Fig. 4.1
Pathologic features of pilocytic astrocytoma (PA). PA is histologically characterized by the presence of neoplastic bipolar astrocytes in compact areas (a). Rosenthal fibers are a frequent feature of pilocytic astrocytomas and may be numerous (b). The second architectural pattern of PA is characterized by loose stroma, containing microcysts (c) and occasionally multinucleated cells and clusters of small nuclei (d). Microvascular hyalinization is frequent in PA (e), as it is typical of long standing, slow growing tumors. A variable component of round cells with perinuclear halos may be present in a subset of PA (f), and raises the important differential diagnosis with oligodendroglial neoplasms.
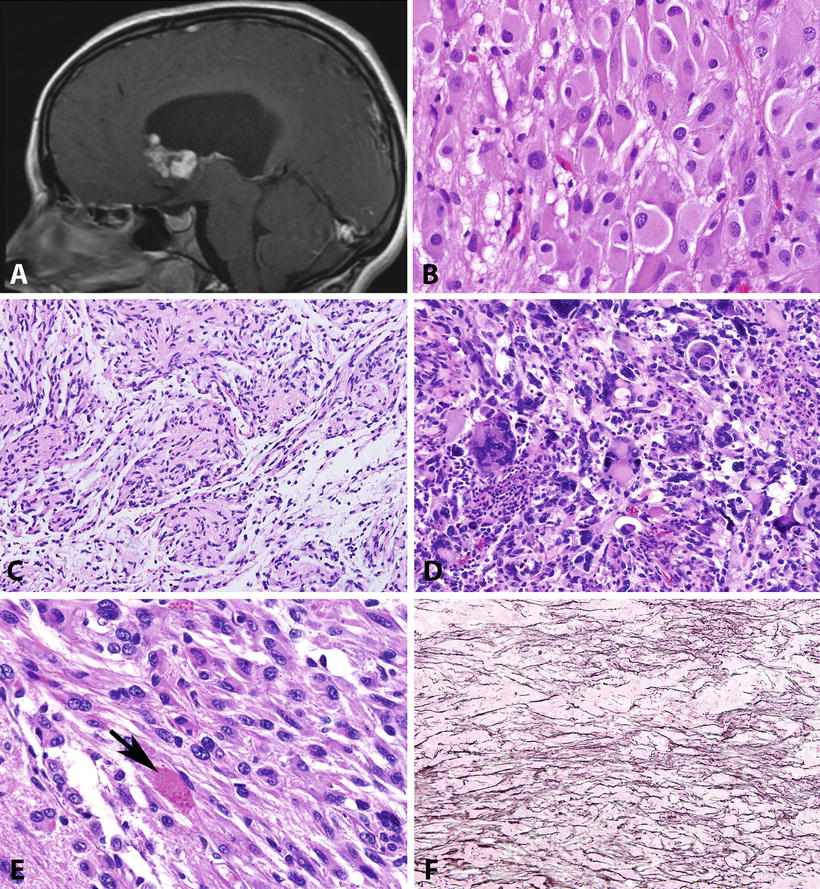
Fig. 4.2
Pathologic features of circumscribed gliomas. Subependymal giant cell astrocytoma (SEGA) develops within the lateral ventricle, almost always near the Foramen of Monro (T1-weighted MR image post-contrast) (a). The histology of SEGA is distinctive, containing large cells with voluminous eosinophilic cytoplasm and prominent nucleoli (b). Pilomyxoid astrocytoma is a distinctive variant of pilocytic astrocytoma, usually developing in the hypothalamus of young children, characterized by perivascular arrangements in a loose myxoid stroma (c). Pleomorphic xanthoastrocytoma is another distinctive astrocytoma characterized by conspicuous pleomorphic cells (d), usually with low proliferative activity, as well as a fascicular arrangement and eosinophilic granular bodies (arrow) (e). Unlike other gliomas, PXAs tend to be reticulin-rich, particularly in superficial regions juxtaposed to the leptomeninges (f).
Table 4.1
Histopathology and molecular features of low-grade gliomas.
