Extraovarian primary examples of this neoplasm do not occur in our opinion. These are aggressive neoplasms with a poor prognosis, and the majority of patients die of their disease, usually within 2 years (39).
Gross Features
The tumors usually form large, predominantly solid, cream-colored to gray masses, often with areas of softening, necrosis, and hemorrhage. Cystic degeneration may be seen but is only uncommonly conspicuous. They may closely resemble ovarian lymphomas and dysgerminomas when uniformly solid and cream-colored.
Microscopic Features
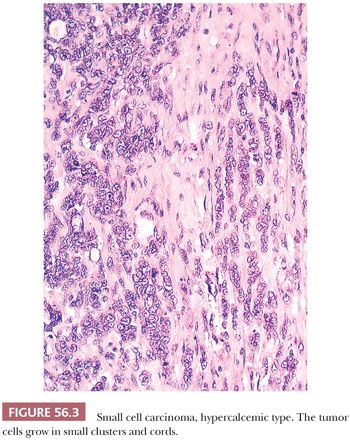
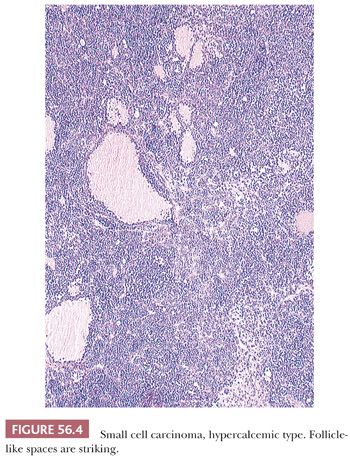
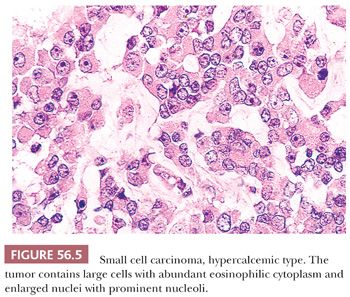
Diffuse sheets of small, closely packed, round to occasionally spindle-shaped cells with scanty cytoplasm and nuclei containing single nucleoli are typical. Mitotic figures are common. The tumor cells also often form small islands, cords (Fig. 56.3), and trabeculae. Follicle-like structures lined by tumor cells are present in 80% of cases (Fig. 56.4). These spaces typically contain eosinophilic, but occasionally basophilic, fluid. In 40% of tumors, a variable proportion of large cells is present. The large cells have abundant eosinophilic cytoplasm, which may have a globular, hyaline quality (Fig. 56.5), and large nuclei with prominent nucleoli. Rarely, small foci of tumor cells may have clear cytoplasm.
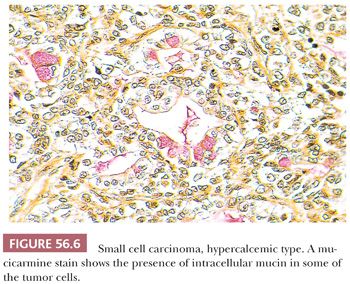
Intracellular mucin is encountered in 10% of cases (Fig. 56.6), either focally within otherwise typical tumor cells, as groups of signet ring cells, or as columnar cells lining glands or cysts that are indistinguishable from those of mucinous cystadenomas. The stroma of the tumor is typically scanty and fibrous, but occasionally, it is edematous or even myxoid, particularly in regions where there is a conspicuous component of the larger cells. A scattering of chronic inflammatory cells may be seen in the stroma, but this is not a prominent feature. Vascular invasion is common.
The tumor cells are diffusely positive for Wilms tumor 1 with antibodies directed against the N-terminus, and most are immunoreactive for cytokeratin, epithelial membrane antigen, vimentin, calretinin, CD10, and p53 (40,41). In most cases, electron microscopy shows nonspecific epithelial features. Abundant rough endoplastic reticulum is the most characteristic finding (42); large cells may contain whorls of microfilaments. Convincing dense-core granules have not been seen. Flow cytometry on paraffin-embedded material has shown a diploid pattern of DNA (43). A low frequency of chromosomal abnormalities has been observed in the small number of tumors that have been genotyped (44). Frequent inactivating mutations of the SWI/SNF chromatin-remodeling gene SMARCA4 have recently been identified in small cell carcinomas of hypercalcemic type, and 80% of these tumors show loss of SMARCA4 protein expression by immunohistochemistry (45, 45a). In contrast, only 0.4% of other primary ovarian tumors show loss of SMARCA4 protein expression (45a).
Differential Diagnosis
Small cell carcinomas may be misinterpreted as many neoplasms, with the presence of follicles making adult or juvenile granulosa cell tumors the most common misinterpretations. From a clinical viewpoint, hypercalcemia strongly favors a diagnosis of small cell carcinoma, and evidence of estrogen excess strongly favors a diagnosis of granulosa cell tumor. Extraovarian spread is much more common for small cell carcinomas than juvenile granulosa cell tumors. The nuclei of small cell carcinomas, compared with those of adult granulosa cell tumors, are more hyperchromatic and mitotically active, and they lack grooves. The occasional predominance of large cells with abundant cytoplasm in small cell carcinoma, particularly if interrupted by follicles, and the young age at which it occurs may suggest the possibility of juvenile granulosa cell tumor, but the differing clinical features and the characteristic microscopic patterns of the two tumors are diagnostically helpful. The follicles of juvenile granulosa cell tumors usually contain mucicarminophilic fluid, in contrast to the follicle-like spaces of small cell carcinomas. The occasional presence of mucinous epithelium is more typical of small cell carcinoma than granulosa cell tumor. Immunohistochemical staining for inhibin supports a diagnosis of granulosa cell tumor.
The dysgerminoma, which occurs in an age group similar to that of the hypercalcemic small cell carcinoma and may be grossly identical to it, is also, treacherously, sometimes associated with hypercalcemia. Rare small cell carcinomas have cells with clear cytoplasm and fibrous stroma that may impart a superficial resemblance to a dysgerminoma on low-power examination. Despite these overlapping features, there are many morphologic differences, and if needed, immunohistochemistry will aid. This potential pitfall should be remembered because of the markedly differing prognosis and treatment of the two neoplasms.
Small cell carcinomas can also be confused with lymphomas and the pulmonary form of small cell carcinoma. A variety of features of these three neoplasms, particularly their cytologic features, should distinguish them, and if necessary, immunohistochemical stains facilitate their distinction. The small cell carcinoma may be considered undifferentiated carcinoma, not otherwise specified, and indeed, regions may have an appearance consistent with that diagnosis. However, in the right age group, search for features indicative of the more specific diagnosis of the small cell carcinoma of hypercalcemic type should be assiduously performed, albeit this is largely an academic distinction at this time. Metastatic tumors such as malignant melanoma and alveolar rhabdomyosarcoma can be problematic because they may have a small cell phenotype, and the follicle-like spaces of some metastatic tumors may enhance their resemblance to small cell carcinoma. Clinical features and immunohistochemical staining, if necessary, will clarify these issues.
