Jeffrey S. Sartin
Liver Diseases
Key Questions
• What clinical and laboratory findings would lead to a diagnosis of liver cirrhosis?
• What treatment modalities are available to patients with end-stage liver failure?

http://evolve.elsevier.com/Copstead/
The liver is a vital but vulnerable organ. Its role in digestion of fats, storage of carbohydrates, detoxification of blood, and production of proteins makes it indispensable; in contrast to the kidney and heart, there are no “artificial livers.” Nevertheless, the liver is susceptible to a wide variety of metabolic, circulatory, toxic, microbial, and neoplastic insults. In some instances the disease is primary to the liver, as in viral hepatitis and hepatocellular carcinoma (HCC). More often the hepatic involvement is secondary, a consequence of some of the more common diseases of humans, such as cardiac decompensation, metastatic cancer, alcoholism, and infections. This chapter will focus on primary diseases of the liver.
Structure and Function of the Liver
The liver, the largest parenchymal organ of the body, averages 1500 grams (g). It is located in the right upper quadrant of the abdomen, beneath the diaphragm, and is anatomically divided into right and left lobes and then further subdivided according to the pattern of its blood supply and biliary drainage. It is covered by a connective tissue capsule, the Glisson capsule, which in turn is covered by visceral peritoneum, reflections of which form the various suspensory hepatic ligaments. These structures demarcate the bare area of the liver directly in contact with the diaphragm (Figure 38-1).1–3
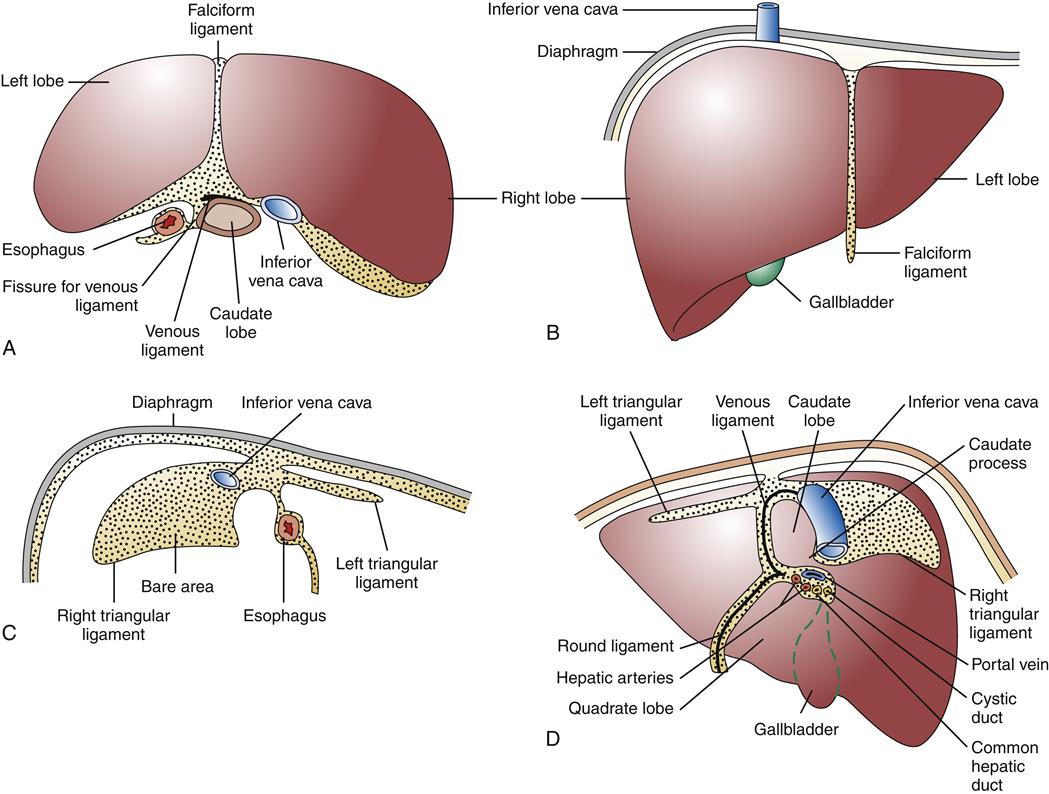
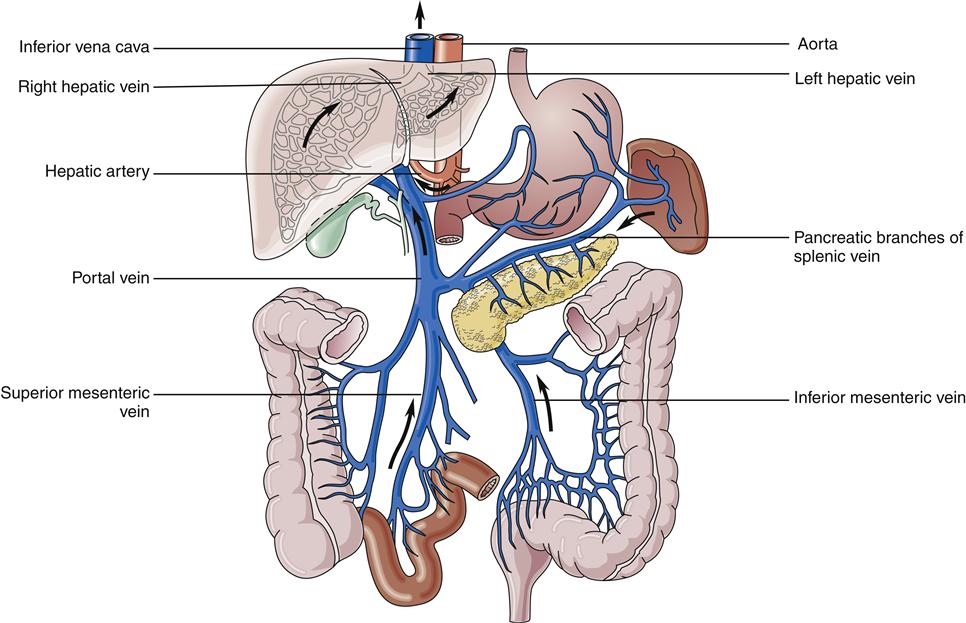
The liver has a dual blood supply. Arterial inflow from the aorta via the celiac trunk and hepatic artery provides 25% of the organ’s blood supply, with the remainder from the portal vein, which drains the capillary bed of the alimentary canal and pancreas (Figure 38-2). This oxygen-depleted venous blood is rich in substances absorbed and secreted by the gut. These afferent blood vessels then branch throughout the liver in association with the bile ducts and form the portal triads (consisting of the portal veins, hepatic arteries, and bile ducts). Eventually, blood from both the hepatic artery and the portal vein drains into the hepatic sinusoids, which surround sheets of liver cells, or hepatic plates (Figure 38-3). The sinusoids are lined by endothelial cells and Kupffer cells (a type of phagocytic macrophage). This blood drains into the central veins, which finally coalesce into the hepatic vein and empty into the inferior vena cava. Any obstruction to the flow of blood may result in a rise in portal venous pressure proximal to the level of blockage. This condition is called portal hypertension and is a central pathophysiologic event in many liver diseases. The liver also has a rich and complex lymphatic drainage system.
