Restrictive Pulmonary Disorders
Lorna L. Schumann
Key Questions
• How do fibrotic lung disorders develop?
• How do abnormal accumulations in the pleural space affect lung function?
• What neuromuscular disorders are associated with reduced lung compliance?
• What is the pathogenesis of tuberculosis?
• What pulmonary function test abnormalities are characteristic of restrictive pulmonary disorders?

http://evolve.elsevier.com/Copstead/
Restrictive pulmonary diseases result from decreased expansion of the lungs attributable to alterations in the lung parenchyma, pleura, chest wall, or neuromuscular function. These disorders may be classified as pulmonary or extrapulmonary and represent acute or chronic patterns of lung dysfunction, rather than a single clinical disease. Table 23-1 lists the various disease processes that can be classified as restrictive. These diseases are characterized by a decrease in total lung capacity (TLC), vital capacity (VC), functional residual capacity (FRC), and residual volume (RV). The greater the decrease in lung volume, the greater the severity of the disease.1 Blood gas analysis often shows decreased arterial partial pressure of oxygen (PaO2) and normal or decreased arterial partial pressure of carbon dioxide (PaCO2), resulting in increased pH (respiratory alkalosis). This chapter presents information related to restrictive pulmonary diseases, including lung parenchyma disorders, pleural space disorders, neuromuscular and chest wall disorders, pneumonia, and tuberculosis. Specific lung parenchyma disorders, including interstitial fibrosis, sarcoidosis, hypersensitivity pneumonitis, and pneumoconiosis, as well as atelectatic disorders, including acute (adult) respiratory distress syndrome (ARDS) and infant respiratory distress syndrome (IRDS), are presented. Pleural space disorders, divided into pneumothorax and pleural effusions, are discussed. The section on neuromuscular and chest wall disorders is divided into neuromuscular weakness, chest wall deformities, and obesity. The final section presents etiologic factors, pathogenesis, clinical manifestations, diagnosis, and management of pneumonia, tuberculosis, and severe acute respiratory syndrome (SARS). Table 23-2 describes variations in respiratory parameters that affect restrictive lung disease in infant and elderly populations.
TABLE 23-1
RESTRICTIVE PULMONARY DISORDERS
| DISORDER TYPE | REPRESENTATIVE EXAMPLES |
| Diseases of the Lung Parenchyma | |
| Neoplastic disease | — |
| Pneumonia | Pneumonia (viral, bacterial, fungal), hypersensitivity pneumonitis |
| Granulomatous disease | Sarcoidosis, tuberculosis, coccidioidomycosis, blastomycosis |
| Pneumoconioses | Occupational lung disease |
| Acute interstitial pneumonitis | — |
| Collagen disease | Rheumatoid arthritis, scleroderma, systemic lupus erythematosus |
| Atelectasis | — |