TUMOR OF PROBABLE WOLFFIAN ORIGIN
Rare ovarian tumors (46,47) are microscopically identical to their more common counterparts originating within the broad ligament that have been designated female adnexal tumors of probable wolffian origin. The ovarian tumors have no unique clinical features.
Gross Features
The tumors average 12 cm in diameter, are solid or focally cystic, and are almost always stage Ia. The solid tissue varies from gray-white to tan or yellow, and it may be rubbery or firm.
Microscopic Features
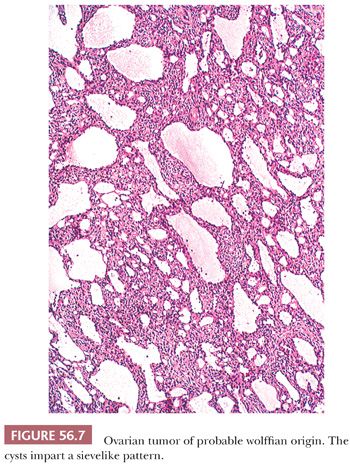
Microscopic examination reveals a cellular neoplasm with tumor cells that grow diffusely or form closely packed solid or hollow tubules. In apparently diffuse areas, a tubular pattern may be unmasked by periodic acid-Schiff (PAS) or reticulin staining. Varying amounts of fibrous stroma may result in a lobulated appearance. Cysts, which can be numerous, often punctuate these background patterns or may represent the dominant pattern and impart a characteristic sievelike appearance (Fig. 56.7). The cysts contain eosinophilic luminal secretions. The tumor cells are oval or spindle shaped, and they have scanty eosinophilic cytoplasm or pale cytoplasm in solid tubular areas. Rarely, a spindle cell pattern dominates (48). Rare tumors contain areas with poorly differentiated cells; such tumors may have malignant behavior. Areas of typical morphology should ideally be present to diagnose a malignant example.
Differential Diagnosis
These tumors are often misinterpreted as sex cord–stromal tumors, particularly Sertoli cell tumors, because the tubules of the two tumors may be indistinguishable. However, the other patterns of the wolffian-type tumor, which are almost always present as well, are incompatible with Sertoli cell tumor. In isolated cases, large hollow tubules resemble the glands of endometrioid adenocarcinoma, but other patterns of the neoplasm and the absence or paucity of luminal mucin help rule out this diagnosis. Wolffian tumors with a diffuse pattern can be misinterpreted as undifferentiated carcinomas on low power, but high-power evaluation generally shows cells with bland cytologic features and few mitotic figures. Rare tumors with a prominent spindle cell component may be difficult to distinguish from a cellular fibromatous tumor if not adequately sampled (48). Wolffian tumors are among the few ovarian tumors not of sex cord–stromal origin that can stain for inhibin (49); they can also show reactivity for calretinin and FOXL-2 (50–52). Accordingly, immunohistochemistry is not always definitive in differentiating wolffian tumors from sex cord tumors.
TUMORS OF THE RETE OVARII
These lesions are uncommon; most are cystadenomas (53), but some are adenomas (54), and rare carcinomas are of rete derivation (53). Rete cystadenomas may be functioning as a result of the presence of steroid cells at their periphery (55).
Gross Features
The cystic tumors range up to 24 cm (average, 8.7 cm) in diameter and have a hilar location; they are either unilocular or multilocular. They typically contain clear or yellow fluid and have thin walls with smooth linings. Adenomas have usually been incidental microscopic findings, and the only rete carcinoma reported had nonspecific gross features.
Microscopic Features
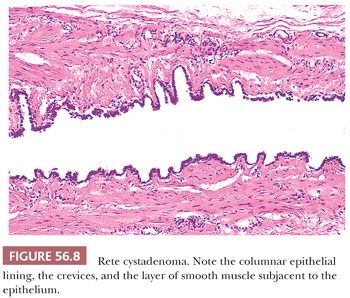
The walls of rete cystadenomas are composed of fibrovascular tissue that frequently contains fascicles of smooth muscle. The luminal surfaces characteristically have an undulating contour with shallow crevices lined by columnar, cuboidal, or flat cells that are only rarely ciliated (Fig. 56.8). Hyperplasia of hilus cells, which is frequently seen in the cyst wall, accounts for the virilization that is occasionally associated with these lesions (55). Intracystic papillae may be prominent. Adenomas are composed of closely packed small tubules within the ovarian hilus, some of which may be dilated and contain simple papillae. A single rete adenocarcinoma was characterized by an irregular network of branching tubules and cysts that contained papillae with fibrovascular or hyalinized cores (53). The tubules, cysts, and papillae were lined by obviously malignant, nonciliated cuboidal cells. Areas of solid growth and extensive transitional cell metaplasia were also observed.
Differential Diagnosis
Rete cystadenomas are often misdiagnosed as serous cystadenomas; this differential diagnosis has been discussed in Chapter 54. Rete adenomas may be confused with female adnexal tumors of probable wolffian origin, but the latter tumors typically have patterns other than tubular or papillary that are incompatible with the diagnosis of rete tumor. The one reported case of adenomatous hyperplasia of the rete differed from rete adenoma by the presence of a poorly defined margin that blended with the adjacent normal rete, a fibromuscular stroma, and tubules that varied in appearance (56). The rete carcinoma is distinguished from pure retiform Sertoli-Leydig cell tumor primarily by its focal solid pattern and transitional cell metaplasia. This unique tumor also occurred in an elderly woman, unlike most of the retiform tumors.
PARAGANGLIOMA
Only five examples of this neoplasm occurring in the ovary have been reported (57). The tumors have occurred in patients 15 to 68 years of age; three patients had a history of hypertension. The lesions have nonspecific gross features and have ranged in size up to 22 cm. Three have been confined to the ovary, but in one case, there were deposits on the opposite ovary and uterine serosa, and in another, para-aortic lymph node involvement and peritoneal nodules were present. Microscopic examination showed that four cases were typical paragangliomas, but one was the gangliocytic variant. For the most part, the tumors are characterized by the typical nested pattern, but a more diffuse growth can occasionally be encountered. That feature and the occasionally moderately conspicuous amount of eosinophilic cytoplasm may bring a steroid cell tumor into the differential diagnosis, and other entities, such as granulosa cell tumor, may not be unreasonable considerations. When paraganglioma is a diagnostic consideration, immunohistochemistry can be used to confirm the diagnosis (58), but it should be noted that two ovarian paragangliomas have been positive for inhibin (57).
WILMS TUMOR
Four examples of primary ovarian Wilms tumor have been reported (59–61); the patient age range was 3.5 to 56 years. One tumor formed a multilocular cystic tumor and contained areas that resembled a metanephric adenoma. None of the tumors was clinically malignant, although follow-up of significant duration was available in only two of the cases. The tumors must be distinguished from the retiform Sertoli-Leydig cell tumor, which can form glomeruloid structures but which almost always contains other patterns of that neoplasm.