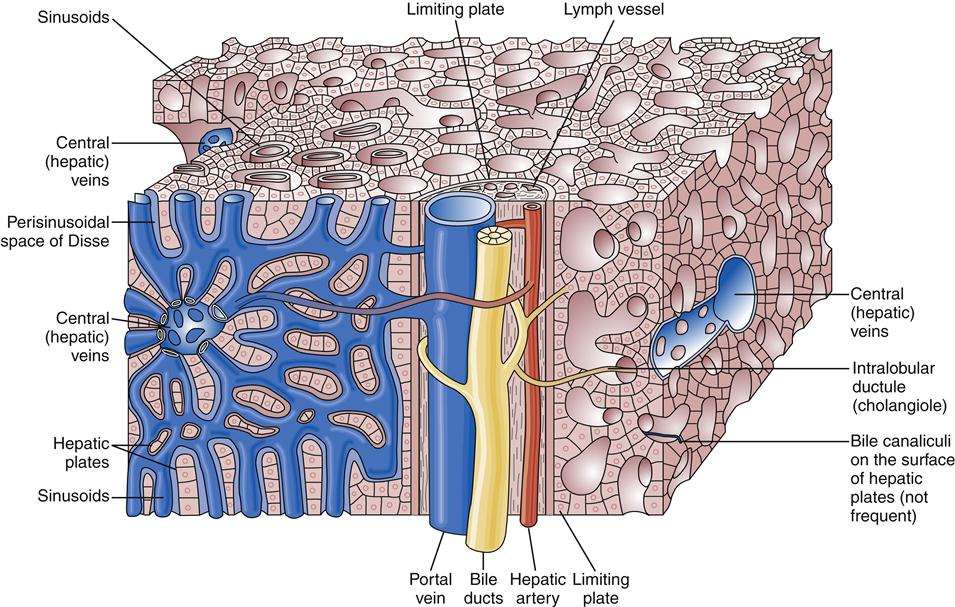
The liver is one of the most metabolically active organs in the body and functions simultaneously as a digestive organ, an endocrine organ, a hematologic organ, and an excretory organ (Box 38-1). All of these functions are elegantly interwoven with such redundancy that more than 80% of the liver may be destroyed before life is threatened.
General Manifestations of Liver Disease
Whether primary or secondary, all hepatic derangements tend to cause similar signs and symptoms that are directly attributable to loss of hepatocellular function or disruption of blood flow through the liver. Because of the liver’s considerable reserve, however, manifestations appear only when the injury is significant and diffuse or so strategically located that it obstructs biliary outflow.
Hepatocellular Failure
Hepatocellular failure results in a number of typical manifestations, including jaundice, muscle wasting, ascites, excessive bleeding, deficiencies of important blood proteins and vitamins, glucose imbalance, and impaired hormone production (Table 38-1). At its most basic the liver is a sophisticated biochemical factory, and these conditions all derive from problems with processing the essential molecules of the body. Inadequate protein metabolism leads to decreased production of clotting factors and hypoalbuminemia. Decreased serum albumin level in turn leads to generalized edema as a result of low serum oncotic pressure. Abnormal storage and release of glucose in the form of glycogen may result in bouts of either hyper- or hypoglycemia. Reduced production of bile salts by the liver impairs absorption of the fat-soluble vitamins A, D, E, and K from the gastrointestinal (GI) tract. Lack of vitamin D may lead to osteomalacia (impaired bone homeostasis); lack of vitamin K contributes to poor blood clotting factor production. Altered lipoprotein processing leads to dyslipidemias, particularly hypertriglyceridemia.
TABLE 38-1
PATHOPHYSIOLOGY UNDERLYING THE SYMPTOMS AND SIGNS OF LIVER DISEASE
| SYMPTOMS/SIGNS | PATHOPHYSIOLOGIC MECHANISM |
| Weakness, fatigue, anorexia, weight loss, muscle wasting | Failure of multiple metabolic functions |
| Fever | Liver inflammation, decreased reticuloendothelial function with increased risk of infection |
| Bruising, increased bleeding | Thrombocytopenia secondary to splenic enlargement, decreased synthesis of clotting factors I, II, V, VII, VIII, IX, and X |
| Palmar erythema, cutaneous spider telangiectases, irregular menses, gynecomastia, impotence, female body hair distribution in men, testicular atrophy | Altered metabolism of sex hormones, chronic debilitation |
| Hepatic encephalopathy | Abnormal protein metabolism |
| Fetor hepaticus | Decreased detoxification |
| Pruritus | Decreased bile salt excretion |
| Cyanosis | Arteriovenous shunts in lungs, liver |
| Jaundice | Biliary obstruction, decreased bilirubin synthesis, decreased bilirubin excretion |
| Hyperdynamic circulation, wide pulse pressure, tachycardia | Generalized vasodilation (? Hormonally mediated) |
| Ascites, peripheral edema | Portal hypertension, sodium and water retention, low serum albumin secondary to decreased hepatic synthesis |
| Splenomegaly | Portal hypertension |
| Hepatomegaly | Cirrhosis (liver may be small), hepatitis, vascular congestion, bile duct obstruction, infection, benign infiltrative disease (e.g., fatty liver, amyloidosis, hemochromatosis), malignant infiltrative disease (e.g., metastatic cancer, lymphoma, large space-occupying lesions such as neoplasm, abscess) |
| Varices (esophageal, gastric, rectal, ectopic) or abnormal abdominal vascular pattern (caput medusae, umbilical bruit) | Portal hypertension with collateral blood flow around hepatic blockage |
| Osteomalacia, hypocalcemia, night blindness, coagulopathy | Fat-soluble vitamin malabsorption and loss of fat-soluble vitamin reserves A, D, and K; loss of vitamin K metabolism (a cofactor for I, II, VII, VIII, IX, and X) |
| Anemia | Multifactorial: blood loss, chronic disease, vitamin B12 deficiency, splenic sequestration |
| Leukopenia | Hypersplenism secondary to portal hypertension |
| Hypoglycemia | Altered glycogenolysis, gluconeogenesis |
| Hyperglycemia | Portosystemic shunting with delayed hepatic uptake of absorbed glucose |
| Hypercholesterolemia | Obstructive jaundice with decreased cholesterol excretion |
Hepatocellular failure is associated with impaired processing of endogenous steroid hormones and the by-products of protein metabolism, as well as decreased clearance of exogenous drugs and toxins. Impaired metabolism of estrogen leads to feminization in men (gynecomastia, impotence, testicular atrophy, female hair distribution), irregular menses in women, palmar erythema, and spider telangiectasia. Impaired conversion of ammonia to urea is associated with hepatic encephalopathy, which will be discussed later in this chapter.
