26
Local Anesthetics
CASE STUDY
A 67-year-old woman is scheduled for elective total knee arthroplasty. What local anesthetic agents would be most appropriate if surgical anesthesia were to be administered using a spinal or an epidural technique, and what potential complications might arise from their use? What anesthetics would be most appropriate for providing postoperative analgesia via an indwelling epidural or peripheral nerve catheter?
Simply stated, local anesthesia refers to loss of sensation in a limited region of the body. This is accomplished by disruption of afferent neural traffic via inhibition of impulse generation or propagation. Such blockade may bring with it other physiologic changes such as muscle paralysis and suppression of somatic or visceral reflexes, and these effects might be desirable or undesirable depending on the particular circumstances. Nonetheless, in most cases, it is the loss of sensation, or at least the achievement of localized analgesia, that is the primary goal.
Although local anesthetics are often used as analgesics, it is their ability to provide complete loss of all sensory modalities that is their distinguishing characteristic. The contrast with general anesthesia should be obvious, but it is perhaps worthwhile to emphasize that with local anesthesia the drug is delivered directly to the target organ, and the systemic circulation serves only to diminish or terminate its effect. Local anesthesia can also be produced by various chemical or physical means. However, in routine clinical practice, it is achieved with a rather narrow spectrum of compounds, and recovery is normally spontaneous, predictable, and without residual effects. The development of these compounds has a rich history (see Box: Historical Development of Local Anesthesia), punctuated by serendipitous observations, delayed starts, and an evolution driven more by concerns for safety than improvements in efficacy.
 BASIC PHARMACOLOGY OF LOCAL ANESTHETICS
BASIC PHARMACOLOGY OF LOCAL ANESTHETICS
Chemistry
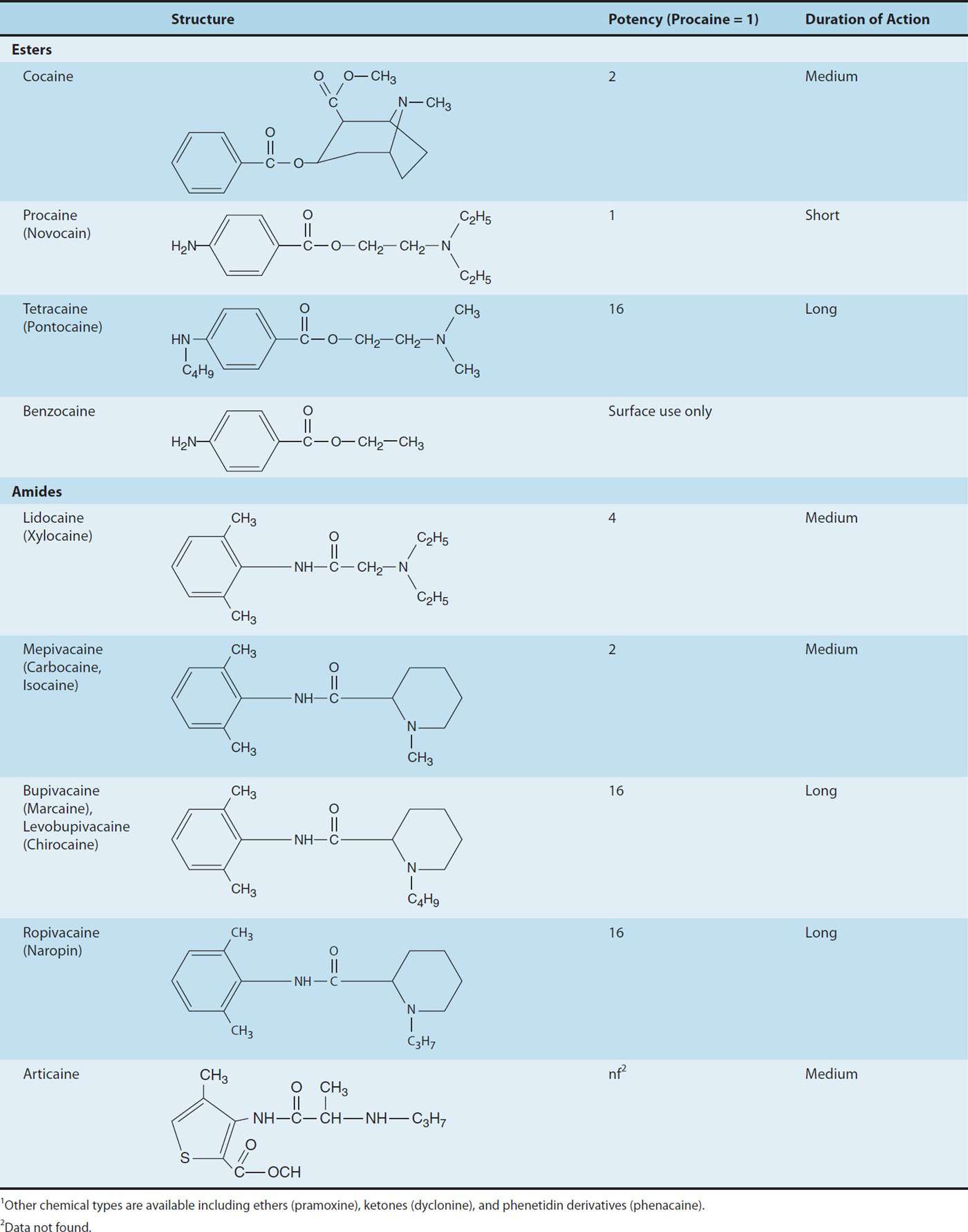
Most local anesthetic agents consist of a lipophilic group (eg, an aromatic ring) connected by an intermediate chain via an ester or amide to an ionizable group (eg, a tertiary amine) (Table 26–1). In addition to the general physical properties of the molecules, specific stereochemical configurations are associated with differences in the potency of stereoisomers (eg, levobupivacaine, ropivacaine). Because ester links are more prone to hydrolysis than amide links, esters usually have a shorter duration of action.
TABLE 26–1 Structure and properties of some ester and amide local anesthetics.1