| Pulmonary resection | — |
| Vascular Diseases | Pulmonary edema, pulmonary embolism |
| Acute respiratory distress syndrome | — |
| Diseases of Extrapulmonary Restriction | |
| Chest wall disease | Kyphoscoliosis, ankylosing spondylitis, obesity |
| Neuromuscular disease | Quadriplegia, hemiplegia, Guillain-Barré syndrome, myasthenia gravis, amyotrophic lateral sclerosis, muscular dystrophy |
| Pleural diseases | Pleural effusion, hemothorax, pneumothorax, chylothorax |
| Other | Abdominal distention, surgery, pregnancy |
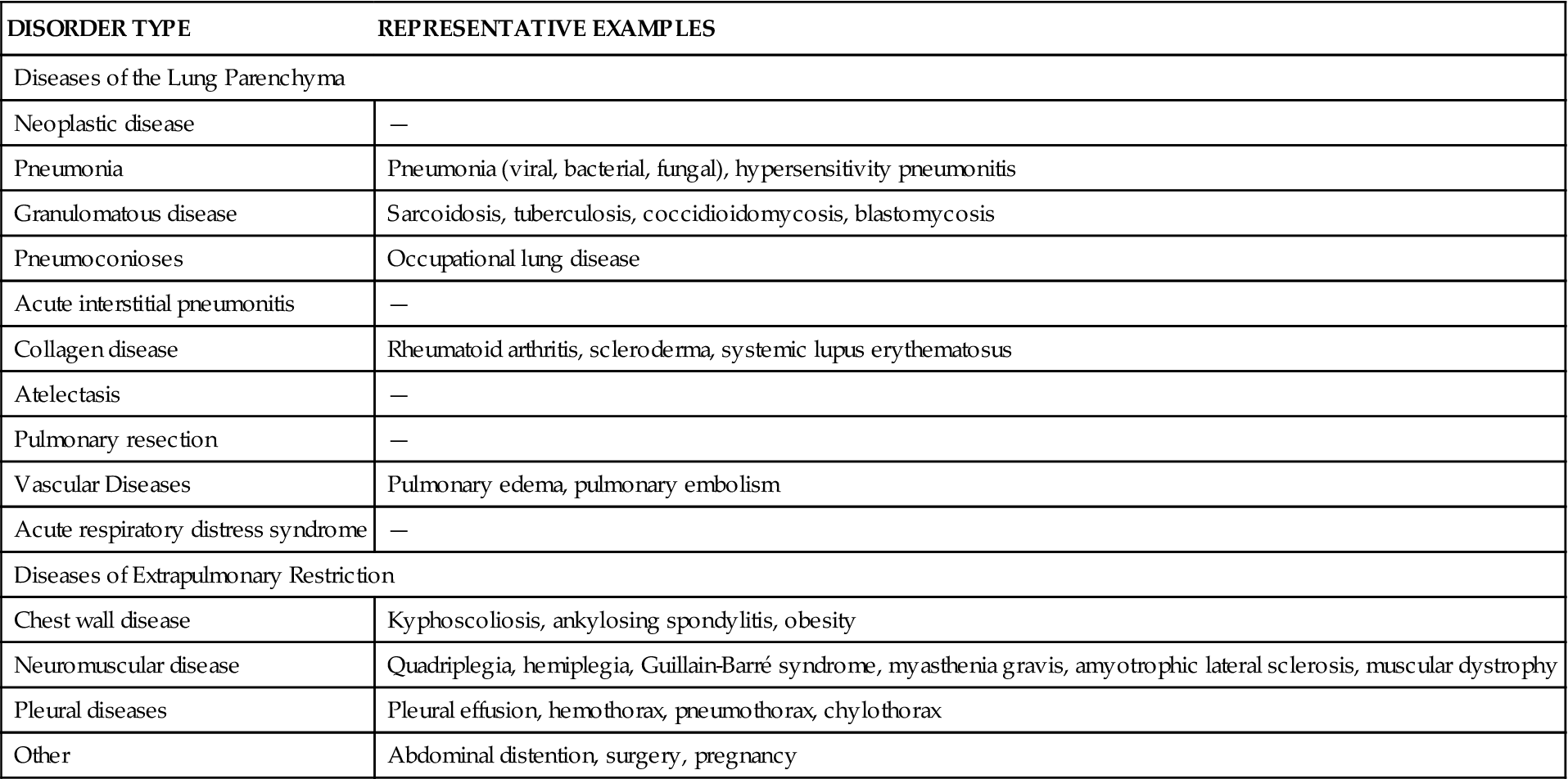
TABLE 23-2
AGE-RELATED FEATURES CONTRIBUTING TO RESTRICTIVE LUNG DISEASE
| ANATOMIC SITE | IMPACT ON RESTRICTIVE DISEASE |
| Infant | |
| Sternum and ribs are cartilaginous with soft chest wall | Diminishes effect of restrictive disease in infants |
| Ribs are horizontally oriented so that ribs move in and out easily | Diminishes effect of restrictive disease in infants |
| Accessory muscles of respiration are poorly developed | Majority of respiratory movement relies on diaphragm; restrictive diseases that compromise diaphragmatic excursion affect respiratory status; e.g., thoracic or abdominal surgery, paralysis, and abdominal masses all affect diaphragmatic excursion |
| Diaphragm rests horizontally and draws lower ribs inward in supine position so that diaphragmatic excursion is decreased | Leads to compromised effort of breathing |
| Cartilage of infant larynx is soft, so airway is compressed when neck is flexed or hyperextended | Increases airway resistance |
| During first month of life, neonate is obligate nose breather | Nasal obstruction may lead to respiratory distress from decreased airflow |
| Small diameter of airway leads to increased resistance to airflow | Mucus or edema in the airway may lead to significant increase in resistance and decrease in airway diameter |
| Fewer alveoli than in adults, leading to decreased radial traction applied to airways | Increased tendency of airways to collapse |
| Pores of Kohn and channels of Lambert are underdeveloped, leading to fewer collateral ventilation pathways | May lead to respiratory compromise, reducing ventilatory support with restrictive diseases |
| Elderly | |
| Decreased ciliary activity | Increased incidence of infection; decreased mucus clearance in all types of respiratory disorders |
| Decreased chest wall compliance and decreased lung elasticity in some areas of lung | Leads to a reduction in lung volume; leads to decreased expansion of lungs and to decreased matching of ventilation and perfusion |
| Decreased stress tolerance | Increased incidence of disease and trauma with age |
| Decreased muscle tone | Decreased physical conditioning |
| Impaired immunity as evidenced by decreased T cell function; increased autoantibodies | Decreased resistance to infection |
| Decreased oxygen uptake | Decreased oxygen level in blood |