Jaundice
Etiology and pathogenesis
Jaundice, the green-yellow staining of tissues by bilirubin, results from impaired bilirubin metabolism and is one of the most characteristic signs of liver disease. A study of the mechanisms of bilirubin metabolism is essential to an understanding of liver disease and may serve as a paradigm for other hepatic processes (Figure 38-4).4–6
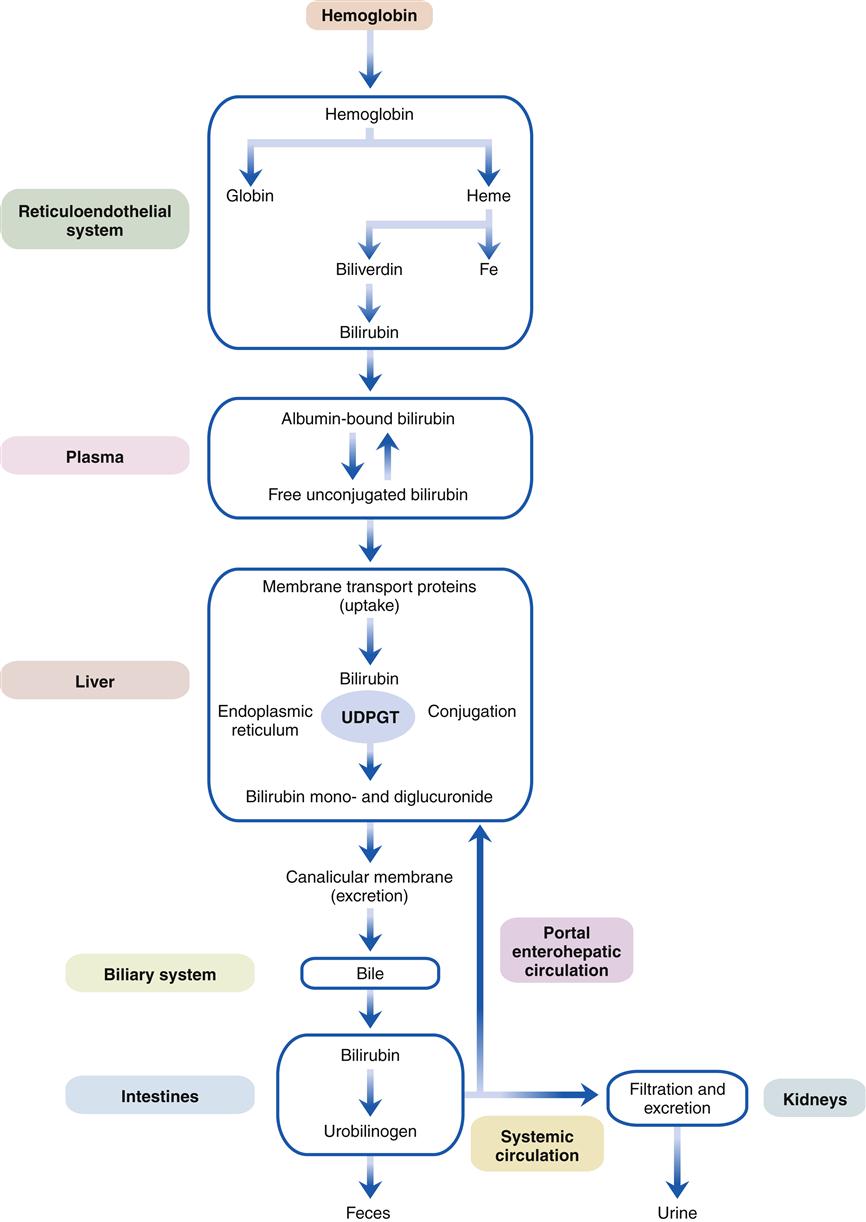
As red blood cells age or are damaged by disease, they lyse and release oxygen-carrying hemoglobin molecules. These are taken up by the reticuloendothelial system, which separates heme from globin, and through the action of heme oxygenase opens the heme ring to release the central iron atom. This process yields biliverdin, which in turn is converted by the enzyme bilirubin reductase to bilirubin. (A small percentage of bilirubin is derived from the premature destruction of immature cells in the bone marrow and spleen and from heme proteins such as myoglobin and the cytochromes in the liver.) Bilirubin is released into the plasma and transported to the liver tightly bound to the plasma protein albumin. The free unconjugated bilirubin is lipid soluble and can be displaced from albumin by fatty acids and certain organic anions (e.g., sulfonamides, salicylates). The neonate is particularly sensitive to free unconjugated bilirubin, which can diffuse into the brain and cause a type of encephalopathy known as kernicterus (see the Liver Diseases and Pediatric Considerations section).
Liver cells are able to extract unconjugated bilirubin from the plasma with special transport proteins. In the cytosol, bilirubin is quickly bound, or conjugated, to water-soluble derivatives of glucuronic acid by the action of the enzyme uridine diphosphate glucuronosyltransferase (UDPGT) located in the endoplasmic reticulum. This process yields water-soluble bilirubin monoglucuronide and diglucuronide, which is then actively excreted into microscopic bile ducts (canaliculi). Bilirubin is then transported through the biliary system as a component of bile to the small intestine. Because it cannot be absorbed in the small intestine, it passes to the colon where bacterial β-glucuronidase enzymes convert it to urobilinogen. A small fraction of urobilinogen is absorbed from the colon and re-excreted by the kidneys and the liver. In the presence of liver disease, the hepatic fraction decreases and the urinary fraction increases, thus accounting for the rise in urinary urobilinogen concentration seen with liver dysfunction. With complete obstruction to bile flow or with intestinal obstruction above the colonic level, urinary urobilinogen level falls to zero, since no bilirubin reaches the colon. (The function of bile salts and the other components of bile flow are discussed in Chapter 37.)
Therefore, jaundice may result from dysfunction anywhere along this complex pathway. Classically, it is divided into prehepatic, hepatic, and posthepatic or cholestatic, but much overlap occurs.
Prehepatic
The most common causes of prehepatic jaundice are hemolysis and ineffective erythropoiesis. The resorption of large hematomas in patients with mild liver disease is a frequent and harmless cause of mild jaundice attributable to unconjugated hyperbilirubinemia.
Hepatic
Dysfunction of each of the hepatic steps in bilirubin metabolism may cause jaundice. In the neonate, immature UDPGT levels may result in physiologic jaundice of the newborn. Various genetic disorders of UDPGT synthesis are characterized by high levels of unconjugated bilirubin in the blood. Mutant UDPGT enzymes can produce the common and benign Gilbert syndrome, in which low levels of unconjugated bilirubin may be increased by fasting or illness (e.g., viral gastroenteritis). Other UDPGT mutations cause the Crigler-Najjar type I and II syndromes with severe neonatal unconjugated hyperbilirubinemia (see the Liver Diseases and Pediatric Considerations section). Most of the liver diseases to be discussed later, such as viral hepatitis, alcoholic liver disease, and autoimmune hepatitis, result in jaundice because dysfunction within the liver cells result in elevated levels of conjugated bilirubin.
Posthepatic
At the level of canalicular bilirubin transport, the rare inherited Dubin-Johnson and Rotor syndromes cause conjugated hyperbilirubinemia. Both conditions have an excellent prognosis. At the canalicular posthepatocytic level, many drugs such as the phenothiazines and the sex hormones may cause jaundice.1 In susceptible women, the high sex hormone levels of normal pregnancy can cause benign cholestasis of pregnancy.1 This condition is also associated with defective transport of bile salts and is characterized by jaundice and intense pruritus (i.e., itching).
Mechanical obstruction of the bile ducts from obstructing tumors, strictures, or gallstones is the most common cause of cholestatic jaundice. Some experts differentiate obstructive jaundice caused by a gross mechanical blockage to bile flow in the biliary tract from intrahepatic cholestatic jaundice, the latter implying a defect at the microscopic level.
Evaluation
Evaluation of a jaundiced patient may be used as a model for investigation of any patient with liver disease. After a complete history and physical examination are obtained and routine laboratory data reviewed, specific liver-related tests may be performed (Table 38-2). The underlying cause, such as alcoholic liver disease, a drug reaction, or metastatic or primary malignancy, is often suggested by the history and physical examination. Physical stigmata of chronic liver disease include telangiectasia, ascites, palmar erythema, gynecomastia, testicular atrophy, hair loss (in men), and central obesity with peripheral muscle wasting.