SOLID PSEUDOPAPILLARY NEOPLASM OF PANCREATIC TYPE
This recently described primary ovarian tumor is morphologically and immunophenotypically identical to its pancreatic counterpart. Only six examples have been reported (62–65). Patients ranged in age from 17 to 57 years and presented with nonspecific symptoms of an abdominal mass. The tumors, which have ranged in size from 3 to 25 cm, are solid and cystic with white-tan to red, friable cut surfaces. Hemorrhage is often present, and necrosis has been described in one malignant example. All but one of the reported tumors was confined to the ovary; the sixth was associated with widespread intra-abdominal metastases. Histologically, diffuse and pseudopapillary growth patterns predominate, with occasional nests and small cysts filled with colloid-like material. The tumor cells are cytologically bland with eosinophilic to clear vacuolated cytoplasm. Mitotic activity is absent to minimal, except in the one reported malignant example that had 62 mitotic figures per 50 hpf as well as mild cytologic atypia, extensive necrosis, and lymphovascular invasion (65). Immunohistochemical studies show nuclear positivity for β-catenin and lack of membranous E-cadherin staining. The differential diagnosis includes sex cord–stromal tumors, steroid cell tumors, and struma ovarii. Awareness of this tumor’s occurrence in the ovary, as well as ancillary immunohistochemical studies, will aid in its recognition. It must also be distinguished from an ovarian metastasis of solid pseudopapillary neoplasm of pancreatic origin (66).
MELANOTIC Xp11 TUMOR OF RENAL TYPE
A single nonrenal case of this tumor has been described, occurring in the ovary of a 15-year-old female (67). The morphologic, immunohistochemical, and molecular features of the tumor were identical to those of the rare melanotic Xp11 translocation renal cancer.
ADENOMATOID TUMOR
Rarely, this benign tumor of mesothelial origin is located in or immediately adjacent to the ovary. In one series of six cases, the patients ranged from 23 to 79 years of age (68). Four tumors were less than 1 cm, and one was 5 cm in diameter. Five tumors were predominantly hilar, with focal medullary extension. The tumors are identical at the microscopic level to their counterparts in the fallopian tube and uterus. Because of their rarity in the ovary, diagnostic errors are sometimes made. One example was initially considered a yolk sac tumor because it had a focal reticular pattern and contained hyaline bodies; a lack of significant nuclear atypia and a negative result with α-fetoprotein immunostaining excluded this diagnosis. One strikingly oxyphilic example that required consideration of other oxyphilic ovarian tumors has been reported (69).
MALIGNANT MESOTHELIOMA
Peritoneal mesotheliomas are discussed in more detail in Chapter 58. Ovarian involvement by these tumors is usually limited to the ovarian surface and occasionally the superficial cortex, but rarely, the ovary is extensively involved, with a clinical picture similar to that of ovarian cancer. In one study, ovarian involvement was present in 10 of 13 patients with peritoneal mesothelioma (70), and in another series, all nine tumors presented as ovarian masses (71); two were considered primary ovarian tumors because of confinement to the ovary. Malignant mesotheliomas, whether primary or secondary, are characterized on microscopic examination by tubular, papillary, and solid patterns and relatively uniform cells with eosinophilic cytoplasm. In routinely stained sections, these tumors are usually readily distinguishable from serous carcinoma by their pattern of growth and typically bland cytologic features as discussed in detail by Baker et al. (72). Histochemical or immunohistochemical stains are rarely necessary to confirm the diagnosis but will often aid should they be deemed necessary.
INTRA-ABDOMINAL DESMOPLASTIC SMALL ROUND CELL TUMOR
This tumor is discussed in detail in Chapter 58. Patients with this neoplasm may have prominent ovarian involvement, leading to confusion with primary ovarian neoplasms. The patients are typically teenagers or young adults (73); ovarian involvement is often bilateral. Laparotomy typically reveals extensive extraovarian disease. Because many ovarian tumors are characterized by small cells, the differential diagnosis is wide. A prominent nesting pattern of small cells in a desmoplastic stroma, as well as the young age of the patient, should lead to consideration of this tumor. Immunohistochemical positivity for both cytokeratin and desmin almost always establishes the diagnosis.
SECONDARY TUMORS
This is an important area of ovarian tumor interpretation with much new information in the past two decades. For more detailed coverage than space constraints allow here, historical perspective, and more complete referencing, the reader is referred to two reviews by one of us (74,75).
Certain clinical and operative findings should alert the pathologist to the possibility of metastatic involvement of the ovary. These features include the previous or concurrent existence of a primary tumor elsewhere and a pattern of extraovarian spread that is atypical for ovarian cancer. For example, hepatic or pulmonary metastases at the time of presentation, especially in the absence of extensive peritoneal spread, would be unusual for ovarian cancer. Because metastases to the ovary are bilateral in two-thirds to three-fourths of the cases, metastasis should be considered in the evaluation of bilateral disease, especially if the tumor is not undifferentiated or of serous type. Approximately 10% to 20% of surgically encountered bilateral ovarian cancers are metastatic. Two other gross findings suggestive of metastasis are the presence of several nodules and the involvement of the ovarian surface without generalized peritoneal spread. Single or multiple cysts, even when they are thin-walled, do not exclude metastasis. Prominent vascular space invasion is more typical of metastasis.
A study (76) comparing primary and metastatic mucinous carcinomas in the ovary found that the following features strongly favored metastasis: nodular invasive pattern, ovarian hilar involvement, single-cell invasion, signet ring cells, vascular invasion, and microscopic surface mucin. Features strongly favoring a primary ovarian mucinous carcinoma were an expansile pattern of invasion and a complex papillary pattern. Findings that were less frequent but that also favored a primary tumor were as follows: a size greater than 10 cm, a smooth external surface, benign-appearing and borderline-appearing areas, microscopic cystic glands, and necrotic luminal debris. In a subsequent study looking at the aid provided by simple parameters such as size and laterality, particularly in the frozen section setting, it was found that 90% of neoplasms would be correctly classified regarding their primary or metastatic nature by simply following the rule that bilateral tumors less than 10 cm are metastatic and unilateral tumors greater than 10 cm are primary (77). This certainly can be helpful as a guide, but unfortunately, there are sufficient exceptions to make this of limited applicability, as has been pointed out in several subsequent publications (78–80). As an example, in one of these studies (78), it was found that 60% of metastatic mucin-producing adenocarcinomas were 10 cm or greater.
In the following sections, metastatic tumors are largely discussed according to their source, but some, such as the Krukenberg tumor, may have several origins.
KRUKENBERG TUMOR
Krukenberg tumors are metastatic carcinomas with a component of signet ring cells comprising at least 10% of the tumor (81–83). They usually originate in the stomach. The primary tumor may be very small, escaping detection at operation or even for 5 or more years after surgery. Occasional tumors originate in other portions of the gastrointestinal tract (particularly the appendix and large bowel) or its associated structures, gallbladder and pancreas. Breast, uterine cervix, urinary bladder, and renal pelvis are other rare possibilities. Women with Krukenberg tumors tend to be unusually young for patients with metastatic carcinoma. Many are between 40 and 50 years of age, and a sizable number are in their 20s, and some are even teenagers. The symptoms are usually nonspecific, but endocrine manifestations, particularly virilization during pregnancy, may be seen due to stromal luteinization.
