Chapter 54 Mechanisms of Action of Antineoplastic Drugs
| Abbreviations | |
|---|---|
| Ara-C | Cytarabine |
| DNA | Deoxyribonucleic acid |
| FH4 | Tetrahydrofolate |
| 5-FU | 5-Fluorouracil |
| IV | Intravenous |
| MESNA | Sodium 2-mercaptoethane sulfonate |
| 6-MP | 6-Mercaptopurine |
| MTX | Methotrexate |
| RNA | Ribonucleic acid |
| 6-TG | 6-Thioguanine |
Therapeutic Overview
Antineoplastic agents are used to treat more than 100 types of neoplastic diseases, with the goal of destroying malignant cells. Additional drugs (see Chapter 55) are used to enhance host defense mechanisms to eradicate those tumor cells not killed by the antineoplastic drugs. In clinical practice nearly all neoplastic diseases are treated by using multiple drugs, although only individual drugs, which form the basis for multiple drug therapy, are discussed in this chapter; multiple-drug protocols are described in Chapter 53.
The effectiveness of antineoplastic drugs varies greatly with the following:
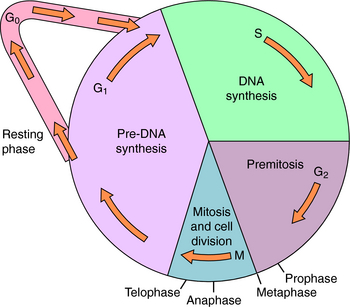
The end point used to evaluate effectiveness (e.g., tumor response, patient survival) is also important. Most antineoplastic agents, particularly chemotherapeutic agents, are more effective destroying cells that are progressing through the cell cycle (Fig. 54-1) than destroying cells that are resting in the G0 phase. The “growth fraction,” defined in Figure 54-1, is the fraction of cells progressing through the cycle. Besides tumor cells that may be proliferating, there are also some non-neoplastic cells undergoing division, particularly those of hair follicles, bone marrow, and intestinal epithelium. These rapidly dividing cells are especially sensitive to antineoplastic drugs and account for many of their undesirable side effects. It is believed that most, if not all, anticancer drugs kill cells primarily through a programmed, energy-dependent process called apoptosis, rather than through necrosis.
The number of cultured neoplastic cells that survive exposure to each drug typically shows a first-order relationship to the drug concentration (Fig. 54-2). This means that the same fraction of cells is killed with each drug dose and that a series of several doses does not kill 100% of them. This “log-cell kill” hypothesis is compatible with the clinical observation that a functional host immune system is needed for killing all neoplastic cells and curing a patient.
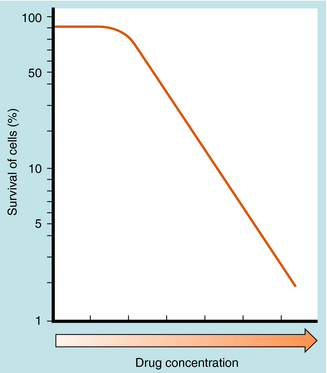
Endogenous cellular defenses, such as thiols or deoxyribonucleic acid (DNA) repair enzymes, however, can necessitate a threshold of drug concentration, or “shoulder,” in the survival curves of patients receiving these drugs (see Fig. 54-2).
| Therapeutic Overview |
|---|
| Goal |
| Uses |
| Effects |
| Considerations |
Mechanisms of Action
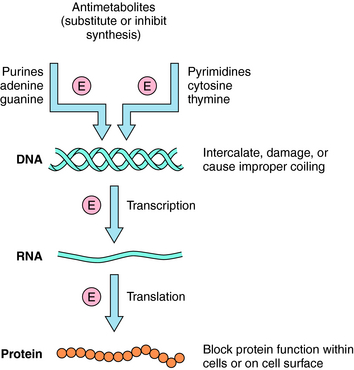
The basic mechanisms by which antineoplastic drugs kill tumor cells are summarized in Figure 54-3. Only compounds that show some selectivity for neoplastic cells are used clinically. In general, antimetabolites inhibit DNA synthesis, whereas alkylating agents, intercalators, and antibiotics damage or disrupt DNA, interfere with topoisomerase activity, or alter ribonucleic acid (RNA) structure. Steroids interfere with transcription, several plant alkaloids disrupt mitosis, agents such as asparaginase destroy essential amino acids needed for translation, and other drugs act through important growth factor signal transduction pathways. Many clinically used antineoplastic drugs must undergo either chemical or enzymatic modification to become actively cytotoxic.