TABLE 38-2
COMMON LABORATORY TESTS IN LIVER DISEASE
| TEST | NORMAL RANGE | SIGNIFICANCE |
| AST/SGOT | 5-40 units/ml | Elevated levels indicate hepatocellular inflammation or necrosis |
| ALT/SGPT | 5-35 units/ml | AST much greater than ALT in alcoholic liver disease |
| AST less specific; may be of skeletal muscle, myocardial, kidney, or liver origin | ||
| ALT more specific for liver disease | ||
| Alkaline phosphatase | 35-150 units/ml | Elevated in cholestasis, infiltrative liver disease (e.g., cancer, granulomas) |
| May be of bone origin | ||
| γ-Glutamyltranspeptidase | 10-48 units/ml | Elevated in cholestasis and hepatocellular disease |
| Used to confirm that elevated alkaline phosphatase is of hepatic origin | ||
| Disproportionately elevated in alcoholic liver disease | ||
| May be induced by many drugs (e.g., phenobarbital) | ||
| 5′-Nucleotidase | 2-11 units/ml | Elevated in cholestasis |
| Very specific to liver | ||
| Total bilirubin | <1.0 mg/dl | Elevated levels diagnose jaundice |
| Indirect bilirubin | <0.8 mg/dl | Elevated in hemolysis, Gilbert disease |
| Prothrombin time | 11.5-14 sec | Prolongation suggests decreased hepatic synthetic function |
| Serum albumin | 3.5-5.5 gm/dl | Decreased level suggests decreased hepatic synthesis |
| Serum globulin | 2.5-3.5 gm/dl | Elevated in autoimmune hepatitis |
| Urine bilirubin | 0 | Increased with elevation of serum conjugated (direct) bilirubin, zero in unconjugated (indirect) hyperbilirubinemia |
| Urinary urobilinogen | 0-4 mg/24 hr; spot test ± on urine dipstick | Zero in complete biliary or proximal bowel obstruction Increase may suggest liver disease |
| Nonspecifically insensitive | ||
| Primary utility because its presence on urine dipsticks allows simple office/bedside testing or screening with one-time urinalysis |
Diagnostic tests
Biochemical test abnormalities usually fall into one of several categories. A significant elevation in the levels of transaminases out of proportion to the other liver enzymes indicates a primarily hepatocellular disorder (i.e., hepatitis). Alcoholic and other toxic hepatitides virtually always show the aspartate aminotransferase (AST) concentration markedly elevated in comparison with the alanine aminotransferase (ALT) concentration, whereas in viral hepatitis the reverse is usually true. Predominant elevations of alkaline phosphatase (ALP) level indicate intrahepatic cholestasis and are often due to an infiltrative process (e.g., metastatic carcinoma, sarcoidosis). Elevated levels of bilirubin can result from either direct (conjugated) or indirect (unconjugated) causes. Although an overwhelming liver process in an adult will produce elevations of both forms of bilirubin, as a practical matter unconjugated hyperbilirubinemia points to significant hemolysis. Predominant conjugated bilirubin level elevation points to extrahepatic cholestasis attributable to biliary obstruction. It is noteworthy that jaundice in patients with cirrhosis often shows elevations in all parameters, reflecting the widespread liver dysfunction and obstruction of the bile canals and small vessels caused by scarring.
Evaluation of hepatocellular and cholestatic disorders may include assessment for viral hepatitis (Table 38-3), performance of various biochemical assays (see the specific disorders discussed later), or conduction of a needle biopsy of the liver. Needle biopsies may be carried out “blind” or may be directed by ultrasound or computed tomography (CT), allowing examination of a specific target such as a mass lesion.
TABLE 38-3
IMMUNOLOGIC MARKERS IN VIRAL HEPATITIS
| MARKER | DESCRIPTION |
| Hepatitis A | |
| Anti-HAV IgM | Acute infection with HAV, but may persist for months |
| Anti-HAV IgG | Past infection with HAV |
| Implies immunity to the virus | |
| Hepatitis B | |
| Hepatitis B surface antigen (HBsAg) | Surface protein coat of HBV |
| Implies active infection | |
| Detectable 2-6 wk after infection | |
| Remains present as long as infection is active | |
| Hepatitis B surface antibody IgM (HBsAb IgM) | Antibody to surface protein of HBV |
| Detectable shortly after or with clearance of HBsAg | |
| Implies resolution of infection and immunity to HBV | |
| Hepatitis B core antibody IgM (HBcAb IgM) | Antibody to inner core protein of HBV |
| Detectable 3-5 wk after infection | |
| Implies recent infection | |
| Hepatitis B core antibody IgG (HBcAb IgG) | Same as for HBcAb IgM, but implies past infection |
| Hepatitis Be antigen (HBeAg) | Soluble fraction of HBV |
| Detectable 2-6 wk after infection | |
| Implies ongoing infection with high infectivity | |
| May resolve independently of HBsAg | |
| Hepatitis Be antibody (HBeAb) | Antibody to soluble fraction of HBV |
| Detectable when HBeAg clears | |
| Implies decreased infectivity | |
| HBV DNA polymerase activity | Same significance as HBV DNA |
| Hepatitis C | |
| Anti-HCV | Antibody to HCV antigens |
| May not be detectable early in infection | |
| Does not indicate immunity to the virus | |
| Rapidly evolving area; many commercially available assays of different sensitivity and specificity | |
| Many false-positive and false-negative results | |
| HCV RNA by PCR | Assay for level of viremia |
| Correlates positively with activity of infection | |
| Clears with resolution of infection | |
| Technically difficult; available through reference laboratories | |
| Hepatitis D (Delta) | |
| Hepatitis delta antigen (HDAg) | Assay for 35-nm RNA virus |
| Detectable 2-10 wk after infection | |
| Implies early infection | |
| Anti-HDV | Implies past or chronic infection |
| Does not indicate immunity to the virus | |
| Hepatitis E | |
| HEV RNA by PCR | Detects presence of virus |
| HEV antibody IgG and IgM | Not well standardized |
| Hepatitis G | |
| HGV by PCR | Measures level of viremia |
Radiologic imaging with ultrasonography is often helpful for significant liver disease. This is particularly true given the fact that structural liver abnormalities such as tumors may present with any of the aforementioned enzyme patterns or with a mixed picture. CT provides more information and has a special role in evaluating the content of iron in the liver in cases of suspected hemochromatosis. Specific visualization of the bile ducts may necessitate percutaneous transhepatic cholangiography or endoscopic retrograde cholangiopancreatography (ERCP; see Chapter 37), although magnetic resonance cholangiopancreatography (MRCP) has rapidly gained acceptance as a noninvasive way of evaluating the liver and biliary tree in great detail.