| Decreased vital capacity | Decreased alveolar expansion |
| Decreased cough reflex | Impaired ability to clear secretions and inhaled particulate matter |
Lung Parenchyma Disorders
Fibrotic Interstitial Lung Diseases
The term interstitial lung disease describes a group of more than 180 disorders characterized by acute, subacute, or chronic infiltration of alveolar walls by cells, fluid, and connective tissue.1–4 If left untreated, the inflammatory process may progress to irreversible fibrosis.4 The incidence of interstitial lung disease is 20 cases per 100,000 persons in the general population and 175 per 100,000 in people more than 75 years of age.3
Diffuse Interstitial Lung Disease
Etiology
Diffuse interstitial lung disease (diffuse interstitial pulmonary fibrosis) is the name typically used for restrictive diseases characterized by thickening of the alveolar interstitium.2 Synonyms frequently presented in the literature include interstitial pneumonia, diffuse parenchymal lung disease, Hamman-Rich syndrome, intrinsic fibrosing alveolitis, cryptogenic fibrosing alveolitis, and idiopathic pulmonary fibrosis.
Pathogenesis
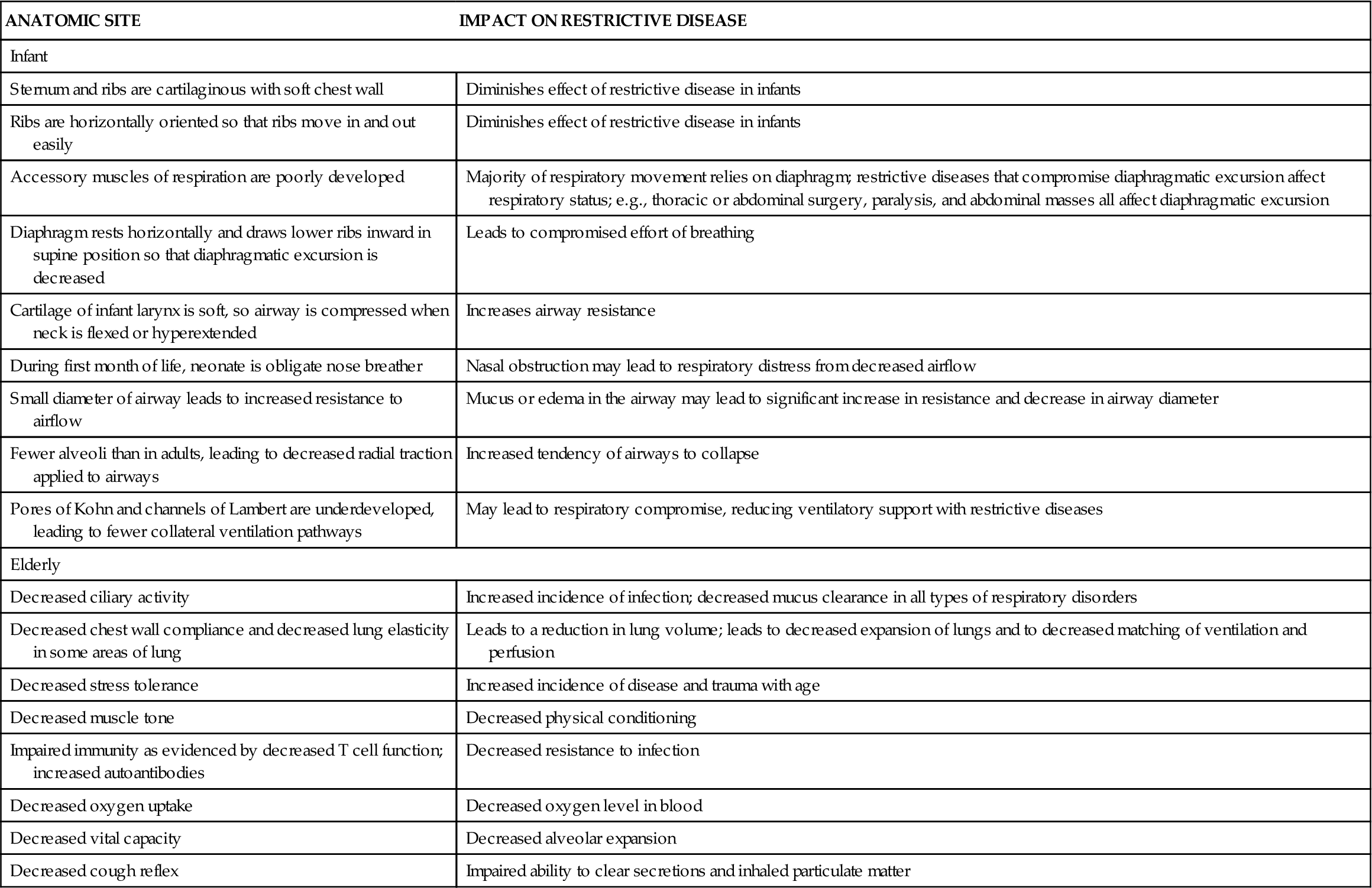
Pathogenesis of the disease is not well understood, but is possibly related to an immune reaction that usually begins with injury to the alveolar epithelial or capillary endothelial cells.1,2 Pathophysiologic changes may include interstitial and alveolar wall thickening and increased collagen bundles in the interstitium (Figure 23-1). Lung tissue becomes infiltrated by lymphocytes, macrophages, and plasma cells. Persistent alveolitis may lead to obliteration of alveolar capillaries, reorganization of the lung parenchyma, and irreversible fibrosis.2 These changes in turn lead to the formation of large air-filled sacs (cysts) accompanied by dilated terminal and respiratory bronchioles. The immune response noted in interstitial lung disease is characterized by three pathologic patterns in the alveoli: inflammation, fibrosis, and destruction.
The inflammatory pattern occurs early and is potentially reversible.4,5 The triggering event (occupational exposure, tobacco abuse, drug ingestion, connective tissue disease) causes an inflammatory response leading to increased numbers of inflammatory cells (neutrophils, lymphocytes, macrophages).1,2,4 An associated injury to the alveolar capillary basement membrane from the triggering event leads to increased membrane permeability and movement of fluid and debris into the alveoli. The initial injury, in association with the inflammatory pattern, leads to fibroblastic proliferation and deposition of large amounts of collagen. The fibrotic pattern is manifested by increases in the number of mesenchymal cells and fibroblasts in the interstitium, and alveolar walls become thickened with increased amounts of fibrous tissue.5,6 Physiologic restriction leads to reduced compliance and increased elastic recoil. The lung destruction pattern is manifested by loss of alveolar walls. Radiographically, this appears as a “honeycomb lung” and indicates end-stage disease.2 Ground-glass appearance on chest radiograph is often an early finding.3 The fibrotic and honeycomb patterns respond poorly to treatment.5