Tumor type | Location | Histology | Grade | IHC | Cytogenetics | Molecular genetics | Signaling pathways |
|---|---|---|---|---|---|---|---|
Pilocytic astrocytoma | Cerebellum > optic pathways, brainstem, supratentorial, spinal cord | Bipolar cells, compact and microcystic areas, Rosenthal fibers, eosinophilic granular bodies | I | GFAP, OLIG2+, p53− | 7q34 duplication, whole Ch7 or 8 gain | BRAF–KIAA1549 fusion, BRAF (V600E) mut, RAF1 fusions, NF1 inactivation (syndrome associated cases) | MAPK, mTOR |
Pilomyxoid astrocytoma | Hypothalamic region > brainstem, hemispheres, spinal cord | Monomorphic spindle cells, myxoid stroma, perivascular pseudorosettes | II | GFAP+ | Similar to PA | Similar to PA | Similar to PA |
SEGA | Lateral ventricle (near foramen of Monro) | Large cells, round nuclei, macronucleoli, amphophilic cytoplasm | I | S100, GFAP+++, synaptophysin, neurofilament+ | – | TSC1 and TSC2 mutations | mTOR |
Angiocentric glioma | Cortex (temporal lobe) | Monotonous spindle cell with perivascular arrangement | I | GFAP+, EMA + (dot-like) | 6q23.3 gain/deletion | MYB alterations | Cell cycle regulation |
Pleomorphic xanthoastrocytoma | Hemispheric (temporal lobe) | Cellular pleomorphism, xanthic change, fascicular arrangement, eosinophilic granular bodies | II | S100 > GFAP+, CD34, synaptophysin +/−, p53− | – | BRAF (V600E) mut; mTOR pathway (NF1, TSC2, PI3R1), TP53 (rare) | MAPK, mTOR |
Diffuse astrocytoma | Throughout CNS | Infiltrative pleomorphic cells, rare to absent mitotic activity | II | GFAP+++, OLIG2+++, IDH1 (R132H) (frequent), p53 (frequent) | 7q, 8q gain | IDH1/2, TP53, ATRX, PTEN, CDKN2A, mutations | MAPK, PI3K/mTOR, cell cycle regulation |
Low-grade oligodendroglioma/oligoastrocytoma | Hemispheric (frontal > temporal, parietal, occipital) | Round cells with perinuclear halos, chicken-wire microvasculature | II | S100+++, OLIG2+++, IDH1 (R132H) (frequent), p53− | 1p19q co-deletion | IDH1/2, CIC, FUBP1 mutations; TERT promoter mutations | |
Pediatric diffuse astrocytoma | Same as adult | II | GFAP, OLIG2+++, IDH (R132H) (usually −), p53+ | 8q13.1 gain/deletion; 6q23.3 gain/deletion | FGFR1, MYB, MYBL1, BRAF (V600E) alterations | MAPK, PI3K/mTOR, cell cycle regulation | |
Pediatric low-grade oligodendroglioma | Hemispheric (frontal–temporal lobes) | Same as adult | II | S100, OLIG2+++, IDH1 (R132H) (usually −), p53− | 1p19q co-deletion (rare) | FGFR1, MYB alterations; IDH1 (R132H) (rare) | MAPK, PI3K/mTOR, cell cycle regulation |
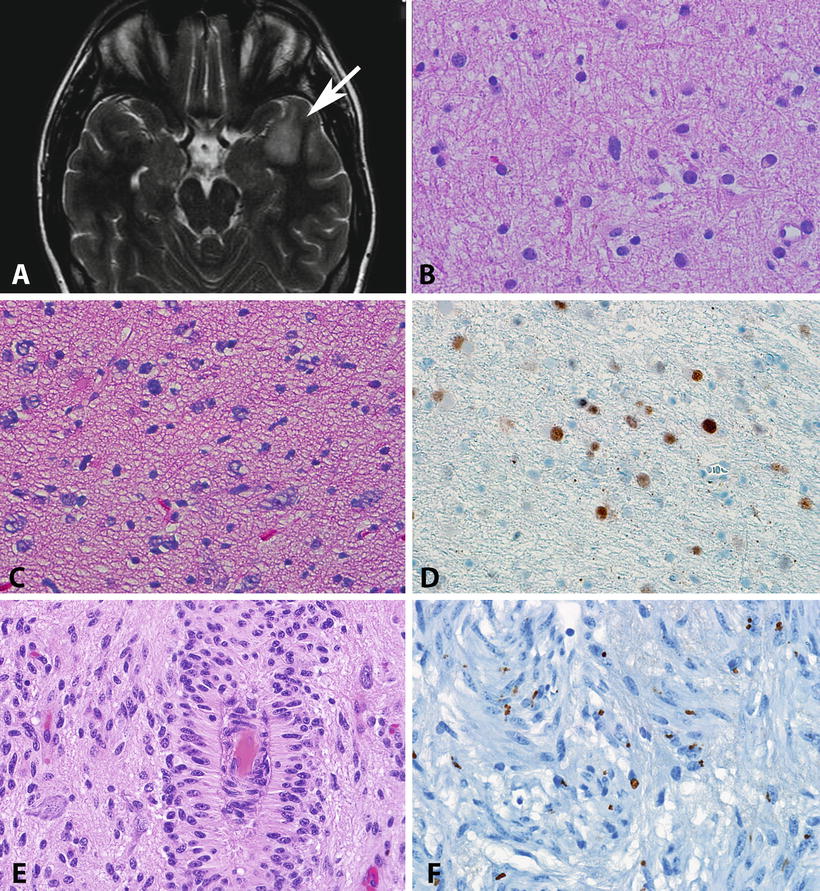
Fig. 4.3
Pathologic features of diffuse astrocytomas and angiocentric gliomas. Diffuse astrocytomas (WHO grade II) present as areas of hyperintensity in T2 weighted MR images (arrow, a). As the histologic level, diffuse astrocytomas usually have low cellularity but have definite atypia manifesting by irregular nuclear contours and hyperchromasia (b). Some diffuse astrocytomas may show obvious hypercellularity, but by definition mitotic activity is rare to absent (c). Immunohistochemistry frequently demonstrates strong p53 immunolabeling, a surrogate for TP53 mutations (d). Angiocentric glioma is a distinctive low-grade neoplasm characterized by monotonous cells with elongated nuclei infiltrating cortex but also arranged around vessels (e). Despite their infiltrative nature, angiocentric glioma shares biologic properties with ependymomas, including the presence of microlumens highlighted by EMA immunohistochemistry (f).