Gross Features
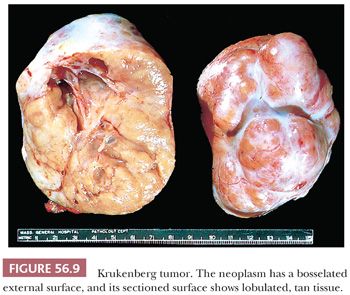
Krukenberg tumors are bilateral in approximately 90% of cases. They are almost always solid with a smooth or bosselated contour (Fig. 56.9); they rarely contain thin-walled cysts. The cut surfaces vary from firm to soft and white to tan and may resemble a typical or edematous fibroma. Red, fleshy, and gelatinous appearances are quite common. Sometimes, the central region is softer and of different color from the peripheral component, leading one writer, aptly, to liken the appearance to that of a shell in some cases (84). Necrosis and hemorrhage are common.
Microscopic Features
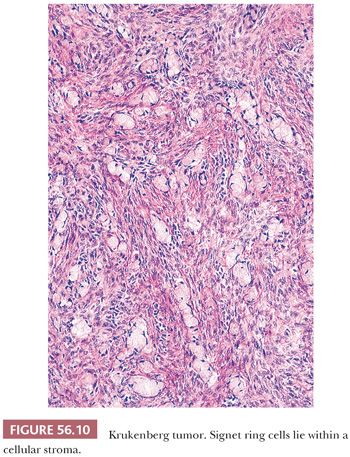
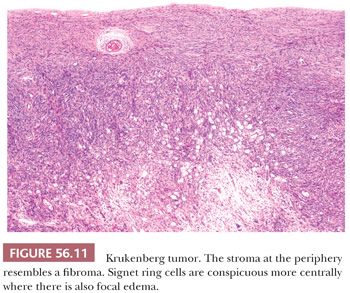
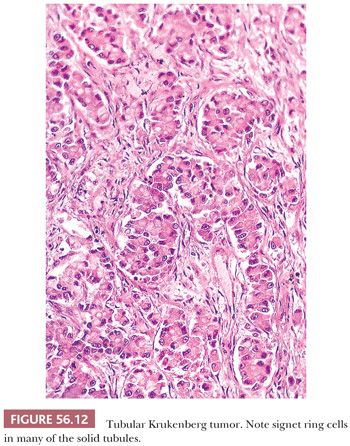
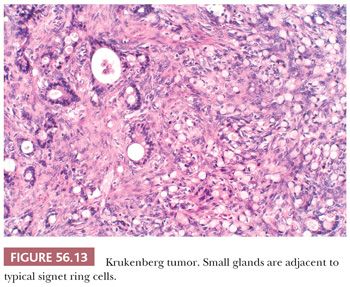
The low-power appearance is variable but often at least somewhat nodular, like many metastatic tumors. The nodules may be confluent or separated by stroma that may be strikingly edematous. Rounded malignant epithelial cells, many of which have a signet ring cell appearance, are embedded as small nests, cords, tiny glands or cysts, or single cells in a typically spindle cell stroma (Fig. 56.10). The relative amounts of tumor and stroma vary considerably; the latter varies from very cellular to markedly edematous and hypocellular, and it may be luteinized, hyalinized, or desmoplastic (83). The stroma at the periphery may differ in appearance from that seen more centrally (Fig. 56.11). In some cases, large rounded islands of signet ring cells are suspended in pools of mucin, or they grow in pseudotubular arrangements. Occasionally, the predominant architecture is characterized by hollow or solid tubular glands that may be Sertoli-like apart from the focal component of signet ring cells lining them (tubular Krukenberg tumor) (Fig. 56.12) (85). Large intestinal-type glands may be seen, as may tiny glands (Fig. 56.13) that may have flattened lining cells. Benign to atypical to frankly malignant columnar mucinous cells may be present, as may foci that resemble mucinous carcinoid; the latter suggest a possible origin of the tumor from the appendix (86), where it may be occult at operation or even on gross examination. Vascular involvement is often conspicuous, particularly at the periphery of the neoplasm.
The cytoplasm of the signet ring cells is typically pale, but it may be eosinophilic or weakly or strongly basophilic. In some areas, the cells have little or no intracellular mucin, or the mucin occupies only a small area of the cytoplasm adjacent to oval or rounded nuclei. Targetoid eosinophilic globules within cells are sometimes striking. Occasionally, diffuse areas of variable extent are composed of cells with clear cytoplasm that are suggestive of clear cell carcinoma. Areas with minimal mucin may resemble Sertoli cell tumor, granulosa cell tumor, transitional cell carcinoma, or dysgerminoma. In most cases, PAS staining with and without diastase and Alcian blue staining are more extensively positive than is mucicarmine staining.
Examination of non–signet ring cell areas in the tumor can provide clues as to the primary site. For example, the presence of intestinal-type glands suggests a gastrointestinal or biliary origin, whereas prominent tumor cell cording suggests a mammary origin.
Differential Diagnosis
Numerous tumors can be misdiagnosed in cases that ultimately are shown to be Krukenberg tumor. Only the most common, or treacherous, are noted here; more detailed discussions are available elsewhere (74,83). The focal dearth of signet ring cells in some cases may make fibroma a reasonable consideration, particularly at the time of frozen section, as emphasized by Holtz and Hart (81). Other stromal neoplasms such as the sclerosing stromal tumor and signet ring stromal tumor may enter the differential, but signet ring–like cells in the former contain fat, not mucin, and, in the latter, are negative for mucin. Like the Sertoli-Leydig cell tumor, some Krukenberg tumors are virilizing, and on microscopic examination, both may be composed of densely cellular lobules. Care must be taken not to confuse luteinized stromal cells of a Krukenberg tumor, particularly one with endocrine manifestations, for the Leydig cells of a Sertoli-Leydig cell tumor. Careful evaluation of the cellular areas should disclose signet ring cells in the case of a Krukenberg tumor; these cells are only present in the rare foci of mucinous carcinoid seen in some cases of Sertoli-Leydig cell tumor with heterologous elements. Signet ring cells, or at least tiny glands that can simulate signet ring cells, can be seen in occasional surface epithelial carcinomas (87). However, well-sampled tumors will usually disclose themselves as being of primary or Krukenberg character, as the case may be. In the differential diagnosis of Krukenberg tumor, the importance of being aware of the spectrum of morphology of various ovarian tumors with somewhat overlapping features cannot be overemphasized nor can the crucial importance of fundamental aspects of ovarian tumor evaluation such as the clinical history, bilaterality, surface involvement, and miscellaneous features already noted.
GASTRIC CARCINOMA, INTESTINAL TYPE
Only a small number of cases of this type have been reported (88). The patients have been in a somewhat older age group than patients with Krukenberg tumors of gastric origin.