Clinical manifestations
The most common patient complaint is progressive dyspnea with nonproductive cough.3,5 Clinical features also include rapid, shallow breathing; dyspnea; clubbing of the nail beds (40% to 80% cases)4; bibasilar end-expiratory3,4 crackles (Velcro rales); and marked dyspnea with exercise. Cyanosis is a late finding. Anorexia and weight loss are noted on physical exam. While the disease progresses, patients exhibit an inability to increase cardiac output with exercise as evidenced by low maximal heart rate and high peripheral vascular resistance. Arterial oxygen desaturation occurs with exercise.4
Diagnosis
Chest radiographs show a honeycomb appearance and a coarse reticular pattern indicating late stage of disease.2,3,5,6 Ground-glass haziness is indicative of the presence of infiltrates. High-resolution computed tomography (HRCT) and bronchoalveolar lavage are the primary diagnostic tests used to evaluate interstitial lung disease.6 Open lung biopsy or transbronchial biopsy and gallium-67 scanning may be used for diagnostic evaluation.3 Results of pulmonary function tests are usually consistent with restrictive lung disease (decreased vital capacity, reduced total lung capacity, and decreased diffusing capacity).3
Treatment
The patient should be encouraged to avoid tobacco use and environmental exposure to the offending agent.2–4 Primary therapy consists of administration of antiinflammatory and immunosuppressive agents. Immunosuppressive agents have been useful in reducing the dosage of corticosteroids required. Oxygen therapy is needed in patients with hypoxemia. Lung transplantation has been successful in selected patients.3,5–9
Sarcoidosis
Etiology
Sarcoidosis is categorized as an acute or chronic systemic disease of unknown cause, although an immunologic basis appears likely.1,9 A common feature of sarcoidosis is the presence of CD4+ T cells.9 Activation of the alveolar macrophage from an unknown antigen trigger is a possible cause.1 The acute process occurs more commonly in women in the second or third decades of life.1,2 The chronic form is seen more commonly in the third to fourth decades of life, with the highest incidence seen in North American blacks (35.5/100,000) and northern European whites (11.9/100,000).2 Having a first-degree relative with sarcoidosis increases the risk for disease fivefold.9
Pathogenesis