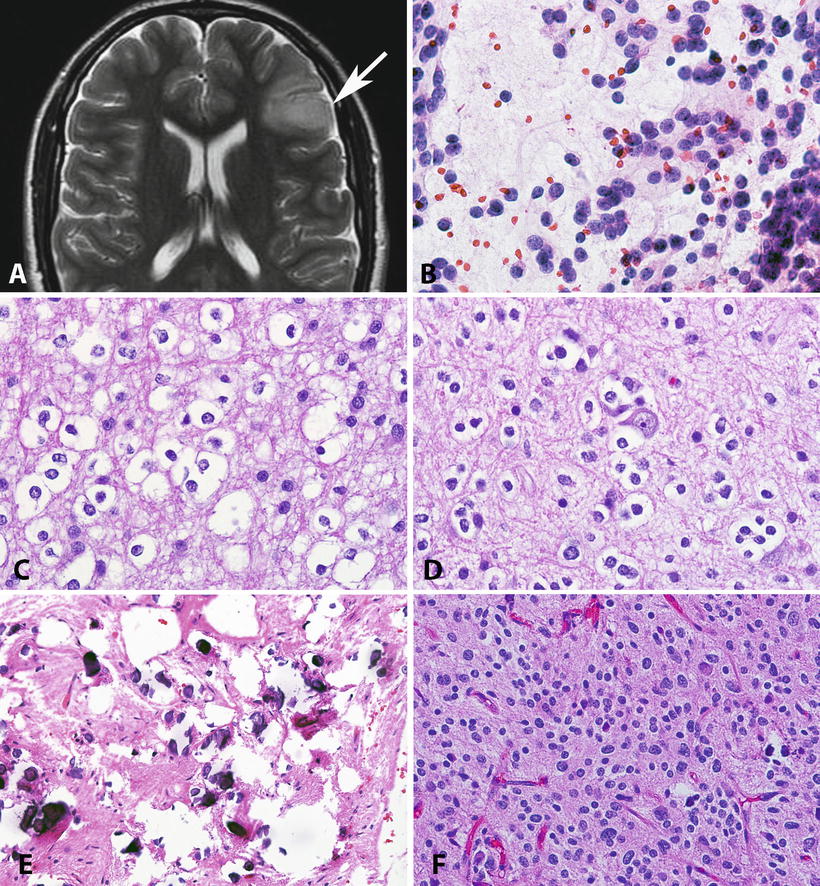
Fig. 4.4
Pathologic features of low-grade oligodendrogliomas. Oligodendrogliomas frequently involve the frontal cortex, with expansion of infiltrated gyri (Axial T2 weighted MR) (a). On cytologic preparations, oligodendrogliomas demonstrate round cells with a small nucleolus (b). The nuclear uniformity and characteristic halos are best appreciated in formalin-fixed paraffin-embedded sections (c). Perineuronal satellitosis is a characteristic feature of infiltrating gliomas, particularly oligodendroglioma (d). Microcalcifications are not uncommon in oligodendroglial neoplasms (e). The designation of oligoastrocytoma is reserved for tumors that have areas with astrocytic and oligodendroglial morphology. Most of these tumors are morphologically ambiguous as this H&E shows (f), and lead to prominent interobserver variability.
Circumscribed Gliomas
Pilocytic Astrocytoma
PAs represent the most frequent glioma subtype in children. They are characterized by elongated, bipolar astrocytes usually with bland nuclear features. The classic architecture is a biphasic pattern, with alternating Rosenthal fiber-rich compact areas and loose, microcyst rich regions. Eosinophilic granular bodies may be present in these regions and even abundant. Additional variable features include hyalinized or glomeruloid vessels, hemosiderin deposition, and degenerative nuclear pleomorphism or multinucleated cells. Monotonous oligodendroglial-like cells may predominate in some examples. Occasional mitoses and non-pseudopallisading necrosis may be present, but they have an inconsistent relation with clinical outcome [6, 7]. As other astrocytomas, these tumors express GFAP and OLIG2. P53 and Ki-67 show low labeling indices. Elevated Ki-67 labeling indices have been associated with worse outcome in some studies [8] but not in others [9, 10]. Unlike the category of diffuse gliomas, the development of anaplasia/histologic malignancy in PA is a very rare event (<2 %), and has been defined as the presence of brisk mitotic activity with or without necrosis [11].
Pilomyxoid Astrocytoma
Pilomyxoid astrocytoma (PMA) is considered a variant of PA, characterized by monophasic morphology within a myxoid background and conspicuous aggregates around blood vessels [12]. Unlike conventional PA, PMA lacks a biphasic pattern and Rosenthal fibers. Eosinophilic granular bodies are rare to absent. The classic presentation of PMA is the hypothalamic region of young children. Because of its higher likelihood for aggressive behavior and leptomeningeal dissemination [13], the WHO assigns a grade II to this variant. Morphologic and molecular overlap occurs with conventional PA, which is supported by the recognition of tumors with intermediate features between PMA and PA or PMAs that mature into PA over time [14].
Subependymal Giant Cell Astrocytoma
SEGA is a unique astrocytoma subtype that arises almost always within the lateral ventricles near the foramen of Monro in tuberous sclerosis patients. It is composed of large cells with voluminous eosinophilic cytoplasm, round nuclei and prominent nucleoli. The mitotic activity is very low. At an immunohistochemical and ultrastructural level, these tumors demonstrate evidence of neuronal differentiation in addition to a glial phenotype, which is more accurately consistent with a glioneuronal neoplasm [15]. This includes immunoreactivity for synaptophysin and neurofilament protein in addition to GFAP.
Pleomorphic Xanthoastrocytoma
PXA is a rare neoplasm with distinctive histology. It is composed of spindle neoplastic cells with often conspicuous pleomorphism, sometimes including giant cell forms. Mitotic activity is very low in most cases, and this disconnect between the cellularity and pleomorphism of the tumor and its relative lack of mitoses is one of the first clues to the diagnosis. Eosinophilic granular bodies, particularly large pale forms, are also frequent.