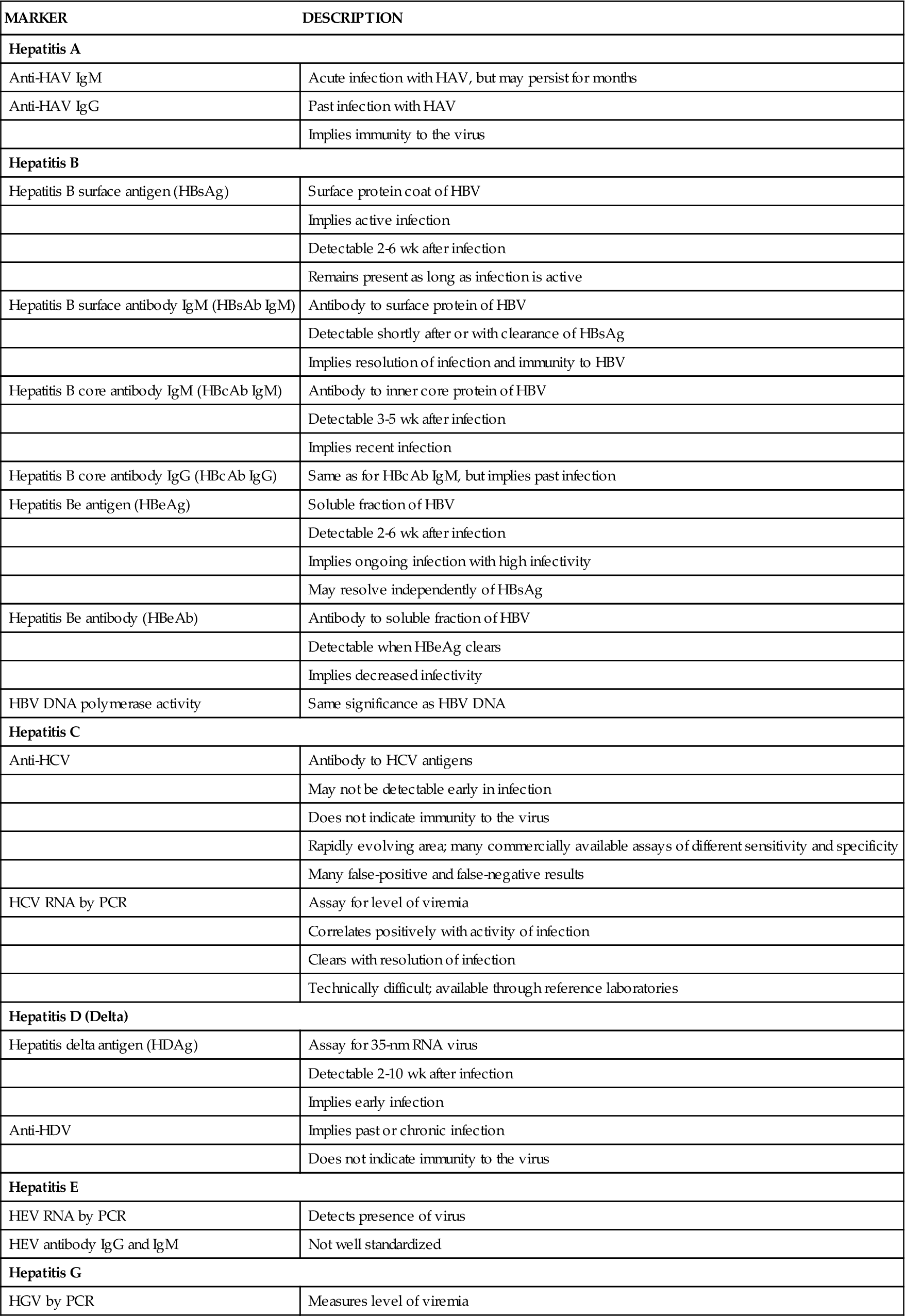
Portal Hypertension
Manifestations of liver disease not attributed to hepatocellular failure are mainly due to impaired blood flow through the liver as a result of increased resistance from fibrosis and degeneration of liver tissue. Sluggish blood flow through the liver results in increased pressure in the portal circulation (portal hypertension) (Figure 38-5). In this condition, venous drainage of much of the gastrointestinal tract is congested. Symptoms are surprisingly few early in the course, but as abnormal vascular patterns progress anorexia may result. The end results of elevated venous pressure are varices, particularly esophageal, but also gastric and hemorrhoidal. A pathognomic feature of advanced liver disease is superficial periumbilical varices, known as caput medusae, or the head of Medusa. Portal hypertension may present as an accumulation of peritoneal fluid, or ascites. A serious consequence of portal hypertension is uncontrolled bleeding from esophageal varices, which are prone to rupture.
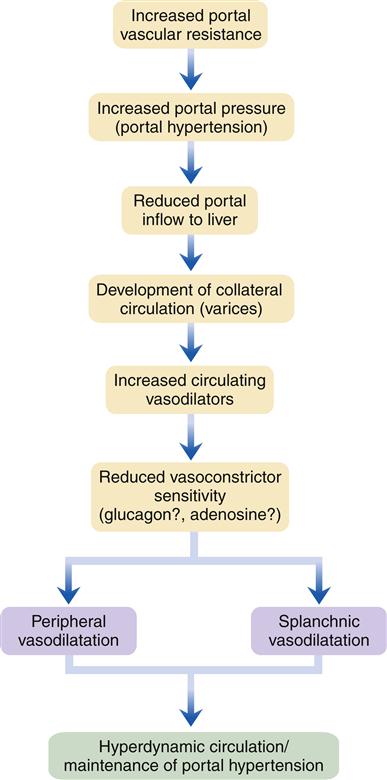
Gastroesophageal Varices
Etiology
Esophageal varices result mainly from portal hypertension, which in Western society is generally the result of cirrhosis attributable to the chronic effects of alcoholism or viral hepatitis. In developing tropical countries, chronic infection with the Schistosoma species of liver fluke is a major cause of portal hypertension. Recently, it has been recognized that vasoactive hormones, as well as increased splanchnic blood flow and increased vascular resistance in the liver, have a prominent role in the formation of variceal esophageal veins.1 Gastric varices may occur in conjunction with or independently from esophageal varices, the latter if splenic vein obstruction or thrombosis occurs.
Pathogenesis
Gastroesophageal varices are merely one of a number of collateral venous pathways that dilate in response to elevated portal pressure in an attempt to transport blood from the splanchnic bed around the cirrhotic obstructed liver and back to the heart. Other common pathways include a variety of spontaneous deep and usually entirely asymptomatic portosystemic shunts, such as splenorenal shunts; dilated veins in the small intestine, colon, and rectum are also not uncommon.1 Unfortunately, part of the very complex venous network that surrounds the proximal part of the stomach and esophagus lies just beneath the mucosa, rendering it especially liable to rupture when portal pressures reach a critical level. Rupture often results in massive, life-threatening upper GI bleeding (Figure 38-6).
Clinical features
Varices will affect more than half of cirrhotic patients, and approximately 30% of them experience an episode of variceal hemorrhage within 2 years of the diagnosis of varices.1 Variceal size is the main determinant of risk for bleeding, which is one of the main causes of death (20% to 33%) in persons with long-standing cirrhosis. The mortality after an episode of significant variceal bleeding is as high as 50%. The diagnosis is made mainly endoscopically, but varices may be seen on CT scans of the abdomen, as well as on upper GI barium examinations.
The initial symptoms and signs of bleeding from gastroesophageal varices include hematemesis, melena, and even bright red rectal bleeding. These characteristics may be associated with profound anemia and symptoms and signs of shock. In most cases, concomitant evidence of chronic liver disease and portal hypertension is seen on physical and laboratory examination. There are two distinct phases of the variceal hemorrhage process: one, coincident with and shortly after the bleeding; and two, a period of 6 to 8 weeks following the initial bleed, when there is a high risk of rebleeding. The greatest risk of rebleeding occurs in the first 72 hours.
Treatment
Initial treatment is directed at performing fluid resuscitation, correcting the coagulopathy, and stopping further bleeding. Large-bore intravenous lines are placed, and fluid resuscitation is carried out with normal saline. Blood components and clotting factors are replaced as needed. Any coagulopathy may necessitate administration of parenteral vitamin K, fresh frozen plasma, and platelet infusions if profound thrombocytopenia is present. Recombinant factor VIIa is helpful in reversing the coagulopathy associated with advanced liver disease and may be considered for patients whose prothrombin time (PT) fails to normalize after the previously stated measures have been performed.7–9
Primary acute pharmacologic management rests on drugs that can effectively lower portal pressure by dilating alternative collateral pathways, reducing splanchnic blood flow, or both. Until recently, the agent of choice in the United States was vasopressin, an analogue of antidiuretic hormone, administered by continuous intravenous infusion along with nitroglycerin. Although effective in controlling variceal bleeding, vasopressin use may be associated with angina pectoris, severe abdominal cramping, and hyponatremia, side effects that limit its usefulness.3
In recent years, octreotide acetate, a synthetic analogue of the naturally occurring hormone somatostatin, has been effectively used as a replacement for vasopressin.1 It is administered as an initial intravenous bolus followed by continuous infusion, which may be administered for as long as 3 to 5 days. In the doses used, the drug is remarkably free of side effects and more effective than vasopressin. However, it should be noted that no drug treatment has shown a mortality benefit for this condition.