The disease is characterized by the development of multiple, uniform, noncaseating epithelioid granulomas that affect multiple organ systems, most commonly lymph nodes and lung tissue. Noncaseating granulomas are fibrotic and surrounded by large histiocytes.1,2,9 Sarcoid granulomas may also develop in the bronchial airways. Abnormal T cell function is noted with this disease.9 Other systems/organs frequently involved are the skin, eyes, spleen, liver, kidney, and bone marrow.1,9
Clinical manifestations
Sarcoidosis is characterized by malaise, fatigue, weight loss, fever, chest discomfort, dyspnea of insidious onset, and a dry, nonproductive cough.2,9 Other features include erythema nodosum (lesions marked by the formation of painful nodes on the lower extremities); macules, papules, hyperpigmentation, and subcutaneous nodules; hepatosplenomegaly; and lymphadenopathy. Patients with acute disease usually present with enlarged lymph nodes and arthritis, although some patients experience no symptoms.9 Skin lesions and lacrimal and parotid gland involvement are also noted in the acute process.1 Iritis, uveitis (65% of patients), blurred vision, conjunctivitis, and ocular discomfort may develop.9
Diagnosis
Common laboratory findings in patients with sarcoidosis include leukopenia, anemia, increased eosinophil count, elevated sedimentation rate, and increased calcium levels (seen in 11% of patients).2,9 Serum levels of liver enzymes may also be elevated. Approximately 70% of patients exhibit anergy (decreased sensitivity to specific antigens such as Trichophyton, Candida, mumps virus, and tuberculin).2 Patients with active disease also demonstrate elevated levels of angiotensin-converting enzyme (40% to 80% of cases).2 Chest radiographs can be used to differentiate stages of the disease process: stage 0, normal; stage I, hilar adenopathy alone; stage II, hilar adenopathy and bilateral pulmonary infiltrates; and stage III, pulmonary infiltrates without adenopathy. Stage IV is characterized by advanced fibrosis with evidence of honeycombing, hilar retraction, bullae, cysts, and emphysema.7 Gallium-67 scans will localize areas of granulomatous infiltrates. Pleural effusion is noted in 10% of cases of sarcoidosis.1,2,9 Pulmonary function test results may be normal or show evidence of restrictive disease and/or obstructive disease.9 Transbronchial lung biopsy demonstrates noncaseating granulomas, thus providing a definitive diagnosis (75% to 90% cases). Bronchoalveolar lavage may be used to monitor cell content in patients with sarcoidosis.1,9 The lavage fluid is characterized by increased lymphocytes and a high CD4/CD8 cell ratio.2
Treatment
Administration of corticosteroids and management of symptoms is the mainstay of treatment for patients whose disease process does not resolve spontaneously and in whom progressive lung disease or evidence of extrapulmonary sarcoidosis develops. For patients with progressive disease that does not respond to corticosteroids, immunosuppressive agents may be used.9 Hydroxychloroquine is effective for treatment of disfiguring skin lesions, hypercalcemia, and neurologic involvement.9 The prognosis is best for stage I disease. Death attributable to pulmonary insufficiency occurs in about 5% to 7% of patients.2,9
