1 Introduction
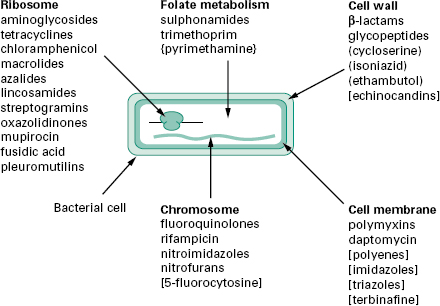
The antibiotics and synthetic anti-infective agents described in Chapter 11 are used to treat infections caused by bacteria, fungi and protozoa. Most exert a highly selective toxic action on their target microbial cells but have little or no toxicity towards mammalian cells. They can therefore be administered at concentrations sufficient to kill or inhibit the growth of infecting organ-isms without damaging mammalian cells. By comparison, the disinfectants, antiseptics and preservatives described in Chapter 17 are too toxic for systemic treatment of infections. Figure 12.1 illustrates the five broad target areas of the major groups of antibiotics and synthetic agents used to treat microbial infections. Note that most of the agents are selective antibacterials; relatively few agents are available for treatment of fungal or protozoal infections. Study of the mechanism of action reveals the basis for the selective toxicity.
Figure 12.1 Schematic diagram of a typical bacterial cell showing the ites of action of the major classes of antibiotics and antimicrobial agents used to treat infections. Agents listed without brackets are used to treat bacterial infections. Agents used against mycobacterial, fungal or protozoal infections are indicated by the use of (), [] and {} brackets, respectively
2.1 Peptidoglycan biosynthesis in bacteria and its inhibition
Peptidoglycan is a vital component of the cell wall of virtually all bacteria. About 50% of the weight of the wall of Gram-positive bacteria is peptidoglycan; smaller amounts occur in mycobacterial walls (30%) and Gram-negative bacterial cell walls (10-20%). It is a macromol-Figure 12.1 Schematic diagram of a fusidic acid short peptide chains (Figure 12.2). The glycan chains contain alternating units of N-acetylmuramic acid and N-acetylglucosamine. Each N-acetylmuramic acid contains a short peptide substituent made up of four amino acids (the stem peptides). A key feature of peptidoglycan is the occurrence of the d-isomers of some amino acids in the stem peptides (particularly d-alanine and d-glutamic acid) and unusual amino acids such as meso-diaminopimelic acid which are not found in proteins. In some organisms (e.g. Escherichia coli) cross-linking of the stem peptides involves a direct peptide bond between the fourth amino acid of the stem peptide on one chain and the third amino acid in the stem peptide on an adjacent chain. In other organisms (e.g. Staphylococcus aureus) the linkage is made by a short peptide bridge (e.g. five glycines) between the stem peptides. The precise composition of peptidoglycan varies between different organisms but the overall structure is the same.
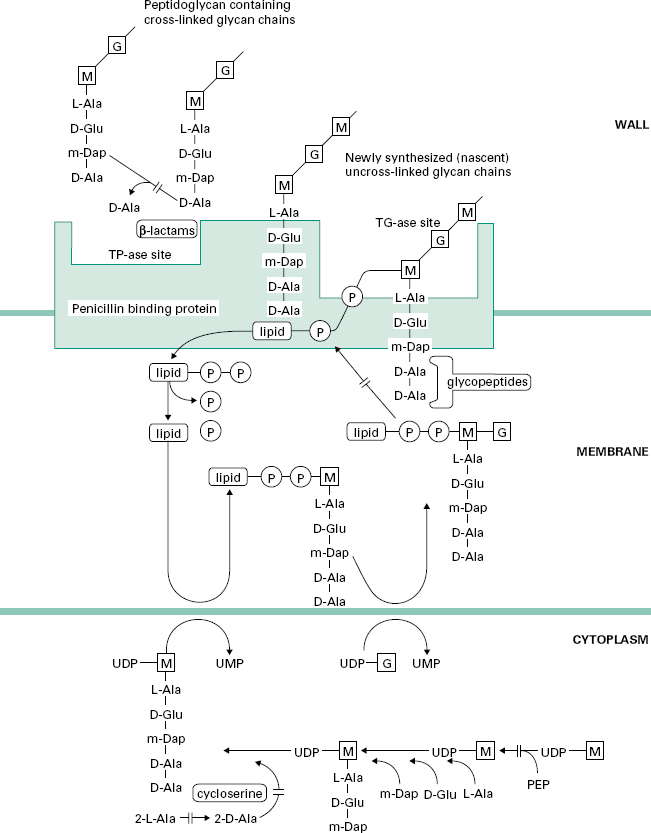
Figure 12.2 Pathway for the biosynthesis of peptidoglycan in bacterial cells showing the sites of action of cycloserine, glycopeptide and β-lactam antibiotics.