Metoclopramide and β-blockers have been used as ancillary treatments in the past and may be considered for selected patients. Intravenous H2-blockers or proton pump inhibitors are also often administered. The use of prophylactic antibiotics is recommended by the American Association for the Study of Liver Diseases; intravenous ceftriaxone or an oral quinolone for a 1-week course is the preferred regimen.8
Emergency esophagogastroduodenoscopy (EGD) is crucial in determining the site of bleeding, as well as excluding other causes of upper GI bleeding. In addition to its diagnostic role, EGD actively addresses bleeding varices. Endoscopic sclerosis of esophageal varices is accomplished by passing a flexible needle through the gastroscope and injecting various sclerosant solutions into and around the bleeding varix (Figure 38-7). Such treatment results in initial thrombosis of the vein with hemostasis. Repeated injections cause fibrosis and obliteration of the varix and fibrosis of the overlying mucosa. This process can effectively obliterate all of the varices at risk of bleeding. Unfortunately, this treatment may be associated with a variety of acute and chronic complications, including drug reactions to the sclerosing solutions, exacerbation of bleeding, perforation, ulceration, infection, and stricture formation.5
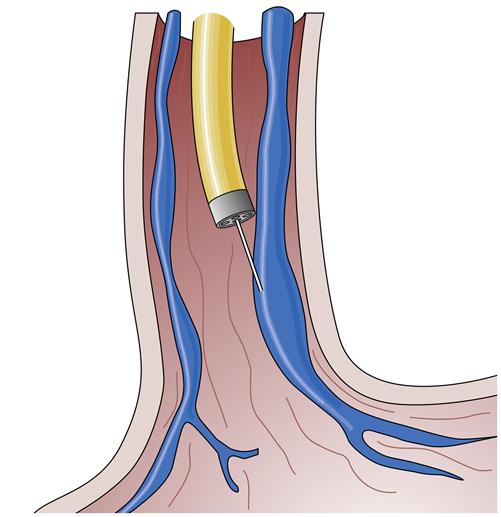
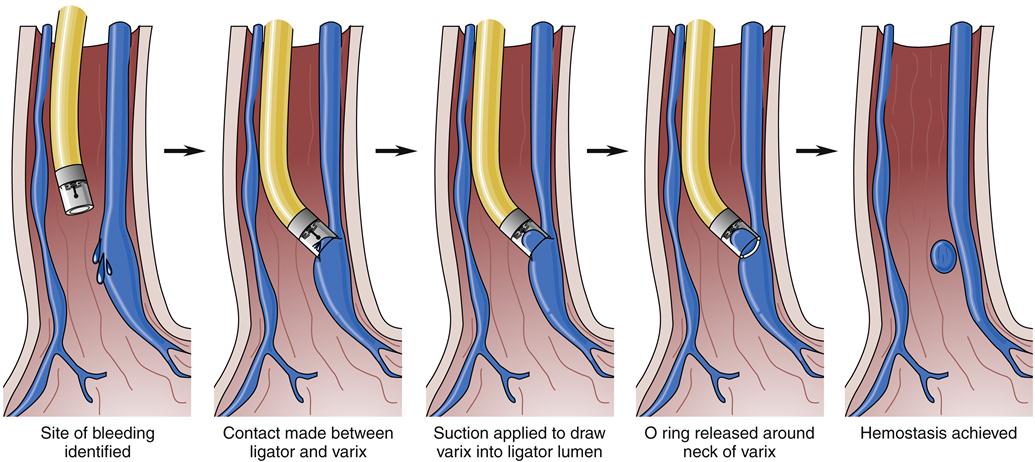
An alternative treatment method is endoscopic ligation of esophageal varices (Figure 38-8). In this technique, a special apparatus is preloaded onto the gastroscope so that the endoscopist can suction a varix into a special chamber at the end of the gastroscope and then ligate the varix with a small rubber band. This technique also results in immediate loss of flow in the varix and eventually leads to thrombosis and fibrosis. The area ligated simply sloughs off over the next few weeks without significant residual ulceration or scarring. This method requires fewer sessions than endoscopic sclerosis to completely obliterate the varices, seems to be associated with a lower complication rate, and may be more effective in the management of bleeding gastric varices.1 However, it is technically more challenging and is generally reserved for elective use.
Endoscopic techniques have shown a mortality benefit relative to treatment with drugs alone but fail to control acute bleeding in 10% to 20% of patients. Unfortunately, both of these methods may result in an increase in venous pressure proximal to the area treated, perhaps resulting in an increased risk of bleeding from congestive gastroenteropathy (a diffuse venous congestion that may result in both chronic and acute severe blood loss).
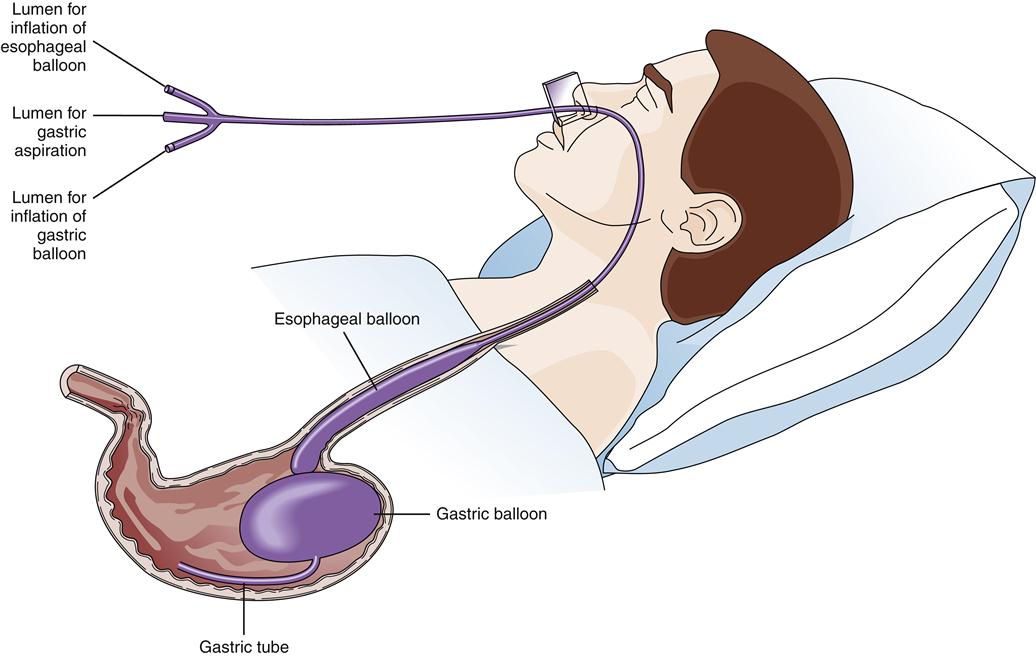
Balloon tamponade of varices was widely used before the availability of endoscopic treatment.1 This treatment consists of a gastric balloon that is passed orally or transnasally into the stomach, inflated, and held in gentle traction against the gastric varices in the fundus, thus tamponading bleeding vessels and restricting blood flow from the fundus up into the esophageal varices (Figure 38-9). Suction of oropharyngeal and gastric secretions is accomplished with integral or separate drainage tubes. Balloon tamponade carries frequent risks, including aspiration of stomach contents, migration of the tube with airway compression, pressure necrosis of the esophagus and stomach, rupture of the balloon, and rebleeding after the maximal inflation period of 24 hours. These limit the usefulness of balloon tamponade to a temporizing role until definitive treatment.
Chronic pharmacologic management of portal hypertension is frequently successful with nonselective β-blockers such as propranolol or nadolol. The drug is carefully titrated to reduce the initial resting heart rate by 25%. These drugs may be used prophylactically in patients with known portal hypertension and are often prescribed following endoscopic therapy as part of a combined approach to variceal bleeding prevention. It should be noted that studies have not yet shown a consistent survival benefit for this intervention, however. In addition to β-blockers, oral long-acting nitrates such as isosorbide mononitrate act synergistically with β-blockers to reduce portal pressure and have been studied for prevention of variceal hemorrhage.9,10 Side effects limit use of nitrates as primary prophylaxis, but these agents could be considered for secondary prophylaxis in patients who do not respond to β-blockers alone.