Gross Features
The tumors are typically large and solid and cystic. The cysts contain mucinous secretions or hemorrhagic fluid. Necrosis is often seen.
Microscopic Features
On microscopic examination, there is characteristically an appearance of variably sized tubular glands (Fig. 56.14), imparting a pseudoendometrioid appearance that is more commonly associated with metastatic intestinal adenocarcinoma. Dirty necrosis is common, and the epithelium lining the glands is often high grade. Less commonly, the tumors have a mucinous phenotype and may have deceptively benign foci, as is the case with other metastatic mucinous carcinomas. Unusual patterns include papillary, trabecular, and nested.
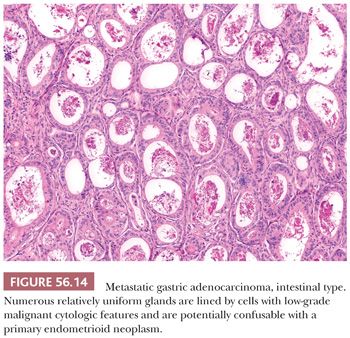
Differential Diagnosis
The differential diagnosis is with primary endometrioid and, less often, mucinous tumors and is similar to that discussed under several headings that follow.
INTESTINAL CARCINOMA
From 2% to 10% of women with intestinal cancer have ovarian metastases during the course of their disease (89–93). The patients are, on average, older than those with Krukenberg tumor, with only rare examples in the first two decades; we have seen one case in which the patient was 12 years old. Should a patient have intestinal carcinoma, ovarian metastasis is more likely to occur if the woman is younger than 40 years of age, presumably because of the greater vascularity of the ovary in that age group. The patients fall into the following three categories: those with an intestinal carcinoma who subsequently have ovarian metastasis (approximately 70% of cases), those with ovarian involvement discovered during resection of intestinal carcinoma (approximately 20% of cases), and those in whom the diagnosis of the ovarian tumor precedes that of the primary tumor (approximately 10% of cases). Patients with ovarian metastases from undiagnosed colorectal carcinomas tend to be younger than those with known colorectal carcinomas (48 years vs. 61 years) (94). Most ovarian metastases of intestinal carcinoma originate from the large intestine, but a few examples are from the small bowel (95). Occasional patients have endocrine manifestations (96).
Gross Features
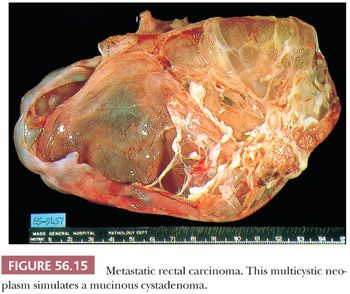
Approximately two-thirds of the tumors are bilateral. Sectioning of smaller tumors generally reveals firm or soft, solid tissue, whereas larger tumors are composed of friable yellow, red, or gray tissue with cysts that contain necrotic tumor, mucinous or clear fluid, or blood. Rare examples are composed of several thin-walled cysts and resemble a mucinous cystadenoma (Fig. 56.15).
Microscopic Features
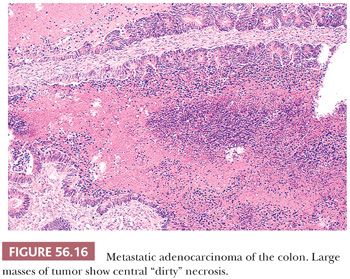
The metastases resemble, to varying degrees, primary intestinal carcinomas, typically forming glands lined by poorly differentiated stratified epithelial cells lacking mucin and containing numerous mitotic figures. A cribriform pattern of small- to medium-sized glands is common, and large cystically dilated glands are occasionally conspicuous. Necrosis is often present and can be extensive. It is often of the “dirty necrosis” type (Fig. 56.16), with central large, round aggregates composed of eosinophilic material containing abundant nuclear debris and viable glands that surround the necrotic material in a garland arrangement (90). Occasionally, cystic glands lined by well-differentiated mucin-rich cells are prominent. Rare tumors are colloid carcinomas. Occasional tumors have a prominent micropapillary pattern. The stroma varies from negligible to abundant. It may be desmoplastic, edematous, or myxoid, but it often resembles ovarian stroma. Luteinized stromal cells are present in approximately a third of cases. Indeed, metastatic bowel cancers are among those most frequently associated with stromal luteinization and endocrine manifestations (96). The rare clear cell adenocarcinoma of the intestine may also spread to the ovary and simulate secretory endometrioid carcinoma or clear cell adenocarcinoma (95); although the numbers are small, a disproportionate number of these cases are from the small bowel compared to metastatic intestinal cancer overall.
Differential Diagnosis
The most difficult tumors to exclude on microscopic evaluation are primary endometrioid and mucinous carcinomas, the differential diagnoses of which have been discussed under those headings (Chapter 54), as has the distinction of metastatic intestinal clear cell adenocarcinoma from primary clear cell adenocarcinoma.
CARCINOID TUMORS
Carcinoid tumors account for approximately 2% of ovarian metastases. Almost all patients are older than 40 years of age; approximately half have been reported to have the carcinoid syndrome. The ileum is usually the primary site, but the cecum, jejunum, appendix, colon, stomach, pancreas, and bronchus are rare sources (97–99).
Gross Features
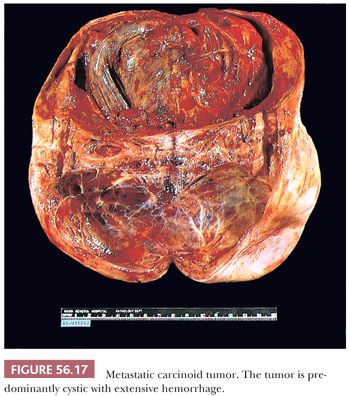
Metastatic carcinoids are usually bilateral. They vary in size, and they are usually solid, with smooth or bosselated serosal surfaces. Sectioning demonstrates single or confluent, firm, white or yellow nodules that may resemble fibromas, thecomas, or Brenner tumors. In some cases, the presence of numerous cysts filled with watery fluid causes the tumor to resemble a cystadenofibroma. Rare tumors are predominantly cystic (Fig. 56.17). Necrosis and hemorrhage may develop in solid tumors, and some of the cysts may contain blood.
Microscopic Features
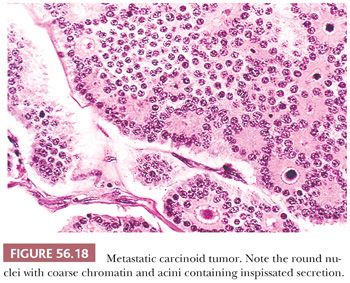
An insular pattern is most common (Fig. 56.18), but trabecular and sometimes solid tubular patterns are also encountered. The nests are usually uniformly solid, but occasionally, small acini punctuate them or are more typically arranged at their periphery. The acini often contain a homogeneous, eosinophilic secretion, which may calcify, sometimes forming psammoma bodies. Larger glands and cysts lined by one or a few layers of neoplastic cells are sometimes seen. The latter, when striking, may produce a follicle-like pattern. Carcinoid is the ovarian metastatic tumor that most typically elicits a fibroma-like stromal proliferation that may become extensively hyalinized. Metastatic “goblet cell carcinoids” to the ovary are reported, but classification of the primary tumors in these cases as carcinomas with neuroendocrine differentiation is probably most appropriate (86).