In all cases the peptidoglycan plays a vital role: it is responsible for maintaining the shape and mechanical strength of the bacterial cell. If it is damaged in any way, and particularly if its synthesis is inhibited, then the shape of the cells becomes distorted, they swell and will eventually burst (lyse) as a result of the high internal osmotic pressure. Mammalian cells do not possess a cell wall and contain no other macromolecules resembling peptidoglycan. Consequently, antibiotics which interfere with the synthesis and assembly of peptidoglycan show excellent selective toxicity.
2.1.1D-Cycloserine
D-Cycloserine interferes with the early stage of synthesis of peptidoglycan involving the assembly of the dipeptide d-alanyl-D-alanine. This occurs inside the cytoplasm and involves a racemase enzyme which converts l-alanine to d-alanine and a ligase which couples two d-alanines together (Figure 12.2). Both of these enzymes are inhibited by d-cycloserine, which bears some structural similarities to D-alanine. The antibiotic binds to the pyridoxal phosphate cofactor of the enzymes, effectively preventing them from forming d-alanyl-D-alanine. Subsequent stages of peptidoglycan synthesis, involving coupling of the dipeptide to three other amino acids forming the stem peptide on UDP-N-acetylmuramicacid,areblocked. Note that initially the peptide contains five amino acids, terminating in d-alanyl-d-alanine.Theterminal D-alanine is removed on insertion into the cell wall during the final step in which the cross-links are formed between stem peptides on adjacent glycan strands.
2.1.2 Glycopeptides—vancomycin and teicoplanin
The peptidoglycan macromolecule is assembled in the cell wall by the sequential action of two enzymes (transglycosylases and transpeptidases) which are located on the outer face of the cytoplasmic membrane. To reach the assembly site (i.e. a region of cell wall growth) the precursors, which are assembled in the cytoplasm, must cross the cell membrane. They do this linked to a lipid, undecaprenylphosphate, which acts as a carrier molecule, cycling between the inner and outer faces of the membrane. The biochemical details of this process are outlined in Figure 12.2. Antibiotics interfering with this stage of peptidoglycan synthesis have been identified, e.g. bacitracin, but they have not found major applications in the treatment of infections.
The glycopeptides vancomycin and teicoplanin act at the stage where the peptidoglycan precursors are inserted into the cell wall by the transglycosylase enzyme on the outer face of the cell membrane. This enzyme assembles linear glycan chains that are not initially cross-linked to the existing peptidoglycan in the cell wall. The linear glycan chains are assembled by the transglycosylase by transfer of the growing glycan chain to the disaccharide peptidoglycan precursor on the lipid carrier as it crosses the cell membrane. Glycopeptides block this process by binding, not to the enzyme itself, but to the disaccharide peptidoglycan precursor, specifically to the d-alanyl-d-alanine portion on the stem peptide. The presence of the bulky glycopeptides tightly bound to each d-alanyl-d-alanine residue prevents the transglycosylase from carrying out the transfer reaction. Binding involves formation of a network of five hydrogen bonds between amino acid residues on the glycopeptide antibiotics and d-alanyl-d-alanine. Resistance to this unusual mecha-nism of enzyme inhibition can result from alteration in the d-alanyl-d-alanine substrate to d-alanyl-d-lactate, which occurs in glycopeptide (e.g. vancomycin)-resistant enterococci (VRE). Vancomycin does not penetrate the cell membrane of bacteria and is thought to bind to the disaccharide-pentapeptides on the outer face of the cytoplasmic membrane. It has been suggested that two vancomycin molecules form a back-to-back dimer which bridges between pentapeptides on separate glycan chains, thus preventing further peptidoglycan assembly. Teicoplanin also binds tightly to the d-alanyl-d-alanine region of the peptidoglycan precursor. However, as a lipoglycopeptide it may act slightly differently from vancomycin, by locating itself in the outer face of the cytoplasmic membrane and binding the pentapeptide as the precursors are transferred through the membrane. Glycopeptides must cross the cell wall to reach the outer face of the cell membrane where transglycosylation takes place. They are too large to penetrate the outer membrane of most Gram-negative bacteria and are consequently used for treatment of serious Gram-positive infections.