Alkylation refers to the covalent attachment of alkyl groups to other molecules. Alkylating agents came to be used for cancer therapy as a result of observations of the effects of the mustard gases on cell growth. Although these compounds are too toxic for clinical use in cancer, the first effective antineoplastic agents, including mechlorethamine, were developed from related nitrogen mustard alkylating agents and are still used today.
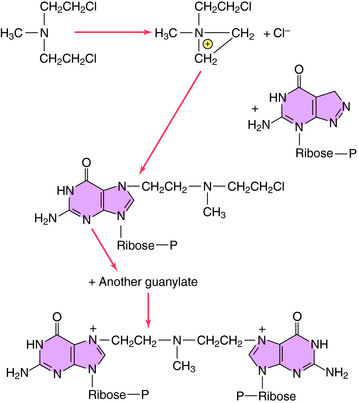
Alkylation takes place through chemical formation of a positively charged carbonium ion that reacts with an electron-rich site, particularly on DNA or RNA, to form modified nucleic acids. Most clinically used alkylating drugs have two active groups, which enable them to form covalent links between adjacent nucleic acid strands that are more difficult to repair than monofunctional adducts. These cross-links also prevent separation of the dual strands of DNA during cell cycling. For maximal kill, it is important to administer the maximally tolerated dose. The alkylation sequence for mechlorethamine reacting with the N-7 position of deoxyguanylate is shown in Figure 54-4. Although many other nucleophilic constituents, including RNA, proteins, and membrane components, become alkylated within cells, it is generally believed that the primary cytotoxic events occur through alkylation of DNA, especially by coupling to the N-7 position of the deoxyguanylates of either single- or double-stranded DNA.
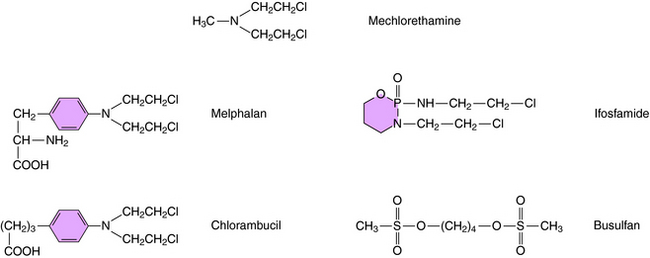
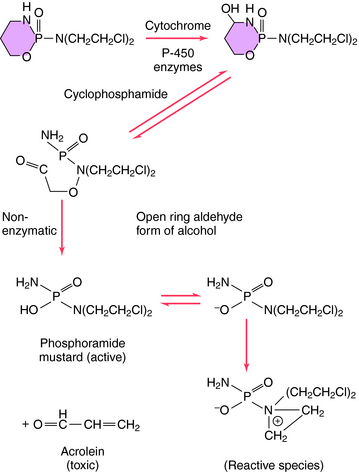
Structures of several clinically used alkylating agents are shown in Figure 54-5. Cyclophosphamide undergoes a combination of enzymatic and chemical activation to form the active phosphoramide mustard alkylating agent (Fig. 54-6). Exposure of cells to cyclophosphamide and other alkylating agents can also lead to carcinogenesis. For example, leukemia is a well-known long-term complication in patients with Hodgkin’s disease treated with a regimen including mechlorethamine.
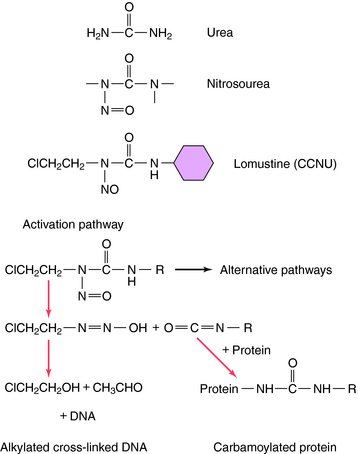
Another group of antineoplastic alkylating agents in clinical use is the nitrosoureas. The structures and primary mechanisms of activation for these compounds are shown in Figure 54-7. In addition to alkylation of DNA, the nitrosoureas also cause carbamoylation of proteins, which may play a role in cytotoxicity. The alkylation route, however, is the major source of cytotoxicity. Nitrosoureas are lipophilic and can cross the blood-brain barrier, so they are often used to treat brain tumors.