Differential Diagnosis
Metastatic carcinoids may be confused with primary carcinoid tumors (see “Carcinoids” in Chapter 55), granulosa cell tumors, Sertoli tumors or Sertoli-Leydig cell tumors, Brenner tumors, adenofibromas, cystadenofibromas (benign, borderline, and malignant), and adenocarcinomas of various types. These differential diagnoses have been discussed under the relevant headings.
BREAST CARCINOMA
Approximately one-third of patients with breast cancer have ovarian metastases during the course of their disease (100–103). The involvement is bilateral in approximately two-thirds of the cases, and a similar number of affected ovaries appear normal grossly (103). Rarely, metastatic breast carcinoma initially appears with signs or symptoms of an ovarian tumor (104). Only a small minority of prophylactic oophorectomy specimens from patients with breast cancer contain metastases, and particularly in the setting of patients with BRCA mutations, when neoplasia is seen in such specimens, it is statistically more likely to be an independent primary serous or undifferentiated carcinoma of the ovary rather than metastatic breast cancer (105).
Gross Features
The ovaries often have irregular, nodular surfaces and typically contain firm or gritty white nodules of various sizes; cysts may be present, but only rare specimens are entirely cystic. Nonspecific appearances consistent with the appearance of many other ovarian tumors may be seen.
Microscopic Features
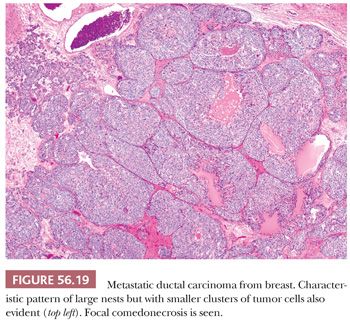
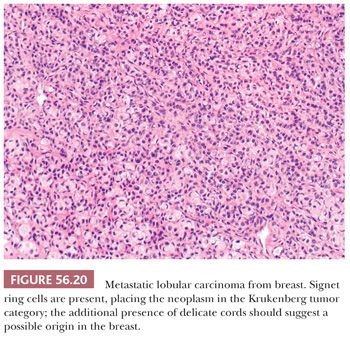
Lobular carcinomas spread to the ovary much more frequently than ductal carcinomas (106) (Fig. 56.19). In many cases of lobular carcinoma, a diffuse pattern is conspicuous at low magnification, but small clusters, cords, or ribbons of cells are usually apparent on high-power examination. Acini, larger glands with a cribriform pattern, and rarely papillae may be conspicuous in cases of ductal carcinoma. Rare neoplasms have a prominent component of large cells with abundant eosinophilic cytoplasm. The stroma varies from sparse to abundant; stromal luteinization is uncommon in contrast to its frequency in metastatic intestinal carcinomas. Rarely, the features are those of a Krukenberg tumor in that signet ring cells are present, but these tumors generally look different from Krukenberg tumors of gastrointestinal origin due to the presence of admixed histologic patterns more typical of breast carcinoma (Fig. 56.20).
Differential Diagnosis
Predominantly glandular, papillary, or poorly differentiated metastases may resemble primary epithelial carcinomas, particularly serous or endometrioid carcinomas; an insular pattern may mimic that of a carcinoid tumor; and a diffuse pattern may suggest a lymphoma. The presence of background endometriosis or an underlying adenofibroma can help support a diagnosis of a primary epithelial carcinoma, whereas bilaterality, surface involvement, multinodular growth, and prominent lymphatic invasion are features more common to tumors metastatic to the ovary. Metastatic breast carcinomas have also been misinterpreted as granulosa cell tumors, but the contrasting features of the neoplastic cells, especially their nuclei; the differing patterns of growth; and the clinical findings facilitate this distinction in almost all cases.
A panel of immunohistochemical stains can aid in differentiating metastatic breast carcinoma from primary ovarian carcinoma (107,108). Immunoreactivity for gross cystic disease fluid protein-15 (GCDFP-15) strongly suggests metastatic breast carcinoma; however, rare ovarian carcinomas (<5%) may show GCDFP-15 positivity, and in one study (109), only 43% of breast cancer metastases to the ovary were positive. Recent data suggest that GATA3, which is expressed in most breast carcinomas, may be another helpful marker in this differential diagnosis because ovarian carcinomas in general are only rarely positive (110–112). Reactivity does vary by subtype of ovarian epithelial tumor, however, as GATA3 is expressed in one-third of ovarian transitional cell tumors (113); this is not surprising as most urothelial neoplasms are positive for this marker. GATA3 positivity can also be seen in germ cell tumors (112). Although mammaglobin is considered a marker of breast origin, focal or patchy expression is frequently seen in ovarian carcinomas, and therefore, it is of limited use in this differential diagnosis (114). Reactivity for breast cancer–associated markers and lack of expression of other markers common in ovarian neoplasia, such as WT1 and PAX8, can help to establish a diagnosis of metastatic breast cancer. The utility of these different immunostains, however, depends on the particular subtypes of carcinoma being considered (108).
TUMORS OF THE APPENDIX
Metastatic appendiceal tumors (other than carcinoids—see previous section) can be considered in three groups: those that result from spread of low-grade appendiceal mucinous neoplasms, including mucinous carcinomas (115); those that fall in the Krukenberg tumor family because of an appreciable content of signet ring cells (83,86); and those of intestinal type, either in pure form or sometimes associated with another component (116). Appendiceal primary tumors are often overlooked at operation because the ovarian tumors are typically much larger (117).
The association between low-grade mucinous cystic tumors of the appendix and coexisting, microscopically similar tumors of the ovary in cases of pseudomyxoma peritonei (118) has been acknowledged for many years and has been controversial (115,119–121), but most investigators now believe that the appendiceal tumor is primary and the ovarian tumor is secondarily involved in most cases. The appendiceal lesion has been variously designated over the years, but we and many others use the classification of Misdraji et al. (122), in which “low-grade appendiceal mucinous neoplasm” is used for the most common primary appendiceal morphology in this situation.
Gross Features
The ovarian metastases of appendiceal low-grade mucinous tumors are often bilateral, large, and multilocular, and they typically contain abundant thick, jelly like mucin. Surface involvement is common. Those of Krukenberg type and intestinal type have gross features similar to those already described for those categories of neoplasms.