2.1.3 β-Lactams—penicillins, cephalosporins, carbapenems and monobactams
The final stage of peptidoglycan assembly is the cross-linking of the linear glycan strands assembled by transglycosylation to the existing peptidoglycan in the cell wall. This reaction is catalysed by transpeptidase enzymes, which are also located on the outer face of the cell membrane. They first remove the terminal d-alanine residue from each stem peptide on the newly synthesized glycan chain. The energy released from breaking the peptide bond between the two alanines is used in the formation of a new peptide bond between the remaining D-alanine on the stem peptide and a free amino group present on the third amino acid of the stem peptides in the existing cross-linked peptidoglycan. In many organisms, including E. coli, this acceptor amino group is supplied by the amino acid diaminopimelic acid. In other organisms, e.g. Staph. aureus, the acceptor amino group is supplied by the amino acid l-lysine. Although there is considerable variation in the composition of the peptide cross-link among different species of bacteria, the essential transpeptidation mechanism is the same. Therefore, virtually all bacteria can be inhibited by interference with this group of enzymes.
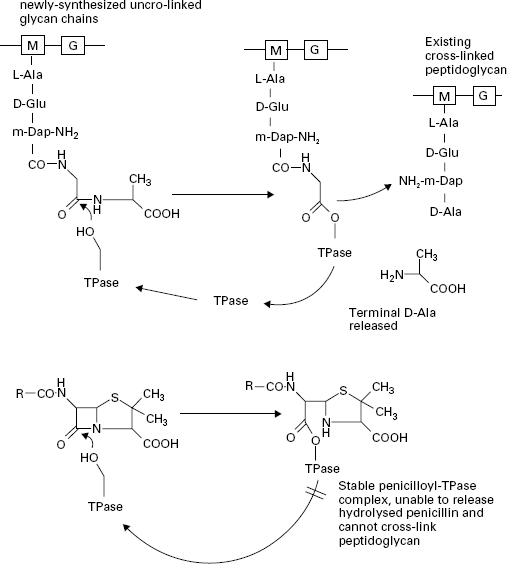
The β-lactam antibiotics inhibit transpeptidases by acting as alternative substrates. They mimic the d-alanyl-d-alanine residues and react covalently with the transpeptidases (Figure 12.3). The β-lactam bond (common to all members of the β-lactam antibiotics) is broken but the remaining portion of the antibiotic is not released immediately. The half-life for the transpeptidase-antibiotic complex is of the order of 10 minutes; during this time the enzyme cannot participate in further rounds of peptidoglycan assembly by reaction with its true sub-strate. The vital cross-linking of the peptidoglycan is therefore blocked while other aspects of cell growth continue. The cells become deformed in shape and eventually burst through the combined action of a weakened cell wall, high internal osmotic pressure and the uncontrolled activity of autolytic enzymes in the cell wall. Penicillins, cephalosporins, carbapenems and monobactams all inhibit peptidoglycan cross-linking through interactionof the common β-lactam ring with the transpeptidase enzymes. However, there is considerable variation in the morphological effects of different β-lactams owing to the existence of several types of transpeptidase. The transpeptidase enzymes are usually referred to as penicillinbinding proteins (PBPs) because they can be separated and studied after reaction with 14C-labelled penicillin. This step is necessary because there are very few copies of each enzyme present in a cell. They are usually separated according to their size by electrophoresis and are numbered PBP1, PBP2, etc., starting from the highest molecular weight species. In Gram-negative bacteria most of the high molecular weight transpeptidases also possess transglycosylase activity, i.e. they have a dual function in the final stages of peptidoglycan synthesis with the transglycosylase and transpeptidase activities located in separate regions of the protein structures. Furthermore, the different transpeptidases have specialized functions in the cell; all cross-link peptidoglycan but some are involved with maintenance of cell integrity, some regulate cell shape and others produce new cross wall between elongating cells, securing chromosome segregation prior to cell division. The varying sensitivity of the PBPs towards different β-lactams helps to explain the range of morphological effects observed in treated bacteria. For example, penicillin G (benzylpenicillin),ampicillin and cephaloridine are particularly effective in causing rapid lysis of Gram-negative bacteria such as E. coli. These antibiotics act primarily upon PBP1B, the major transpeptidase of the organism. Other β-lactams have little activity against this PBP, e.g. mecillinam binds preferentially to PBP2 and it produces a pronounced change in the cells from a rod shape to an oval form. Many of the cephalosporins, e.g. cephalexin, cefotaxime and ceftazidime, bind to PBP3 resulting in the formation of elongated, filamentous cells. The lower molecular weight PBPs, 4, 5 and 6, do not possess transpeptidase activity. These are carboxypeptidases, which remove the terminal D-alanine from the pentapeptides on the linear glycans in the cell wall but do not catalyse the cross-linkage. Their role in the cells is to regulate the degree of cross-linking by denying the d-alanyl-d-alanine substrate to the transpeptidases but they are not essential for cell growth. Up to 90% of the amount of antibiotic reacting with the cells may be consumed in inhibiting the carboxypeptidases, with no lethal consequences to the cells.