Hypersensitivity Pneumonitis
Etiology
Hypersensitivity pneumonitis, also called extrinsic allergic alveolitis, is classified as a restrictive and occupational disease. Numerous (greater than 300) inhaled organic agents are responsible for the inflammatory process.10 Table 23-3 lists various allergens related to the disease. Unlike other pulmonary diseases, hypersensitivity pneumonitis has a predominance in nonsmokers (80% to 95% of cases).10
TABLE 23-3
CAUSES OF HYPERSENSITIVITY PNEUMONITIS
| DISEASE | ANTIGEN | ALLERGEN SOURCE |
| Farmer’s lung | Thermophila, Actinomyces | Moldy hay, silage |
| Bird fancier’s lung | Parakeet, pigeon, chicken | Bird excreta, feathers, and animal protein |
| Bagassosis | Thermophilic bacteria | Moldy sugarcane pulp |
| Mushroom, cork, maple bark, or malt hypersensitivity; cheese maker’s lung, redwood lung | Various fungi | Handling moldy products |
| Grain handler’s lung | Wheat weevil | Insect-infected grain |
| Pituitary extract hypersensitivity | Heterologous pituitary and serum proteins | — |
| Fish-meat worker’s lung | Protein, fungi | Animal food |
| Humidifier lung (fever) | Thermophilic bacteria, amoebae, and fungi | Humidifiers and evaporative air coolers |
Pathogenesis
The causative agent is suggested by the patient’s history and confirmed by demonstration of precipitating antibodies in the serum directed to the causative antigen. The causative antigen combines with the serum antibody in the alveolar walls, leading to a type III hypersensitivity reaction. Type III hypersensitivity diseases are caused by the formation of antigen-antibody complexes1,10 (see Chapter 10). These antigen-antibody complexes then elicit the granulomatous inflammation that leads to lung tissue injury, as evidenced by thickening of alveolar walls; formation of exudate in the bronchiolar lumen; and infiltration by lymphocytes, plasma cells, and eosinophils.1,10,11 Fibrotic lung changes occur in advanced cases. Many individuals develop precipitating antibodies (precipitin) from organic dust exposure, but only a few develop pneumonitis.11 Genetic predisposition may be involved in an exaggerated response to the offending agent. Experiments in animals show that a delayed hypersensitivity (type IV) reaction to the antigen is also required before pneumonitis can occur.11
Clinical manifestations
In the acute stage of the disease, symptoms start 4 to 6 hours after exposure and resolve in 18 to 24 hours.10 General symptoms may include chills, sweating, shivering, myalgias, nausea, lethargy, headache, and malaise.1,11,12 The patient may have a fever. Respiratory symptoms may include dyspnea at rest, dry cough, tachypnea, and chest discomfort. Physical findings may include cyanosis (a late sign) and crackles (rales) in the lung bases.1,3