Microscopic Features
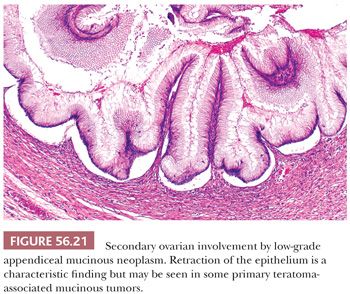
Ovarian involvement by low-grade mucinous tumors often exhibits prominent mucinous cystadenoma-like areas, but when these tumors are thoroughly sampled, they are usually found to contain foci of stratified epithelium. The neoplastic cells are tall columnar and engorged with mucin, the latter often seeming to exude out of the apices of the cells. The glandular structures have scalloped contours and show a distinctive pattern of subepithelial clefting, wherein the base of the epithelium is “lifted off” the underlying stroma (Fig. 56.21) (123). Mucin commonly dissects into the ovarian stroma (pseudomyxoma ovarii). In some cases, all residual ovarian stroma is obliterated by abutting multilocular cysts. In other cases, residual stroma is evident, and the mucinous cysts irregularly infiltrate through it. There is usually surface ovarian involvement. This may be in the form of mucin apparently simply “sitting” on the ovarian surface, or there may be an associated hyaline stromal reaction. The mushroomlike projections with prominent desmoplastic stroma that typify implants in many cases of metastatic mucinous carcinoma are rare.
Metastatic appendiceal tumors of Krukenberg type have the microscopic features of that category of neoplasm, the only comment of note being that those of appendiceal origin tend more often than those primary at other sites to have foci that resemble mucinous (goblet cell) carcinoid (86). The third category of metastatic appendiceal neoplasia is the group that has an intestinal-like morphology (116) and results in a differential diagnosis similar to that of metastatic intestinal adenocarcinoma itself.
Differential Diagnosis
Although distinguishing secondary involvement by low-grade appendiceal mucinous neoplasms from primary ovarian mucinous cystadenomas and cystadenomas of borderline malignancy may be difficult, the former tumors are bilateral much more often, and they usually have a jellylike consistency on gross examination. In addition, their mucinous lining cells are unusually tall and hypermucinous, and these cytologic features in combination with the characteristic glandular scalloping and subepithelial clefting impart a distinctive picture to metastatic low-grade appendiceal mucinous neoplasms. Pseudomyxoma ovarii is frequently present, a finding that is much less common in primary ovarian mucinous tumors. Conversely, histiocytic aggregates (“mucin granulomas”) are generally not seen in appendiceal low-grade mucinous neoplasms but are common in primary mucinous tumors (123). Several investigations of the immunohistochemical profile have shown that the neoplastic epithelium in these cases is typically negative or only focally reactive for CK7 and positive for CK20, CDX2, and MUC2 (123–127).
Rare cases of pseudomyxoma peritonei are associated with primary ovarian mucinous tumors arising in a background of teratoma (123). The morphology is similar to appendiceal low-grade mucinous neoplasms involving the ovaries, including the presence of scalloped glands and subepithelial clefts, and the immunophenotype is similar as well. Teratoma-associated mucinous neoplasms are typically unilateral, however, and accurate diagnosis of such cases requires a careful search for teratomatous components in the ovary and rigorous pathologic evaluation of the appendix.
TUMORS OF THE PANCREAS
Although spread of pancreatic ductal carcinoma to the ovary has historically been considered uncommon, current evidence suggests that it occurs much more frequently than once thought (128). Indeed, it is considered in retrospect to account for the poor prognosis in some older series of primary mucinous cancers of the ovary in that some of the tumors were actually metastatic to the ovary. Furthermore, pancreatic cancer spreading to the ovary is considered the prototypical situation in which a metastatic mucinous cancer in the ovary may simulate a primary neoplasm both grossly and microscopically. The usual bilateral ovarian involvement is a helpful clue to a metastasis because primary ovarian mucinous tumors are bilateral in less than 10% of cases. Additional features suggesting metastasis include extensive extraovarian tumor, ovarian surface implants (Fig. 56.22), and prominent vascular invasion within the ovary. Often, the metastases contain large areas simulating borderline or benign mucinous ovarian neoplasia or, less often, endometrioid adenocarcinoma. Rare metastases from the pancreas are Krukenberg tumors (83). Loss of Dpc4 staining favors the diagnosis of metastatic pancreatic carcinoma over that of primary ovarian mucinous carcinoma (129,130).
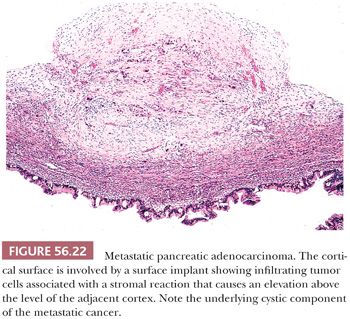
Vakiani et al. (131) reported four cases of acinar cell carcinoma metastatic to the ovary (Fig. 56.23A). The patients were adults, and in three patients, the ovarian tumors were detected prior to the pancreatic primary. In some cases, determining that there was a pancreatic primary was difficult. The ovarian tumors were large and bilateral in three cases. Microscopic examination showed highly cellular neoplasms with scant fibrous stroma. In two cases, there was a predominant acinar growth, and in the other two cases, the patterns were predominantly solid to cribriform with comedo-like necrosis. The cells generally had brightly eosinophilic granular cytoplasm. The major differential is with carcinoid tumor, and positive immunostaining with antibodies against chymotrypsin and trypsin and negative immunostaining with antibodies against synaptophysin and chromogranin aid in this distinction but, of course, can only be applied if the differential diagnosis is considered based on suspicion provoked by the routine microscopic findings. One example of an adrenocorticotropic hormone (ACTH)–secreting pancreatic neuroendocrine tumor presenting with bilateral ovarian metastases and Cushing syndrome has been reported (132), and rare other cases of metastatic pancreatic neuroendocrine tumor to the ovary are documented (Fig. 56.23B,C).
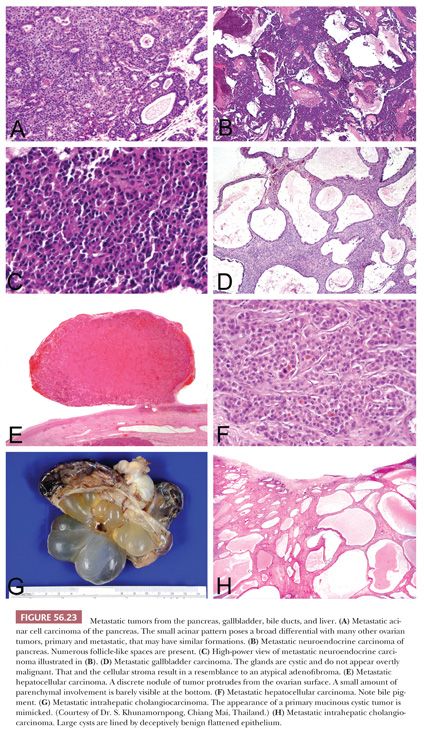
TUMORS OF THE LIVER, GALLBLADDER, AND BILE DUCTS
The recent literature suggests that metastatic hepatocellular carcinoma spreads to the ovary more often than the earlier literature would indicate, although clearly, it is still uncommon (133,134). The tumors may be encountered throughout adult life but have most often been seen in the young, and the ovarian mass may be the presenting manifestation. Bilaterality and the presence of canaliculi or bile on microscopic examination are very helpful features pointing to metastatic hepatocellular carcinoma (Fig. 56.23E,F). The microscopic patterns encountered range from diffuse to various epithelial-like formations including insular, trabecular, and pseudoglandular.