Local anesthetics are weak bases and are usually made available clinically as salts to increase solubility and stability. In the body, they exist either as the uncharged base or as a cation (see Chapter 1, Ionization of Weak Acids and Weak Bases). The relative proportions of these two forms are governed by their pKa and the pH of the body fluids according to the Henderson-Hasselbalch equation, which can be expressed as:
pKa = pH – log [base]/[conjugate acid]
If the concentration of base and conjugate acid are equal, the second portion of the right side of the equation drops out, as log 1 = 0, leaving:
pKa = pH (when base concentration = conjugate acid concentration)
Although the numbing properties of cocaine were recognized for centuries, one might consider September 15, 1884, to mark the “birth of local anesthesia.” Based on work performed by Carl Koller, cocaine’s numbing effect on the cornea was demonstrated before the Ophthalmological Congress in Heidelberg, ushering in the era of surgical local anesthesia. Unfortunately, with widespread use came recognition of cocaine’s significant CNS and cardiac toxicity, which along with its addiction potential, tempered enthusiasm for this application. As the early investigator Mattison commented, “the risk of untoward results have robbed this peerless drug of much favor in the minds of many surgeons, and so deprived them of a most valued ally.” As cocaine was known to be a benzoic acid ester, the search for alternative local anesthetics focused on this class of compounds, resulting in the identification of benzocaine shortly before the turn of the last century. However, benzocaine proved to have limited utility due to its marked hydrophobicity, and was thus relegated to topical anesthesia, a use for which it still finds limited application in current clinical practice. The first useful injectable local anesthetic, procaine, was introduced shortly thereafter by Einhorn, and its structure has served as the template for the development of the most commonly used modern local anesthetics. The three basic structural elements of these compounds can be appreciated by review of Table 26–1: an aromatic ring, conferring lipophilicity, an ionizable tertiary amine, conferring hydrophilicity, and an intermediate chain connecting these via an ester or amide linkage.
One of procaine’s limitations was its short duration of action, a drawback overcome with the introduction of tetracaine in 1928. Unfortunately, tetracaine demonstrated significant toxicity when employed for high-volume peripheral blocks, ultimately reducing its common usage to spinal anesthesia. Both procaine and tetracaine shared another drawback: their ester linkage conferred instability, and particularly in the case of procaine, the free aromatic acid released during ester hydrolysis of the parent compound was believed to be the source of relatively frequent allergic reactions.