PXA may also demonstrate immunoreactivity for neuronal markers in addition to S100 and GFAP. P53 immunolabeling is typically weak or negative, while CD34 expression is relatively frequent [16]. An additional characteristic finding is the presence of pericellular staining with reticulin special stains, particularly in areas juxtaposed to leptomeninges.
Although PXA is low grade (WHO grade II) by definition, among the circumscribed gliomas it has the highest propensity for recurrence and biologic aggressiveness. A subset develops anaplastic features in the form of brisk mitotic activity and/or necrosis and microvascular proliferation [17]. However, these changes do not consistently predict an adverse outcome, and pending more definite data, a grade III is not yet allowed under the WHO classification.
Diffuse Gliomas
Angiocentric Glioma
Angiocentric glioma is a distinctive neoplasm that was added to the WHO 2007 classification [18, 19]. It is slow growing, frequently associated with epilepsy and therefore shares many clinical properties with dysembryoplastic neuroepithelial tumor. Despite gross circumscription, it is histologically infiltrative. The most characteristic feature of angiocentric glioma is the presence of thin elongated nuclei with little pleomorphism and a perivascular aggregation in a parallel and perpendicular pattern (Fig. 4.3). GFAP expression is a constant feature by immunohistochemistry, but in addition EMA frequently stains in a dot-like fashion, and microlumina may be present on electron microscopy, which suggests a dual astrocytic/ependymal phenotype.
Diffuse Astrocytoma
Diffuse astrocytoma (DA) is a specific subtype characterized by neoplastic astrocytes with nuclear hyperchromasia, atypia, usually low cellularity and rare to absent mitotic figures (Fig. 4.3). Unlike the circumscribed glioma group, DA demonstrates an exquisite infiltration of underlying brain parenchyma, making them surgical challenges. They also have a strong tendency for progression to higher grade neoplasms (anaplastic astrocytoma, glioblastoma), particularly in adults.
Oligodendroglial Tumors
Oligodendroglial tumors include oligodendrogliomas and mixed oligoastrocytomas. They may be low (grade II) or high grade (grade III) (Fig. 4.4). Oligodendroglial morphology is defined by round nuclei with perinuclear halos and little internuclear variability. A delicate “chicken-wire” type microvasculature may be present. Limited mitotic activity may be present, but brisk mitotic activity, endothelial hypertrophy and necrosis define higher grade tumors. Adult oligodendroglioma in particular has classic molecular alterations involving chromosomal arms 1p and 19q, and has almost become a combined pathologic/molecular diagnosis.
Pediatric Diffuse Gliomas
Prior observations have hinted that morphologically similar neoplasms in the adult and pediatric populations may have different clinical behavior. DA, for example, may not always have the same predictable progression to anaplasia in children as in adults. This has been confirmed by molecular studies [20]. Diffuse intrinsic pontine gliomas represent a distinct subset of diffuse gliomas in children, and are essentially defined by their anatomic development within the brainstem. Although in theory, a subset of these may be identified as grade II early in their course, high-grade histologic features are almost always present postmortem [21]. Oligodendrogliomas in children for example lack 1p19q co-deletion and IDH1/2 mutations in contrast to adult tumors [22–24]. Furthermore, the mutational landscape uncovered by recent, large scale sequencing efforts is different, and simpler, in pediatric diffuse gliomas.
New and Unclassifiable Entities
After accounting for well-defined histopathologic categories, a subset of low-grade gliomas remain diagnostic challenges, particularly in the pediatric population. In fact, approximately 20 % of pediatric brain tumors are unclassifiable according to traditional schemes, most of which involve low-grade gliomas (Burger PC, personal communication). One particular entity that has been recognized for a while, but has been the recent focus of several larger series is a low-grade oligodendroglioma-like tumor that remains indolent despite widespread superficial CNS dissemination [25–28]. This tumor has frequent 1p chromosome arm loss but lacks IDH1 (R132H) mutations. There is also a rare subset of pediatric low-grade tumors that have been termed descriptively “massively calcified low-grade glioma” that lack alterations associated with other low-grade gliomas [29].
Gene Expression Profiling
Global gene expression profiling studies have provided important biologic insights into the biology of gliomas in general and low-grade gliomas in particular. In PA, gene expression profiling studies have highlighted differential gene expression signatures related to anatomic regions and NF1 vs. sporadic occurring tumors [30]. These studies have also uncovered possible biomarkers associated with worse clinical outcome in PA including overexpression of Matrilin–2 [31], and underexpression of ALDH1L1 [32] and myelin basic protein (MBP) [33].
Analogous studies focusing on DA have also provided important insights particularly highlighting phenotypic and genetic differences between adult and pediatric DA. These included expression changes in genes involved in neural stem cell maintenance, CNS development, DNA replication, and cell cycle [20], which may explain in part the more aggressive behavior of these tumors when occurring in adults. Integration of mutation, copy number, and transcriptome analysis has also allowed the separation of distinct molecular subgroups of grade II and III diffuse gliomas with biological and prognostic relevance. Using this approach, Gorovets et al. classified diffuse astrocytic tumors into three molecular classes denoted “preglioblastoma” (PG), “neuroblastic” (NB), and “early-progenitor-like” (EPL) [34]. The NB and EPL subclasses were associated with a higher frequency of IDH and TP53 mutations and 8q gains, as well as better clinical outcome compared with the PG class. Interestingly, 8q gain is one of the most frequent cytogenetic abnormalities of diffuse astrocytoma [35].