In the chronic form, progressive diffuse pulmonary fibrosis develops in the upper lobes—the hallmark of the disease.1 In the intermediate form, the disease may manifest with acute febrile episodes and progressive pulmonary fibrosis with cough, dyspnea, fatigue, and, eventually, cor pulmonale (right-sided heart enlargement attributable to lung disorders).10
Diagnosis
During the acute/subacute phase, transient bilateral pulmonary infiltrates or increased bronchial markings with alveolar nodular infiltrates may be found on chest radiographs. In the chronic phase, diffuse reticulonodular infiltrates and fibrosis are present.11 Skin testing with the causative antigen may produce a red, indurated, hemorrhagic reaction 4 to 12 hours after injection that lasts several days.1 This reaction suggests precipitin-mediated sensitivity. Skin testing for most precipitating antigens is impractical because most produce irritating reactions before the precipitin reaction occurs, and many individuals without the disease have precipitating antibodies. Common laboratory findings include an increased white blood cell count and a decreased PaO2. Elevations in erythrocyte sedimentation rate and the level of C-reactive protein are often present. Hypoxemia worsens with exercise. Pulmonary function tests show decreased lung volumes, diffusing capacity, and static compliance.1,10
Treatment
The goal of therapy is to identify the offending agent and prevent further exposure. This may require a change in environment or occupation. Oral corticosteroids may be used to decrease the inflammatory process.10,12
Occupational Lung Diseases
Etiology
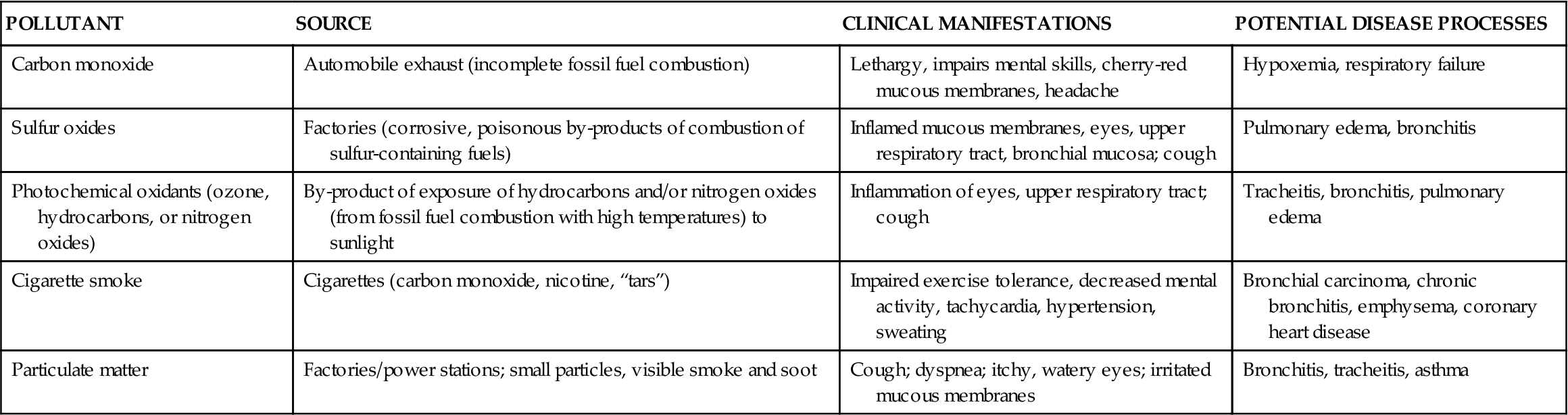
Occupational lung diseases result from the inhalation of toxic gases or foreign particles. Traditionally, occupational lung diseases included pathologic conditions that were associated with the effects of exposure to inhaled dusts. However, a holistic approach to these diseases requires consideration of atmospheric pollutants as well as natural genetic resistance and compliance with health maintenance behaviors. The distinction between occupational and environmental respiratory diseases is becoming increasingly difficult. The integration of multiple environmental areas (home, work, and leisure) further compounds the complexity of defining occupational respiratory diseases. Although atmospheric pollutants (toxic gases) are not discussed in detail here, their impact on occupational respiratory diseases must not be minimized. The sources, potential clinical manifestations, and potential pathologic processes associated with common atmospheric pollutants are presented in Table 23-4.