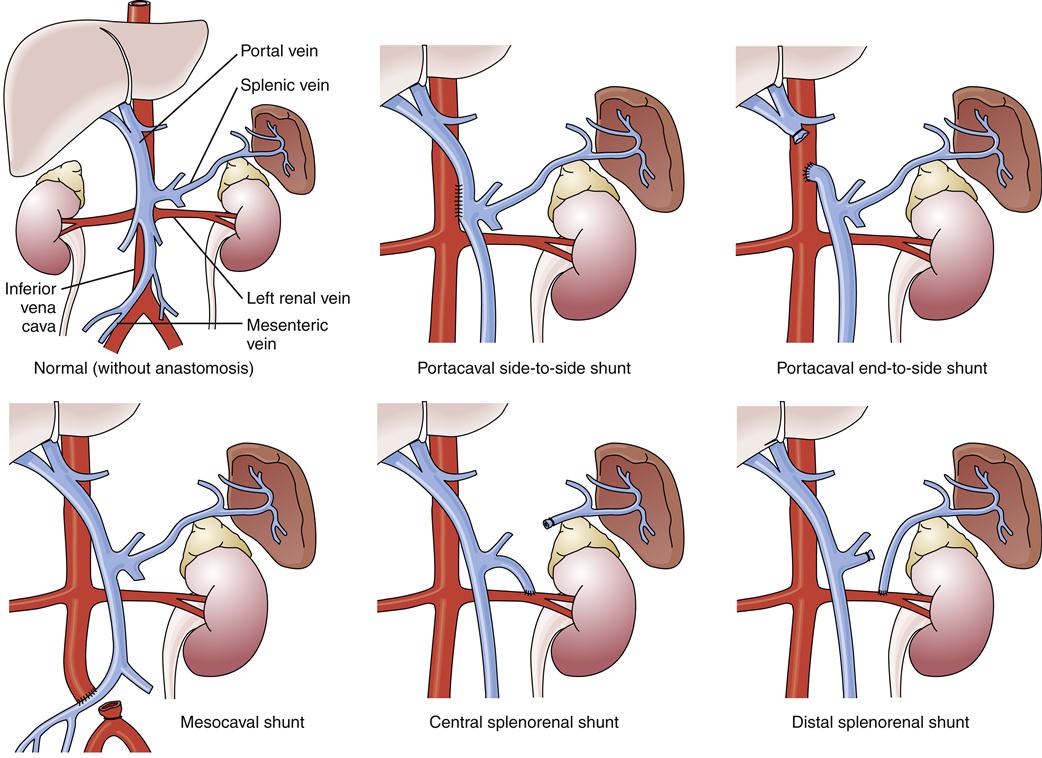
If the aforementioned measures are ineffective, a number of surgical and radiologic procedures are possible. Although rarely used in the United States, esophageal transection and reanastomosis with ligation of other collateral channels has been used in other countries.1 Surgery to reduce portal pressure is very effective in decreasing the rate of rebleeding, but it may not alter overall survival. A variety of surgical techniques are used, all of which create an alternative connection between the splanchnic and systemic circulations. These techniques include portacaval, mesocaval, splenorenal, and distal splenorenal shunts (Figure 38-10). A discussion of the specific indications and technical aspects of these shunts is beyond the scope of this text, but each has certain specific indications, advantages, and disadvantages.1
In recent years a radiographic procedure called transjugular intrahepatic portosystemic shunting (TIPS) has been developed that combines angiographic and ultrasonographic techniques.1 The hepatic vein is cannulated by the transjugular route. A needle is then passed into a main portal vein branch. Catheters are passed over this guidewire along with balloon dilation of the tract just created. This step is then followed by placement of an expandable metallic stent, thus creating a portosystemic shunt (portal vein to hepatic vein) within the liver itself. This procedure is technically very demanding and may be complicated by hemorrhage, infection, stent migration, stent stenosis, and occlusion, both acute and chronic. In addition, hepatic encephalopathy and congestive heart failure may result. The primary use of this modality is as a bridge to allow stabilization of patients who are candidates for liver transplantation.17
Treatment of esophageal varices is often unsatisfactory. Ideally, the underlying condition for varices (i.e., portal hypertension) should be reversed. The only consistently effective way to accomplish this goal is by liver transplantation (see the Transplantation section), which is limited in its application to a select group of patients.
Portal Systemic Encephalopathy
Hepatic Encephalopathy
Pathogenesis
Hepatic encephalopathy is a complex neuropsychiatric syndrome characterized by symptoms ranging from mild confusion and lethargy to stupor and coma. Some patients exhibit dementia, psychotic symptoms, spastic myelopathy, and cerebellar or extrapyramidal signs. The classic physical finding is asterixis, or “liver flap,” a spastic jerking of the hands held in forced extension. Hepatic encephalopathy is associated with fulminant hepatic failure or severe chronic liver disease, conditions in which liver function is severely depressed and blood is shunted around the liver. The arterial ammonia level correlates positively with the level of encephalopathy in most patients, consistent with its central role in the pathogenesis of hepatic encephalopathy as one of the primary causes of neuronal dysfunction. The exact cause is unclear, and other contributing factors such as elevated mercaptan levels, enhanced activation of certain neurotransmitter receptors (including γ-aminobutyric acid and benzodiazepine receptors), and elevated levels of aromatic amino acids (false neurotransmitters) remain under investigation.1,18
Clinical manifestations
Hepatic encephalopathy is usually precipitated by certain well-defined clinical developments, including hypokalemia, hyponatremia, alkalosis, hypoxia, hypercarbia, infection, use of sedatives, GI hemorrhage, protein meal gorging, renal failure, and constipation. In some patients, progressive liver failure leads to chronic encephalopathy without other exacerbating factors.
Hepatic encephalopathy is graded 1 to 4:
Treatment
The first step in treatment of hepatic encephalopathy consists of correcting any identifiable precipitating factors, such as gastrointestinal bleeding. Restriction of dietary protein to 60 g or less daily is indicated for patients with chronic encephalopathy, along with enhanced elimination of the toxic nitrogenous substances produced by intestinal digestion (see following paragraph). Critically ill patients should receive peripheral or central glucose infusions along with vitamins, especially thiamine. As the patient’s ammonia levels drop, protein may be reintroduced into the diet. The initial amount of 20 g/day is increased by 10 or 20 g/day every few days to an ultimate 0.75 to 1.0 g of protein per kilogram of body weight daily.13 Observation for worsening encephalopathy is crucial at this time. When protein is restricted, it is essential to provide at least 400 g of carbohydrate daily. Vegetable protein may be better tolerated than animal protein. High dietary fiber intake may help by decreasing constipation. If dietary measures fail, oral defined-formula feedings containing essential amino acids and enriched with branched-chain amino acids may be indicated.
Osmotic diuretics or antibiotics are used to enhance elimination of nitrogenous wastes. Lactulose is the standard osmotic cathartic and may be given orally or rectally by enema. (Standard precautions must be taken before any cathartic is administered, including ruling out bowel obstruction and monitoring electrolyte levels, particularly in patients with renal insufficiency.) Some evidence suggests that a lactulose-related change in pH also inhibits ammonia production by the gut flora, possibly by selecting for bacterial populations that are less ammoniagenic. No serious adverse reactions have been reported with lactulose therapy, although flatulence and abdominal cramping may occur. The dosage should be individually titrated so that two soft, acidic stools are passed daily.