Molecular Genetics and Signaling Pathways
Genetic Predisposition to Low-Grade Glioma
The earliest insights into the molecular alterations contributing to glioma formation have evolved from the study of inherited tumor syndromes. Neurofibromatosis type 1 (NF1) is associated with germline mutations in the NF1 gene that encodes for the protein neurofibromin, a negative regulator of RAS signaling. These patients are predisposed to gliomas of various grades, particularly PA of the optic pathways. Furthermore, biallelic NF1 inactivation is a feature of these patients tumors [36]. Tuberous sclerosis complex is associated with germline mutations in TSC1 or TSC2, which leads to increased mTOR activity and a predisposition to SEGA. Conversely, Li–Fraumeni syndrome is associated with TP53 mutations, which predisposes to a variety of tumors, including infiltrating astrocytomas [37]. These early observations suggested that the pathways deregulated by these germline alterations were important for gliomagenesis.
More recent studies have also highlighted a role for germline polymorphisms that predispose to diffuse glioma development. For instance, genotyping efforts have uncovered single nucleotide polymorphisms (SNP) variants at 8q24.21 (near the MYC gene) to be associated with an increased risk for gliomas with IDH1/2 mutations, both astrocytic and oligodendroglial [38].
Circumscribed Gliomas
BRAF and MAPK Alterations
One of the most remarkable discoveries in pediatric neurooncology in the recent years has been the identification of BRAF duplications in the majority (53–77 %) of PA tumors [39–44]. Subsequent studies demonstrated that this tandem duplication always involves the kinase domain of BRAF and leads to a novel fusion (usually BRAF–KIAA1549) [42, 45]. This novel fusion has oncogenic properties resulting in ERK/MAPK pathway activation (Fig. 4.5). Furthermore, it induces glioma-like lesions in mice when introduced into neural stem cells [46]. This alteration is almost always restricted to PA, particularly those arising in the cerebellum or optic pathways [40, 41]. Conversely, BRAF fusions are less frequent in PA arising in the cerebral hemispheres. However, a small subset of unclassifiable low-grade astrocytomas and neuroepithelial/glioneuronal tumors may also have it [47]. In addition, it appears that the frequency of BRAF–KIAA1549 fusion varies by age, being less frequent in PA of adults [48].
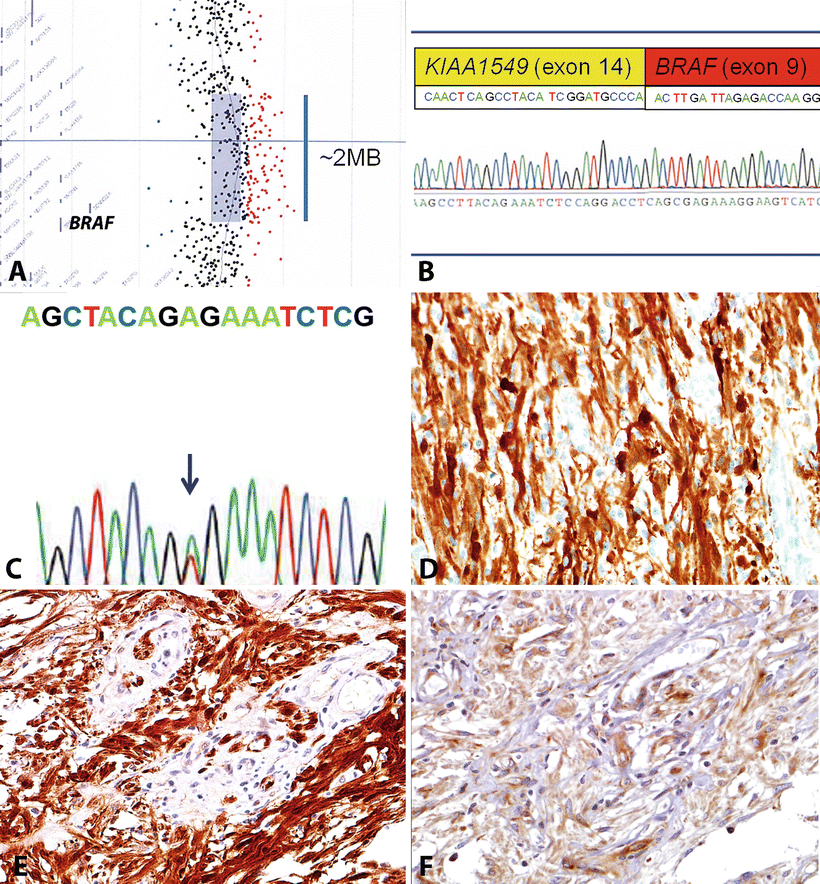
Fig. 4.5
Molecular features of circumscribed gliomas. The most frequent molecular alteration in PA is a BRAF duplication that may be identified by array CGH (a). This duplication usually leads to a gene fusion, usually involving the neighboring gene KIAA1549 (b). In contrast, most pleomorphic xanthoastrocytomas contain a BRAF (V600E) mutation resulting from a single nucleotide change (c). Activating BRAF alterations may result in induction of the phenomenon of oncogene-induced senescence, and associated with increased p16 expression (d), which may explain the low proliferation rates in many of these tumors. The genetic alterations present in PA and other circumscribed gliomas result in near universal activation of the MAPK and mTOR signaling pathways, which may be identified by detection of phospho-ERK (e) and phosphor-S6 (f) protein, respectively.