Löfgren and Lundqvist circumvented the problem of instability with the introduction of lidocaine in 1948. Lidocaine was the first in a series of amino-amide local anesthetics that would come to dominate the second half of the 20th century. Lidocaine had a more favorable duration of action than procaine, and less systemic toxicity than tetracaine. To this day, it remains one of the most versatile and widely used anesthetics. Nonetheless, some applications required more prolonged block than that afforded by lidocaine, a pharmacologic void that was filled with the introduction of bupivacaine, a more lipophilic and more potent anesthetic. Unfortunately, bupivacaine was found to have greater propensity for significant effects on cardiac conduction and function, which at times proved lethal. Recognition of this potential for cardiac toxicity led to changes in anesthetic practice, and significant toxicity became sufficiently rare for it to remain a widely used anesthetic for nearly every regional technique in modern clinical practice. Nonetheless, this inherent cardiotoxicity would drive developmental work leading to the introduction of two recent additions to the anesthetic armamentarium, levobupivacaine and ropivacaine. The former is the S(–) enantiomer of bupivacaine, which has less affinity for cardiac sodium channels than its R(+) counterpart. Ropivacaine, another S(–) enantiomer, shares this reduced affinity for cardiac sodium channels, while being slightly less potent than bupivacaine or levobupivacaine.
Thus, pKa can be seen as an effective way to consider the tendency for compounds to exist in a charged or uncharged form, ie, the lower the pKa, the greater the percentage of uncharged weak bases at a given pH. Because the pKa of most local anesthetics is in the range of 7.5–9.0, the charged, cationic form will constitute the larger percentage at physiologic pH. A glaring exception is benzocaine, which has a pKa around 3.5, and thus exists solely as the nonionized base under normal physiologic conditions.
This issue of ionization is of critical importance because the cationic form is the most active at the receptor site. However, the story is a bit more complex, because the receptor site for local anesthetics is at the inner vestibule of the sodium channel, and the charged form of the anesthetic penetrates biologic membranes poorly. Thus, the uncharged form is important for cell penetration. After penetration into the cytoplasm, equilibration leads to formation and binding of the charged cation at the sodium channel, and hence the production of a clinical effect (Figure 26–1). Drug may also reach the receptor laterally through what has been termed the hydrophobic pathway. As a clinical consequence, local anesthetics are less effective when they are injected into infected tissues because the low extracellular pH favors the charged form, with less of the neutral base available for diffusion across the membrane. Conversely, adding bicarbonate to a local anesthetic—a strategy sometimes utilized in clinical practice—will raise the effective concentration of the nonionized form and thus shorten the onset time of a regional block.
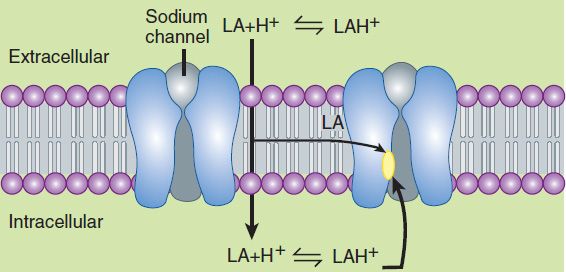
FIGURE 26–1 Schematic diagram depicting paths of local anesthetic (LA) to receptor sites. Extracellular anesthetic exists in equilibrium between charged and uncharged forms. The charged cation penetrates lipid membranes poorly; intracellular access is thus achieved by passage of the uncharged form. Intracellular re-equilibration results in formation of the more active charged species, which binds to the receptor at the inner vestibule of the sodium channel. Anesthetic may also gain access more directly by diffusing laterally within the membrane (hydrophobic pathway).
Pharmacokinetics
When local anesthetics are used for local, peripheral, and central neuraxial anesthesia—their most common clinical applications—systemic absorption, distribution, and elimination serve only to diminish or terminate their effect. Thus, classic pharmacokinetics plays a lesser role than with systemic therapeutics, yet remains important to the anesthetic’s duration and critical to the potential development of adverse reactions, specifically cardiac and central nervous system (CNS) toxicity.