Oral antibiotics have been used for many years to suppress the intestinal flora that break down dietary protein and release ammonia. Amoxicillin and rifaximin are currently used for this purpose. Bacterial overgrowth is one of the complications that limit the long-term use of antibiotics; therefore, this treatment is reserved for persons who cannot tolerate lactulose. Neomycin, the first antibiotic widely used in hepatic encephalopathy, is no longer recommended because of side effects.1
Cerebral Edema
Pathogenesis
Swelling of the brain (cerebral edema) often develops in patients with grade 3 or 4 hepatic encephalopathy and results in an increase in intracranial pressure. Both vascular and toxic mechanisms have been implicated as etiologic factors. With increasing intracranial pressure, blood perfusion of the brain is decreased (cerebral perfusion pressure = carotid artery pressure − intracranial pressure) with resulting cerebral hypoxia. Cerebral edema is a major cause of death in patients with acute hepatic failure.1
Clinical manifestations
Clinically, cerebral edema is suggested by deepening coma, systolic hypertension, and extensor rigidity (decerebrate posture), followed by pupillary dilation and, if brainstem herniation occurs, respiratory arrest. Some highly specialized referral centers monitor patients with advanced hepatic encephalopathy by extradural pressure monitors to permit early detection. Unfortunately, complications of extradural monitors occur up to 20% of the time and include infection and intracranial bleeding.1
Treatment
Cerebral edema is managed primarily by the intravenous infusion of mannitol, which by increasing serum osmolarity draws water from the brain and thus reduces the swelling. Patients should be kept in the semi-Fowler position (head and trunk elevated 30 degrees). The barbiturate sodium pentothal is used as a second-line agent for patients with recalcitrant intracranial hypertension and those who cannot tolerate the fluid volume component of mannitol therapy (e.g., those in heart or kidney failure). Moderate hypothermia with the use of cooling blankets has looked promising in preliminary trials as well.14 Aggressive treatment allows patient survival in 60% of cases, until liver failure resolves or liver transplantation can be accomplished.
Complications of Advanced Liver Disease
Ascites
Etiology, pathogenesis, and clinical manifestations
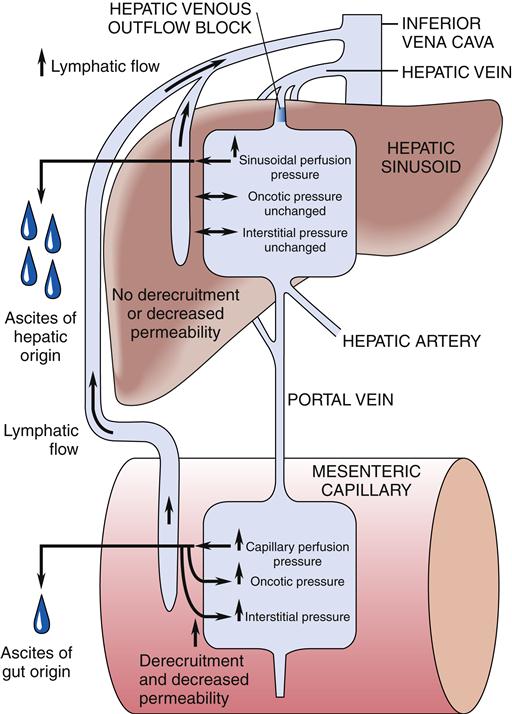
Ascites, or the pathologic accumulation of fluid in the peritoneal cavity, can occur in patients with advanced liver disease complicated by portal hypertension and hypoalbuminemia (Figure 38-11). Abdominal distention results from an inappropriate osmotic gradient across the pleura, with the intraabdominal accumulation of sodium, water, and protein.1,15 Other causes of ascites are malignancy, infection, pancreatitis, hypothyroidism, vasculitis, nephrosis, cardiac failure, constrictive pericarditis, Budd-Chiari syndrome, and portal vein thrombosis. The specific chemical and cellular composition of the ascites varies with its cause.
Abdominal paracentesis should be performed in all patients with new ascites and in those with known ascites who have experienced significant worsening of their condition.1 The fluid should be examined for total protein level, albumin level, and cell count. Optional tests include culture for bacteria, fungi, and mycobacteria; cytologic studies; and measurement of amylase, glucose, and lactate dehydrogenase levels. A small amount of ascites may not require specific therapy. However, with increasing volumes, abdominal discomfort, abdominal or umbilical herniation, respiratory embarrassment, or infection may occur.
Treatment
Dietary sodium should be restricted to 88 mEq (2000 mg) per day in patients with ascites. In motivated patients, this is perhaps the most helpful intervention that can be undertaken. Bed rest is useful, although strict bed rest can result in decubitus ulcers, deconditioning, and other problems. Diuretics are necessary for the majority of patients. The aldosterone antagonist spironolactone works in the distal nephron as a weak diuretic that also inhibits potassium secretion, thus sparing serum potassium. A delay of 2 to 3 days may occur before the full effect of the drug is seen, and the dosage should not be increased more frequently. Many authorities suggest adding a loop diuretic such as furosemide from the beginning and increasing the dose at a ratio of 4:10 with spironolactone.1 It is helpful to monitor both urinary sodium and potassium levels periodically. When the urinary sodium level exceeds the urinary potassium level, spironolactone is exerting its maximal effect. Serum potassium levels must be carefully controlled.
The goal is the loss of approximately 0.5 kg of body weight daily, although in patients with peripheral edema, slightly more rapid weight loss is tolerable. More rapid losses may result in diuretic-induced renal impairment, intravascular volume depletion and severe electrolyte abnormalities, and hepatic encephalopathy. Diuresis should be continued until the ascites is barely detectable. Free water restriction is prescribed if hyponatremia is present or develops during treatment, although compliance with a strict regimen is unlikely.
In patients who do not respond to both diuretic therapy and sodium restriction, the use of 25% albumin infusions may help initiate diuresis. However, the effect of this treatment is often short lived and not without a risk of overexpansion of the intravascular volume, with congestive heart failure, pulmonary edema, and precipitation of variceal hemorrhage all possible. Alternatively, large volumes of ascitic fluid can be removed from the peritoneal space through paracentesis. This “large-volume therapeutic paracentesis” is a very rapid and effective treatment that can be safely instituted if the intravascular volume is maintained by appropriate measures.1 Diuretic and dietary treatment should be continued, but in severe cases, large-volume paracentesis may be repeated as needed.
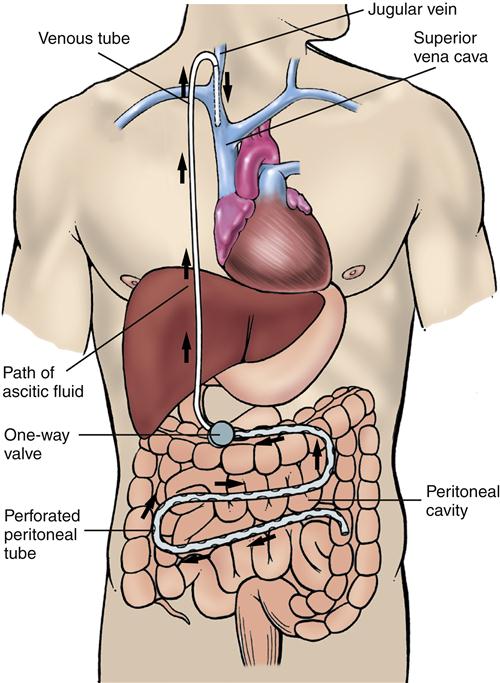
Shunting procedures such as the LeVeen (Figure 38-12) and Denver shunts have been used. These one-way valves connect the peritoneal space with the venous system, usually at the jugular vein. These shunting procedures, although useful, carry significant risks and are best reserved for a small subset of ascites patients who have refractory ascites and are not candidates for liver transplantation.1 The use of transjugular intrahepatic portosystemic stenting (TIPS) has largely replaced traditional surgical procedures for treatment of refractory ascites (see earlier discussion). Diuretic-resistant ascites is also an indication for liver transplantation in the patient who meets other criteria.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