The development of low-grade astrocytomas in patients with germline NF1 loss and the high frequency of somatic BRAF alterations in sporadic circumscribed low-grade gliomas provide strong evidence that the MAPK/ERK signaling pathway is critical for the biology of these tumors. Other studies have shown rarer genetic alterations leading to activation of this pathway, including small BRAF insertions, RAF1–SRGAP3 fusions (at 3p25), activating RAS mutations, and a FAM131B–BRAF fusion mediated by an interstitial deletion [45, 49, 50] (Fig. 4.6). Some of these rearrangements seem to be facilitated by sequence microhomology [51]. A recent whole genome/transcriptome sequencing study of PA identified single MAPK pathway activating alterations, predominantly through BRAF–KIAA11549 fusions, but also novel BRAF fusion partners in very rare cases (i.e., RNF130–BRAF, CLCN6–BRAF, MKRN1–BRAF, and GNAI1–BRAF) [52]. Alterations in other genes not involving BRAF were also identified in a small subset of non-cerebellar PA (NTRK2 rearrangements, as well as FGFR1 and PTPN11 mutations). Of interest, in this study every PA had a genetic alteration in the MAPK pathway, which was almost always exclusive (except for PTPN11 mutations which occurred only in combination with FGFR1 alterations) [52].
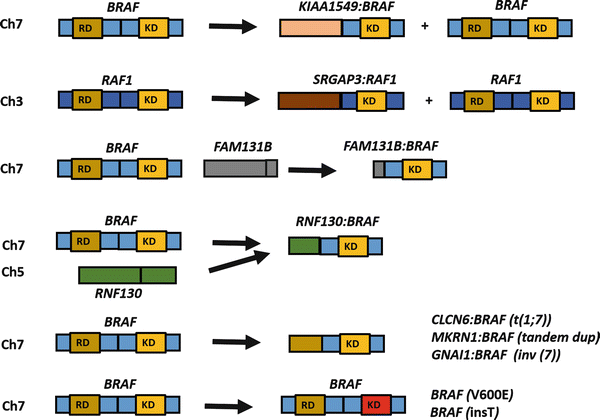
Fig. 4.6
Spectrum of BRAF alterations in pilocytic astrocytoma/circumscribed gliomas. Although tandem duplications involving the BRAF kinase domain (KD), excluding the regulatory domain (RD) and leading to a KIAA1549:BRAF fusion are the most frequent alterations associated with pilocytic astrocytoma in specific, alternative alterations occur in a small proportion of cases. These include an interstitial deletion leading to FAM131B:BRAF fusion, fusion events involving alternative partners, as well as point mutations (e.g., BRAF V600E) and small activating insertions (insT). A similar rearrangement involving the related gene RAF1, also occurs on rare occasions.
BRAF (V600E) mutation, reported in numerous cancer types, is less restricted to histopathology and has been reported to occur with variable frequency in many brain tumor subtypes [53–58], including PXA, gangliogliomas, desmoplastic infantile gangliogliomas, PA, diffuse gliomas and even in dysembryoplastic neuroepithelial tumors. However, the frequency of BRAF (V600E) mutation appears to be higher in PXA, occurring in over half of cases [57, 59, 60].
At the current time, the prognostic significance of BRAF alterations in low-grade gliomas remains unclear. Several studies have not found a significant association with outcome in patients with low-grade gliomas containing BRAF fusions [13, 47, 49]. In a study by Hawkins et al. focusing on a clinically relevant group of 70 pediatric low-grade astrocytoma patients (i.e., sporadic, subtotally resected tumors in non-cerebellar locations), the investigators found BRAF-KIAA1549 fusions to be significantly associated with better clinical outcome [61]. Conversely, Horbinski et al. in a study of 198 cases, found on multivariate analysis midline location and p16 deletion (but not BRAF rearrangement) as independent prognostic factors [62]. In a meta-analysis of BRAF alteration data encompassing approximately 700 pediatric low-grade astrocytomas, specific BRAF–KIAA1549 fusion variants have independent prognostic implications in extra-cerebellar PA, but BRAF fusions in general were not independently associated with outcome (Jones D et al., unpublished data). RT-PCR and FISH based methods that are able to detect this fusion in formalin-fixed paraffin-embedded tissue have been developed [63], and have been increasing applied for clinical use.
Given the high frequency of somatic BRAF genetic alterations in circumscribed low-grade gliomas, the possibility of pharmacologic inhibition as a therapeutic strategy is very appealing. However, targeted therapeutics for BRAF must be taken with caution, since recent pharmacologic evidence suggests that inhibitors that are effective against BRAF (V600E) may have paradoxic pro-growth effects in tumors that are BRAF wild type or contain activating BRAF fusions [64].
Oncogene-Induced Senescence
Senescence, i.e., irreversible growth arrest, is a cellular phenomenon that may occur as a result of oncogene activation. Clinical observations have documented stabilization, or even regression, of a subset of PA. Furthermore, PA shares frequent alterations in the BRAF oncogene with another limited neoplastic proliferation, cutaneous melanocytic nevi, which are known to senesce. Recent studies [65, 66] have shown markers of senescence, including p16 and acidic senescence-associated β-galactosidase, in primary PA and low passage cultures. Furthermore, senescence was also induced after introduction of BRAF (V600E) in neural stem cells, and p16 loss in clinical samples was associated with worse clinical outcome [66].
PI3K/mTOR
PI3K/mTOR signaling has been implicated as a frequent molecular property of a variety of tumor types. This pathway is of great interest for targeted therapeutics, since pharmacologic inhibitors (i.e., rapamycin and its analogs) are widely available. mTOR exists as part of two multiprotein complexes: mTORC1 and mTORC2 (mTORC1 is composed of RAPTOR, mLST8 and GBL), and signaling through this complex leads to increased protein translation, cell growth, and survival. In mTORC2, mTOR interacts with RICTOR, mSin1, and Protor, activation leads to AKT activation (identified by phosphorylation at S473)/PKC signaling, and subsequently increased cell survival and regulation of cytoskeletal dynamics [67].