The major differential diagnosis is with primary and metastatic hepatoid tumors of the ovary. In the young, hepatoid yolk sac tumor is an important consideration, and thorough sampling to rule out areas of typical yolk sac tumor is indicated. In a postmenopausal patient, the differential diagnosis involves hepatoid carcinoma rather than hepatoid yolk sac tumor. Hepatoid carcinomas usually contain foci of more typical surface epithelial carcinoma, typically serous carcinoma. Hepatoid carcinomas may also arise outside the ovary, for example in the stomach and lung, and may potentially metastasize to the ovary. HepPar-1 is not useful in distinguishing among metastatic hepatocellular carcinoma, hepatoid yolk sac tumor, and hepatoid ovarian carcinoma as it is expressed in all (135).
In certain parts of the world such as East Asia, metastatic intrahepatic cholangiocarcinoma is a significant issue in metastatic ovarian neoplasia (Fig. 56.23G,H). This is rarely encountered in other parts of the world where infestation with Opisthorchis viverrini, which is responsible for the high incidence in Asia, is not endemic. Khunamornpong et al. (136) described a series of 16 examples of this phenomenon. Most of the patients presented with signs and symptoms referable to an adnexal mass. The ovarian tumors were typically bilateral and were often grossly indistinguishable from primary mucinous cystic tumors. The general features that aid in the differential of primary versus metastatic mucinous neoplasia, considered earlier, are helpful in this differential.
Khunamornpong et al. (137) also reported their experience with ovarian spread of carcinoma of the extrahepatic bile ducts and gallbladder (Fig. 56.23D) and reviewed the prior literature on this topic. As with ovarian spread of intrahepatic cholangiocarcinoma, this is typically a neoplasm of adults, and the ovarian involvement is characteristically bilateral. Generally, similar microscopic features are seen. A noteworthy finding in this series was the remarkable mimicry in some cases of cystadenoma and cystadenofibroma of nonmucinous type because of flattened epithelium lining the glands and cysts and prominent fibrous stroma. Occasional tumors were in the Krukenberg family, and a colloid-type appearance was observed focally in a few cases. Nonmucinous morphology that resembled high-grade endometrioid carcinoma or papillary carcinoma with nonspecific müllerian-like features and even undifferentiated carcinoma were also encountered.
TUMORS OF THE KIDNEYS AND ADRENAL GLANDS
A small number of striking cases of renal cell carcinoma metastatic to the ovary have been reported (75,138). In just over half of the cases, the ovarian tumor was discovered first, and in one case, the renal primary was not detected until 8 years after the ovarian tumor. Nearly all are renal clear cell carcinomas, and the ovarian tumor in several cases was initially misdiagnosed as primary ovarian clear cell carcinoma. The metastases ranged from 7 to 18 cm in greatest dimension; a minority were bilateral. Microscopic examination shows solid nests composed of clear epithelial cells or tubules lined by clear cells and containing intraluminal eosinophilic material or blood. A prominent sinusoidal vascular pattern is characteristically present. The homogeneous clear cell pattern without hobnail cells or other patterns of ovarian clear cell carcinoma aids in recognizing these tumors as metastatic, but radiologic studies of the kidneys are necessary in some cases to make the diagnosis. We have seen one unpublished case of renal papillary carcinoma with metastasis to the ovary. One renal pelvic urothelial cell carcinoma with gland differentiation spread to the ovary in the form of a Krukenberg tumor (139). Cameron et al. (140) reported that primary ovarian clear cell carcinomas are immunoreactive for CK7 but not for CD10 and only rarely for renal cell carcinoma (RCC) marker. In contrast, renal cell clear cell carcinomas are characteristically positive for CD10 and RCC marker but negative for CK7. Leroy et al. (141) found similar results and also found mesothelin (favoring ovarian origin) to be of some aid.
One rhabdoid tumor of the kidney appeared as an ovarian mass before the renal neoplasm was discovered (142). Among childhood tumors, adrenal neuroblastomas spread to the ovary most frequently; more than 25% are found to involve the ovary at autopsy (142,143). Rare patients present with ovarian metastases (142). These tumors must be distinguished from immature teratomas with a predominant neuroblastoma component or the extremely rare primary pure ovarian neuroblastoma. We have seen one unpublished case of adrenal cortical carcinoma that spread to the ovary, but this is a rare event (144).
TUMORS OF THE URINARY BLADDER, URACHUS, URETER, AND URETHRA
Distinguishing a metastatic urothelial carcinoma of the urinary tract from a borderline or malignant Brenner tumor may be difficult (145). The presence of benign Brenner elements supports an ovarian origin, as does the presence of other forms of surface epithelial carcinoma (often serous). A WT1+/CK20−/uroplakin III- immunophenotype also favors a diagnosis of ovarian Brenner cell tumor rather than metastasis. Three signet ring cell carcinomas metastatic from the bladder have been Krukenberg tumors (145). Rare urachal adenocarcinomas have metastasized to the ovaries and have mimicked mucinous cystic tumors (146).
MALIGNANT MELANOMA
Ovarian involvement is often found at autopsy in patients with malignant melanoma, and clinically evident tumors are also encountered (147–149). Most melanomas metastatic to the ovary have been primary in the skin, but some have arisen in the choroid or elsewhere. The average age of the patients has been only 38 years, and rare patients are teenagers. Approximately 80% of the patients also have metastatic tumor outside the ovary, usually within the pelvis and upper abdomen. The remote history of the primary tumor, over 5 years in 20% of the reported cases, may result in the information not being conveyed to the pathologist.
Gross Features
Both ovaries are involved in approximately one-third of the cases. Tumors average 10 cm in size. Only a minority are black or brown in color. There is commonly a minor cystic component in a specimen that is generally nondistinctive grossly; rare tumors may be predominately cystic.
Microscopic Features
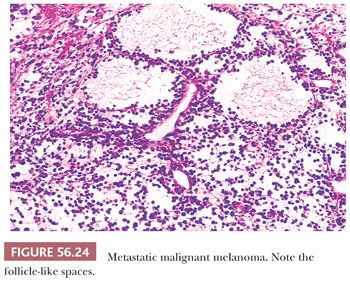
The most common microscopic appearance is that of large cells with abundant eosinophilic cytoplasm growing diffusely or in nodular aggregates. Occasionally, however, small cells with scanty cytoplasm predominate; in some cases, spindle cells are present, but they rarely predominate. Growth in discrete rounded aggregates with a nevoid appearance is helpful diagnostically. A confusing feature is the presence of follicle-like spaces in approximately 40% of tumors (Fig. 56.24). A helpful feature is intracytoplasmic melanin pigment, but melanin is frequently inconspicuous or absent.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