Figure 12.3 The action of transpeptidase (TPase) with its natural substrate (upper) and penicillin G (lower). The-OH group on a serine residue at the active site of the TPase attacks the peptide bond between the terminal d-alanyl-d-alanine residues of the stem peptide in the non-cross-linked glycan chain. The terminal D-alanine is released and a new peptide cross-link is formed with existing peptidoglycan in the cell wall. Penicillin G blocks this process by forming a stable penicilloyl-TPase complex which cannot release the penicillin and therefore cannot form the peptide cross-link in the peptidoglycan.
Gram-positive bacteria also have multiple transpeptidases, but fewer than Gram-negatives. Shape changes are less evident than with Gram-negative rod-shaped organisms. Cell death follows lysis of the cells mediated by the action of endogenous autolytic enzymes (autolysins) present in the cell wall which are activated following β-lactam action. Autolytic enzymes able to hydrolyse peptidoglycan are present in most bacterial walls; they are needed to re-shape the wall during growth and to aid cell separation during division. Their activity is regulated by binding to wall components such as the wall and membrane teichoic acids. When peptidoglycan assembly is disrupted through β-lactam action, some of the teichoic acids are released from the cells, which are then susceptible to attack by their own autolysins.
2.1.4 β-Lactamase inhibitors—clavulanic a cid, sulbactam and tazobactam
Expression of β-lactamase enzymes is the most important mechanism through which organisms become resistant to β-lactams. Over 300 different β-lactamase enzymes have been described and they can be classified either by amino acid sequence or by their biochemical properties. The majority of the enzymes have a serine residue at their active site and bear structural and mechanistic similarities to the carboxypeptidases from which they are thought to have evolved. Unlike the transpeptidases and carboxypeptidases, the β-lactamases hydrolyse β-lactam antibiotics very efficiently, releasing fragments of the antibiotics rapidly instead of remaining bound to the ringopened forms for several minutes. A number of successful inhibitors, including clavulanic acid, sulbactam and tazobactam have been developed for use in combination with susceptible β-lactams (amoxicillin, ampicillin and piperacillin, respectively), protecting them from inactivation by the β-lactamases. The inhibitors are hydrolysed by the β-lactamases in the same manner as susceptible β-lactam antibiotics, the β-lactam ring being broken by attack by a serine residue in the active site of the enzyme. Instead of undergoing rapid release from the active site serine, the inhibitors remain bound and undergo one of several different fates. It is thought that the hydrolysed inhibitors can interact with a second enzyme residue in the active site of the β-lactamase, forming a covalently cross-linked, irreversibly inhibited complex. Other categories of β-lactamase enzymes have zinc atoms at their active sites and hydrolyse the β-lactam ring by a different mechanism to the serinebased enzymes. These metallo-β-lactamases are not inhibited by clavulanic acid, sulbactam and tazobactam.
2.2 Mycolic acid and arabinogalactan biosynthesis in mycobacteria
The cell walls of mycobacteria contain an arabinogalactan polysaccharide in addition to the peptidoglycan, plus a variety of high molecular weight lipids, including the mycolic acids, glycolipids, phospholipids and waxes. The lipid-rich nature of the mycobacterial wall is responsible for the characteristic acid-fastness on staining and serves as a penetration barrier to many antibiotics. Isoniazid and ethambutol have long been known as specific antimycobacterial agents, exerting no activity towards other bacteria, but their mechanisms of action have only recently been established.