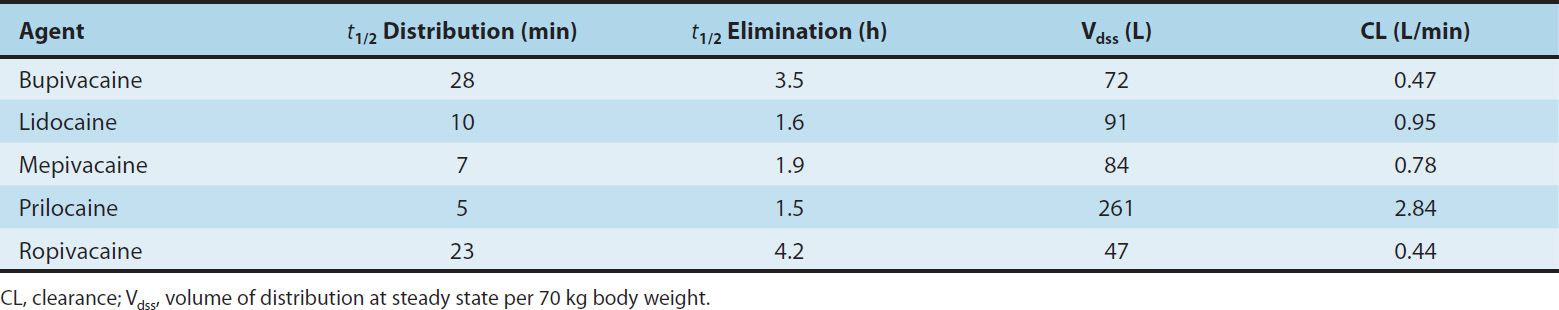
Some pharmacokinetic properties of the commonly used amide local anesthetics are summarized in Table 26–2. The pharmacokinetics of the ester-based local anesthetics has not been extensively studied owing to their rapid breakdown in plasma (elimination half-life < 1 minute).
TABLE 26–2 Pharmacokinetic properties of several amide local anesthetics.
A. Absorption
Systemic absorption of injected local anesthetic from the site of administration is determined by several factors, including dosage, site of injection, drug-tissue binding, local tissue blood flow, use of a vasoconstrictor (eg, epinephrine), and the physicochemical properties of the drug itself. Anesthetics that are more lipid soluble are generally more potent, have a longer duration of action, and take longer to achieve their clinical effect. Extensive protein binding also serves to increase the duration of action.
Application of a local anesthetic to a highly vascular area such as the tracheal mucosa or the tissue surrounding intercostal nerves results in more rapid absorption and thus higher blood levels than if the local anesthetic is injected into a poorly perfused tissue such as subcutaneous fat. When used for major conduction blocks, the peak serum levels will vary as a function of the specific site of injection, with intercostal blocks among the highest, and sciatic and femoral among the lowest (Figure 26–2). When vasoconstrictors are used with local anesthetics, the resultant reduction in blood flow serves to reduce the rate of systemic absorption and thus diminishes peak serum levels. This effect is generally most evident with the shorter-acting, less potent, and less lipid-soluble anesthetics.
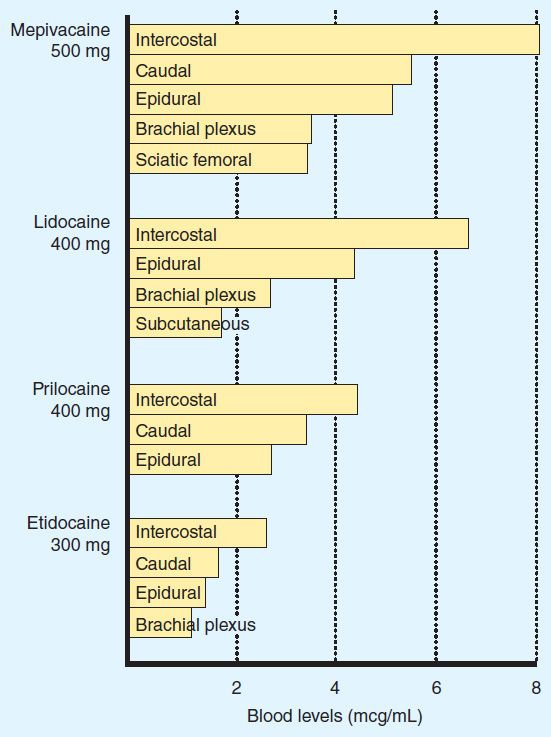
FIGURE 26–2 Comparative peak blood levels of several local anesthetic agents following administration into various anatomic sites. (Adapted, with permission, from Covino BD, Vassals HG: Local Anesthetics: Mechanism of Action in Clinical Use. Grune & Stratton, 1976. Copyright Elsevier.)