Of relevance to low-grade glioma, particularly pediatric, is studies demonstrating increased mTOR signaling in the context of NF1 loss. These include mouse models of NF1-optic glioma [68] and unusual low-grade gliomas in NF1 patients characterized by increased cell size [69]. mTOR activation is also more frequent in rare PA that develop anaplasia [70], and regulates proliferation of murine stem cells containing activating BRAF fusions [46]. A recent study of 177 pediatric low-grade gliomas and PA showed significant mTOR activation (~60 % of cases) as measured by pS6 protein [71]. In addition, mTOR inhibition led to decreased cell growth of two pediatric cell lines in vitro. Of great clinical interest, mTOR inhibitors have pharmacologic efficacy in SEGA and other manifestations of tuberous sclerosis [72], a syndrome essentially defined at the molecular level by constitutive mTOR activation. PI3K/mTOR pathway activation is also a frequent feature of both adult and pediatric diffuse gliomas, through alterations in PTEN, NF1 and genes encoding for receptor tyrosine kinases (e.g., FGFR1) [73].
Diffuse Gliomas
One of the earliest molecular alterations described in diffuse gliomas, with morphologic and prognostic relevance was the identification of 1p19q co-deletion, particularly in tumors with oligodendroglial morphology (Fig. 4.7). Subsequent studies highlighted a strong association between 1p19q co-deletion and therapeutic response, particularly in anaplastic oligodendroglioma [74]. This alteration may be identified by a variety of methods that work in formalin-fixed paraffin-embedded tissue, most commonly FISH [75], but also array based platforms, such as SNP arrays [76]. Although partial deletions involving chromosome arm 1p and/or 19q are not uncommon in gliomas with various histologies, it is whole arm 1p and 19q co-deletion that is most closely associated with oligodendroglial histology, which is mediated by an unbalanced (t1;19) translocation [77, 78]. Subsequent whole exome sequencing studies have identified recurrent mutations in FUBP1 (Ch 1p) and CIC (Ch 19q) as likely tumor suppressor genes inactivated at these locations [79, 80].
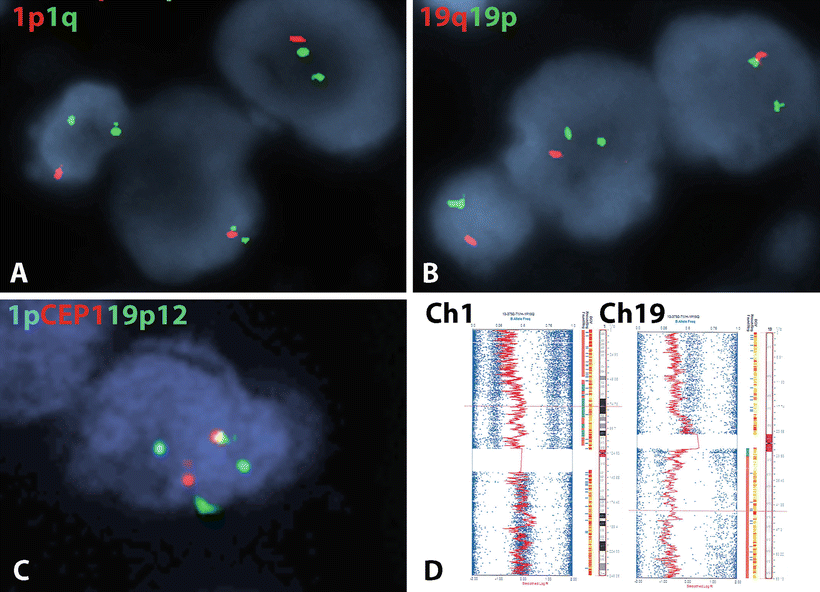
Fig. 4.7
Molecular genetic alterations in oligodendroglial neoplasms. The most characteristic molecular alteration in oligodendroglial tumors is combined deletions of 1p (a) and 19q (b) which is mediated by a t(1;19) translocation (c). The 1p19q co-deletion may also be recognized by array based methods (e.g., SNP platforms) in formalin-fixed tissue. As this case demonstrates, the classic co-deletion of oligodendroglioma involves the whole 1p and 19q chromosomal arms (d) (SNP figure courtesy of Christopher Gocke, MD).
Another remarkable subsequent discovery in the biology of diffuse gliomas was the identification of recurrent point mutations in genes encoding for the cytosolic metabolic enzyme IDH1 (and less frequently IDH2) through exome sequencing efforts. Although initially identified in a subset of glioblastomas, it was subsequently noted that these mutations were highly prevalent (>80 %) in diffuse gliomas grade II and III, both astrocytomas and oligodendrogliomas as well as in secondary glioblastomas [81–83]. These IDH mutations occur almost always at the same site (Arg132 of IDH1 and analogous Arg172 site in IDH2) and result in a neoenzymatic function leading to increased 2-hydroxyglutarate (2HG) [84], which has numerous cellular effects including inhibition of histone and DNA demethylation and global epigenetic alterations (reviewed in [85, 86]). IDH1/2 mutations appear to be early events in tumorigenesis, since they show similar prevalence in grade II astrocytomas and oligodendrogliomas, and may occur earlier than 1p19q loss and TP53 mutations. Diagnostically, an antibody directed against the most frequent IDH1 mutant protein in gliomas (IDH1 R132H), is in clinical use and valuable in differentiating infiltrating gliomas from other tumors and non-neoplastic conditions (i.e., gliosis) [87, 88].