2.2.1 Isoniazid
Isoniazid interferes with mycolic acid synthesis by inhibiting an enoyl reductase (InhA) which forms part of the fatty acid synthase system in mycobacteria. Mycolic acids are produced by a diversion of the normal fatty acid synthetic pathway in which short-chain (16-carbon) and longchain (24-carbon) fatty acids are produced by addition of 7 or 11 malonate extension units from malonyl coenzyme A to acetyl coenzyme A. InhA inserts a double bond into the extending fatty acid chain at the 24-carbon stage. The long-chain fatty acids are further extended and condensed to produce the 60-90-carbon β-hydroxymycolic acids which are important components of the mycobacterial cell wall. Isoniazid is converted inside the mycobacteria to a free radical species by a catalase peroxidase enzyme, KatG. The active free radicals then attack and inhibit the enoyl reductase, InhA, by covalent attachment to the active site.
2.2.2 Ethambutol
Ethambutol blocks assembly of the arabinogalactan polysaccharide by inhibition of an arabinotransferase enzyme. Cells treated with ethambutol accumulate the isoprenoid intermediate decaprenylarabinose, which supplies arabinose units for assembly in the arabinogalactan polymer.
2.3 Echinocandins—caspofungin, a nidulafungin and micafungin
These members of the echinocandin group of antifungal agents have been developed for treatment of serious fungal infections. They interfere with the synthesis of the β-1,3-d-glucan polymer in the fungal cell wall. Without the glucan polymer, the integrity of the fungal cell wall is compromised, yeast cells lose their rigidity and become like protoplasts; the effect is especially pronounced in Candida and Aspergillus species.
3.1 Protein synthesis and its selective inhibition
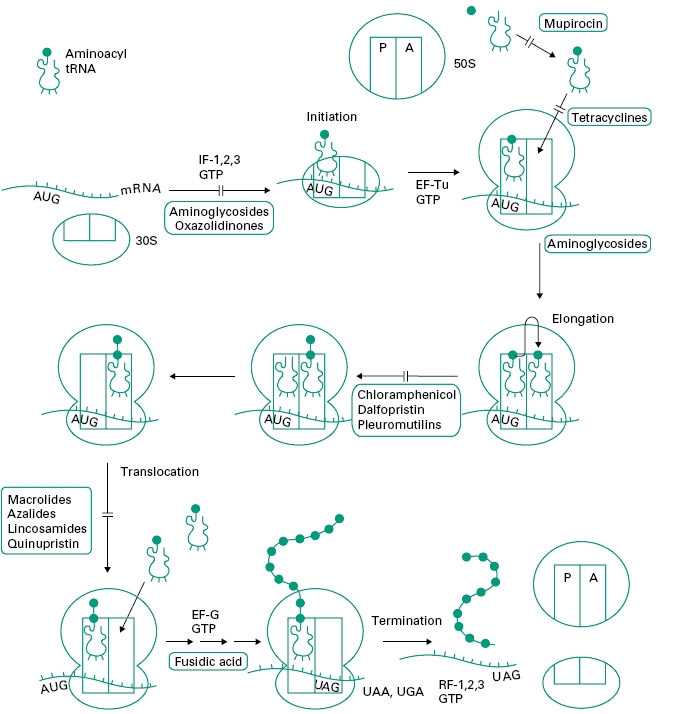
Figure 12.4 outlines the process of protein synthesis involving the ribosome, mRNA, a series of aminoacyl transfer RNA (tRNA) molecules (at least one for each amino acid) and accessory protein factors involved in initiation, elongation and termination. As the process is essentially the same in prokaryotic (bacterial) and eukaryotic cells (i.e. higher organisms and mammalian cells) it is surprising that there are so many selective agents which act in this area (see Figure 12.1).
Figure 12.4 Outline of the process of protein synthesis (translation of messenger RNA) in bacterial cells. The four stages of synthesis are shown: initiation, elongation, translocation and termination with the sites of action of antibiotics. AUG is the start codon on messenger RNA (mRNA) specifying the first amino acid in bacterial proteins, N-formylmethionine. UAG, UAA and UGA are termination codons specifying no amino acid. 30S and 50S are the subunits of the ribosome. Other protein factors involved in protein synthesis are initiation factors (IF-1, 2, 3), elongation factors (EF-Tu and EF-G) and release factors (RF-1, 2, 3).