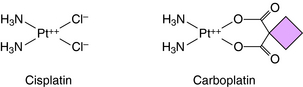
Temozolomide is the first new alkylating agent approved for treatment of malignant gliomas in decades. It has a structure similar to dacarbazine and is rapidly absorbed after oral administration. Unlike many other alkylating agents, temozolomide crosses the blood-brain barrier. It methylates guanine and adenine, resulting in misincorporation of thymidine (across from the methylated guanidine), which cannot be easily corrected by the mismatch repair system. Resistance occurs by up regulation of components of the DNA mismatch repair system. Other compounds that form covalent bonds with DNA are cisplatin and its analog carboplatin, whose structures are shown in Figure 54-8. Cisplatin is a square planar complex of platinum with two ammonia molecules and two chloride ions at the corners of the plane. The reaction sequence of the active species is complex and not completely understood. Replacement of a chloride with a hydroxyl must occur before the platinum-nitrogen bond can interact with DNA. Subsequently, the second chloride is aquated and reacts with DNA. The stereochemistry of the complex enables the cis, but not the trans isomer, to form two covalent platinum-nitrogen bonds, primarily at two adjacent deoxyguanylates of DNA. This intrastrand cross-link prevents DNA replication and is cytotoxic. Cisplatin also forms interstrand cross-links and protein-DNA cross-links. Similar DNA cross-links are formed with carboplatin. Oxaliplatin is an organoplatinum compound constructed to overcome resistance to cisplatin by binding the platinum atom to 1,2 diaminocyclohexane. Oxaliplatin undergoes conversion to reactive metabolites that covalently bind to either adjacent guanines, adjacent adenine-guanines, or guanines separated by an intervening nucleotide. This creates interstrand and intrastrand cross-links and inhibits DNA replication and transcription. Oxaliplatin is approved for treatment of metastatic colorectal carcinoma, has activity in lung cancer, and may have activity in breast and esophageal cancers and lymphoma.
The specific reaction sequences by which dacarbazine or procarbazine alkylate DNA are not well understood. Melphalan is a phenylalanine derivative and is actively transported into the cell by the carriers that transport leucine and glutamine. Melphalan is associated with induction of secondary leukemias. Chlorambucil is structurally similar to melphalan and is used primarily in treatment of chronic lymphocytic leukemia. Ifosfamide, like its analog cyclophosphamide, is activated by hepatic microsomes. Early studies with ifosfamide showed its use was associated with a significant incidence of hemorrhagic cystitis. Ifosfamide is now administered with a systemic thiol, sodium-2-mercaptoethane sulfonate (MESNA). MESNA becomes a free thiol after glomerular filtration and combines with the products responsible for causing the cystitis. Ifosfamide is active against several cancers, including small-cell lung cancer, sarcomas, lymphomas, testicular carcinoma, and gynecological cancers.
The antimetabolites are compounds that mimic the structures of normal metabolic constituents including folic acid, pyrimidines, or purines. The antimetabolites inhibit the enzymes necessary for folic acid regeneration or the pyrimidine or purine activation of DNA or RNA synthesis in neoplastic cells. Antimetabolites frequently kill cells in the S phase (see Fig. 54-1). Methotrexate (MTX), 5-fluorouracil (5-FU), cytarabine (Ara-C), 6-mercaptopurine (6-MP), gemcitabine, and 6-thioguanine (6-TG) are the primary antimetabolites used clinically.
Folic acid is essential for enzymatic reactions that transfer methyl and related groups during purine and pyrimidine synthesis. The antimetabolite MTX competitively inhibits the enzyme dihydrofolate reductase, which catalyzes the reduction of dihydrofolate to tetrahydrofolate (FH4; Fig. 54-9
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