Another molecular property of a subset of infiltrating astrocytomas is the presence of a telomerase independent mechanism of telomere maintenance known as the alternative lengthening of telomeres (ALT) (Fig. 4.8). Although present in a small subset of cancers of various types, this phenotype is enriched in DA (WHO grade II), anaplastic astrocytoma (WHO grade III), as well as secondary and pediatric glioblastoma (WHO grade IV). Subsequent studies found this phenotype to be strongly associated with mutations in the gene encoding the chromatin remodeling protein ATRX [89]. These mutations lead to ATRX protein loss and are strongly associated with IDH mutations [90–92], but are mutually exclusive with 1p19q co-deletion/CIC/FUBP1 mutations [93].
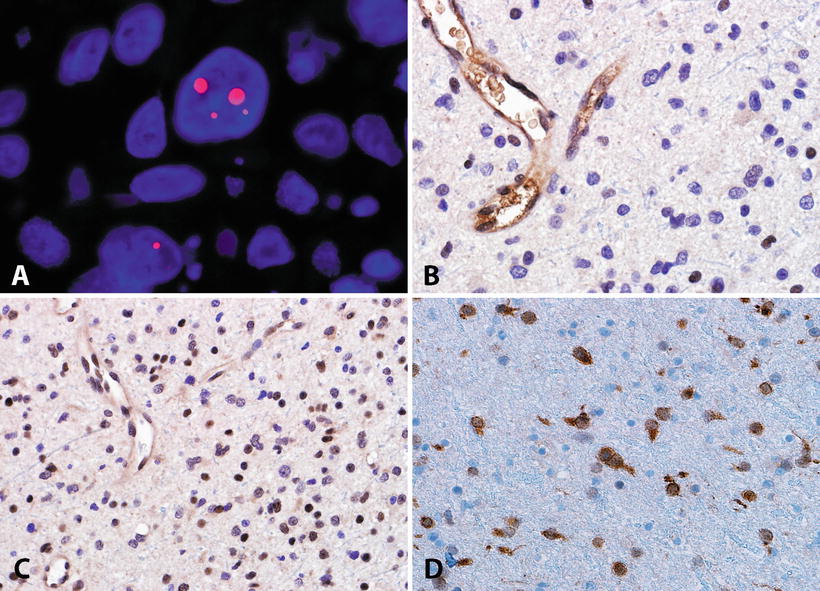
Fig. 4.8
Molecular features of adult diffuse astrocytomas. The alternative lengthening of telomeres (ALT) is a frequent phenotype identifiable in diffuse astrocytomas by telomere specific FISH, which demonstrates ultrabright signals (Courtesy of Christopher Heaphy, Ph.D.) (a). The ALT phenotype is frequently associated with ATRX mutations and protein loss in neoplastic cells (b). Conversely DAXX is usually preserved in most CNS tumors (c). IDH1 mutations are frequent in diffuse gliomas, and may be recognized by an antibody directed against the most frequent mutated protein product (R132H) (d).
All these recent studies highlighted an important role for telomere maintenance in the biology of diffuse gliomas, findings subsequently reinforced by the finding of mutations in the TERT promoter which leads to increased transcriptional activity [94]. Interestingly, among diffuse gliomas, TERT promoter mutations are more frequent in oligodendrogliomas and primary glioblastomas and mutually exclusive with ATRX mutations and the ALT phenotype. An expanding picture is now emerging with distinctive molecular signatures separating various low-grade glioma subtypes (Fig. 4.9).
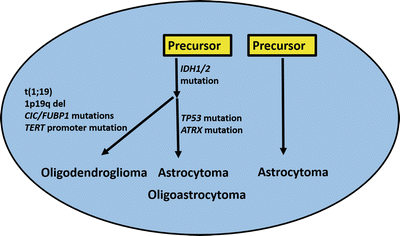
Fig. 4.9
Molecular pathogenesis of adult low-grade diffuse gliomas. Recent studies have also refined our classification of diffuse gliomas in adults. IDH mutations appear to be an early event, shared by oligodendroglial tumors and a subset of diffuse astrocytomas with a relatively better prognosis. Additional alterations (e.g., t(1;19)) are associated with the oligodendroglial subgroup, while ATRX alterations are associated with the astrocytic pathway. Other astrocytomas lack these alterations, and are associated with a worse prognosis. Often, they have molecular alterations more typical of primary glioblastoma. Oligoastrocytoma is an heterogeneous group, and may share molecular properties with oligodendrogliomas or astrocytomas.
Whole Genome Sequencing Studies of Pediatric Low-Grade Glioma
Whole genome/exome sequencing studies have also provided recent, important insights into the molecular genetics important for pediatric low-grade glioma development (Fig. 4.10). Zhang et al. in a whole genome sequencing study of 39 pediatric low-grade gliomas and glioneuronal tumors found very few genetic alterations, with 24 tumors (62 %) containing single relevant (non-silent) somatic alterations [73]. They found rearrangements of MYB and duplications of the gene segments of FGFR1 encoding for the tyrosine kinase domain in approximately half of pediatric DA. Ramkissoon et al. studied 44 pediatric diffuse low-grade gliomas using high resolution copy number analysis and identified 8q13.1 alterations in 28 % of cases leading to MYBL1 gain [95] (Fig. 4.11) The authors also found a similar alteration involving the related gene MYB in two angiocentric gliomas. These alterations frequently result in a duplication as well as truncation of a C-terminal regulatory domain.