B. Distribution
1. Localized—As local anesthetic is usually injected directly at the site of the target organ, distribution within this compartment plays an essential role with respect to achievement of clinical effect. For example, anesthetics delivered into the subarachnoid space will be diluted with cerebrospinal fluid (CSF) and the pattern of distribution will be dependent upon a host of factors, among the most critical being the specific gravity relative to that of CSF and the patient’s position. Solutions are termed hyperbaric, isobaric, and hypobaric, and will respectively descend, remain relatively static, or ascend, within the subarachnoid space due to gravity when the patient sits upright. A review and analysis of relevant literature cited 25 factors that have been invoked as determinants of spread of local anesthetic in CSF, which can be broadly classified as characteristics of the anesthetic solution, CSF constituents, patient characteristics, and techniques of injection. Somewhat similar considerations apply to epidural and peripheral blocks.
2. Systemic—The peak blood levels achieved during major conduction anesthesia will be minimally affected by the concentration of anesthetic or the speed of injection. The disposition of these agents can be well approximated by a two-compartment model. The initial alpha phase reflects rapid distribution in blood and highly perfused organs (eg, brain, liver, heart, kidney), characterized by a steep exponential decline in concentration. This is followed by a slower declining beta phase reflecting distribution into less well perfused tissue (eg, muscle, gut), and may assume a nearly linear rate of decline. The potential toxicity of the local anesthetics is affected by the protective effect afforded by uptake by the lungs, which serve to attenuate the arterial concentration, though the time course and magnitude of this effect have not been adequately characterized.
C. Metabolism and Excretion
The local anesthetics are converted to more water-soluble metabolites in the liver (amide type) or in plasma (ester type), which are excreted in the urine. Since local anesthetics in the uncharged form diffuse readily through lipid membranes, little or no urinary excretion of the neutral form occurs. Acidification of urine promotes ionization of the tertiary amine base to the more water-soluble charged form, leading to more rapid elimination. Ester-type local anesthetics are hydrolyzed very rapidly in the blood by circulating butyrylcholinesterase to inactive metabolites. For example, the half-lives of procaine and chloroprocaine in plasma are less than a minute. However, excessive concentrations may accumulate in patients with reduced or absent plasma hydrolysis secondary to atypical plasma cholinesterase.
The amide local anesthetics undergo complex biotransformation in the liver, which includes hydroxylation and N-dealkylation by liver microsomal cytochrome P450 isozymes. There is considerable variation in the rate of liver metabolism of individual amide compounds, with prilocaine (fastest) > lidocaine > mepivacaine > ropivacaine ≈ bupivacaine and levobupivacaine (slowest). As a result, toxicity from amide-type local anesthetics is more likely to occur in patients with hepatic disease. For example, the average elimination half-life of lidocaine may be increased from 1.6 hours in normal patients (t½, Table 26–2) to more than 6 hours in patients with severe liver disease. Many other drugs used in anesthesia are metabolized by the same P450 isozymes, and concomitant administration of these competing drugs may slow the hepatic metabolism of the local anesthetics. Decreased hepatic elimination of local anesthetics would also be anticipated in patients with reduced hepatic blood flow. For example, the hepatic elimination of lidocaine in patients anesthetized with volatile anesthetics (which reduce liver blood flow) is slower than in patients anesthetized with intravenous anesthetic techniques. Delayed metabolism due to impaired hepatic blood flow may likewise occur in patients with congestive heart failure.
Pharmacodynamics
A. Mechanism of Action
1. Membrane potential—The primary mechanism of action of local anesthetics is blockade of voltage-gated sodium channels (Figure 26–1). The excitable membrane of nerve axons, like the membrane of cardiac muscle (see Chapter 14) and neuronal cell bodies (see Chapter 21), maintains a resting transmembrane potential of –90 to –60 mV. During excitation, the sodium channels open, and a fast, inward sodium current quickly depolarizes the membrane toward the sodium equilibrium potential (+40 mV). As a result of this depolarization process, the sodium channels close (inactivate) and potassium channels open. The outward flow of potassium repolarizes the membrane toward the potassium equilibrium potential (about –95 mV); repolarization returns the sodium channels to the rested state with a characteristic recovery time that determines the refractory period. The transmembrane ionic gradients are maintained by the sodium pump. These ionic fluxes are similar to, but simpler than, those in heart muscle, and local anesthetics have similar effects in both tissues.
2. Sodium channel isoforms—Each sodium channel consists of a single alpha subunit containing a central ion-conducting pore associated with accessory beta subunits. The pore-forming alpha subunit is actually sufficient for functional expression, but the kinetics and voltage dependence of channel gating are modified by the beta subunit. A variety of different sodium channels have been characterized by electrophysiologic recording, and subsequently isolated and cloned, while mutational analysis has allowed for identification of the essential components of the local anesthetic binding site. Nine members of a mammalian family of sodium channels have been so characterized and classified as Nav1.1–Nav1.9, where the chemical symbol represents the primary ion, the subscript denotes the physiologic regulator (in this case voltage), the initial number denotes the gene, and the number following the period indicates the particular isoform.
3. Channel blockade—Biologic toxins such as batrachotoxin, aconitine, veratridine, and some scorpion venoms bind to receptors within the channel and prevent inactivation. This results in prolonged influx of sodium through the channel and depolarization of the resting potential. The marine toxins tetrodotoxin (TTX) and saxitoxin have clinical effects that largely resemble those of local anesthetics (ie, block of conduction without a change in the resting potential). However, in contrast to the local anesthetics, their binding site is located near the extracellular surface. The sensitivity of these channels to TTX varies, and subclassification based on this pharmacologic sensitivity has important physiologic and therapeutic implications. Six of the aforementioned channels are sensitive to nanomolar concentration of this biotoxin (TTX-S), while three are resistant (TTX-R). Of the latter, Nav1.8 and Nav1.9 appear to be exclusively expressed in dorsal root ganglia nociceptors, which raises the developmental possibility of targeting these specific neuronal subpopulations. Such fine-tuned analgesic therapy has the theoretical potential of providing effective analgesia, while limiting the significant adverse effects produced by nonspecific sodium channel blockers.
When progressively increasing concentrations of a local anesthetic are applied to a nerve fiber, the threshold for excitation increases, impulse conduction slows, the rate of rise of the action potential declines, action potential amplitude decreases, and, finally, the ability to generate an action potential is completely abolished. These progressive effects result from binding of the local anesthetic to more and more sodium channels. If the sodium current is blocked over a critical length of the nerve, propagation across the blocked area is no longer possible. In myelinated nerves, the critical length appears to be two to three nodes of Ranvier. At the minimum dose required to block propagation, the resting potential is not significantly altered.
The blockade of sodium channels by most local anesthetics is both voltage and time dependent: Channels in the rested state, which predominate at more negative membrane potentials, have a much lower affinity for local anesthetics than activated (open state) and inactivated channels, which predominate at more positive membrane potentials (see Figure 14–10). Therefore, the effect of a given drug concentration is more marked in rapidly firing axons than in resting fibers (Figure 26–3). Between successive action potentials, a portion of the sodium channels will recover from the local anesthetic block (see Figure 14–10). The recovery from drug-induced block is 10–1000 times slower than the recovery of channels from normal inactivation (as shown for the cardiac membrane in Figure 14–4). As a result, the refractory period is lengthened and the nerve conducts fewer action potentials.
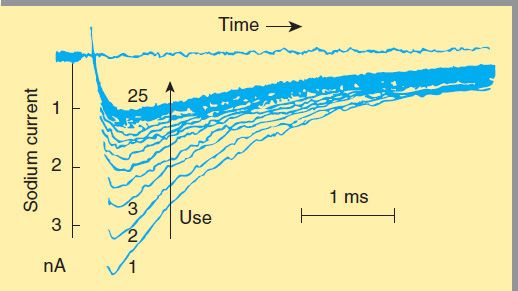
FIGURE 26–3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