TABLE 23-4
COMMON ATMOSPHERIC POLLUTANTS CONTRIBUTING TO LUNG DISEASE
| POLLUTANT | SOURCE | CLINICAL MANIFESTATIONS | POTENTIAL DISEASE PROCESSES |
| Carbon monoxide | Automobile exhaust (incomplete fossil fuel combustion) | Lethargy, impairs mental skills, cherry-red mucous membranes, headache | Hypoxemia, respiratory failure |
| Sulfur oxides | Factories (corrosive, poisonous by-products of combustion of sulfur-containing fuels) | Inflamed mucous membranes, eyes, upper respiratory tract, bronchial mucosa; cough | Pulmonary edema, bronchitis |
| Photochemical oxidants (ozone, hydrocarbons, or nitrogen oxides) | By-product of exposure of hydrocarbons and/or nitrogen oxides (from fossil fuel combustion with high temperatures) to sunlight | Inflammation of eyes, upper respiratory tract; cough | Tracheitis, bronchitis, pulmonary edema |
| Cigarette smoke | Cigarettes (carbon monoxide, nicotine, “tars”) | Impaired exercise tolerance, decreased mental activity, tachycardia, hypertension, sweating | Bronchial carcinoma, chronic bronchitis, emphysema, coronary heart disease |
| Particulate matter | Factories/power stations; small particles, visible smoke and soot | Cough; dyspnea; itchy, watery eyes; irritated mucous membranes | Bronchitis, tracheitis, asthma |
Pneumoconiosis is defined as parenchymal lung disease caused by the inhalation of inorganic dust particles. The greater the exposure to the dust, the worse the pathologic consequences. Anthracosis (coal miner’s lung or black lung), silicosis (silica inhalation), asbestosis (asbestos inhalation), and acute beryllium disease are common examples of occupational lung diseases. However, exposure to several other dusts may also impair respiratory function. Included in this list are antimony ore, barium, iron, tin, fuller’s earth (clay), kaolin (china clay), and talc. Hairdressers exposed to bleach and hair spray are at risk for developing obstructive lung disease. Asthma caused by occupational exposures is seen in 16.3% of all adult-onset asthma.13 Many workers are exposed to “pathogenic dust” through the processing, packaging, or manufacturing of a specific product.10,14 Predisposing factors such as history of preexisting lung disease, exposure to atmospheric pollutants, duration of dust exposure, amount of dust concentration, and size of dust particles affect the onset and severity of the respiratory impairment.
Pathogenesis
The respiratory tract is protected by two interrelated systems: the mucociliary system and alveolar macrophages. The inhalation of inorganic particles has little effect on the mucociliary system. However, atmospheric pollutants (sulfur oxides, nitrogen oxides, and tobacco smoke) interfere with and can paralyze ciliary action.1 As a result, the clearance effect is impaired, and inorganic particles cannot be removed. Alveolar macrophages attempt to engulf and remove inorganic dust by one of the following methods: (1) migrating to small airways to use the mucociliary escalator; (2) engulfing dust and exiting through the lymph and/or blood system; (3) passing through bronchial walls, depositing dust particles in extraalveolar tissue; or (4) destroying the particle (silica).1,14
Macrophage impairment is the primary mechanism through which inorganic particles initiate lung diseases. In an attempt to maintain a sterile alveolar environment, macrophages secrete lysozymes to control foreign particle activity. These enzymes, released in response to the particulate stimuli, eventually damage the alveolar walls, which may cause deposition of fibrous materials. Although the type of inorganic particle inhaled individualizes the pathophysiologic response, the general response is similar in the context of occupational lung diseases. Silica is one of the most toxic particles to alveolar macrophages. Dense deposits of collagen material are formed around the silica particles, resulting in marked fibrotic tissue deposition and restrictive lung disease. Coal dust and asbestos initiate a similar, although less severe, response. The pathologic processes and clinical features for each of the major occupational lung diseases are summarized in Table 23-5.
TABLE 23-5
MAJOR OCCUPATIONAL LUNG PNEUMOCONIOSES
| PNEUMOCONIOSES | PATHOLOGIC FINDINGS | CLINICAL FEATURES |
| Anthracosis (coal miner’s lung) | Early: Collection of coal particles with small amount of dilation of airway | Early: Minimal to no symptoms; may be seen with dyspnea with cough but often due to unrelated bronchitis or emphysema |
| Late: Progressive, massive fibrosis, with condensed areas of black fibrous tissue | Late: Worsening dyspnea on exertion, productive cough, respiratory failure | |
| Silicosis | Dense collagen deposits in respiratory bronchioles and alveoli and along lymphatics | Early: No symptoms noted |
| Late: Productive cough, dyspnea, especially with exercise; increased risk for tuberculosis | ||
| Asbestosis | Fibrous deposits secondary to long, thin fibers, allowing deep lung penetration | Progressive dyspnea on exertion, weakness, clubbing of fingers; pleural thickening with plaque development |
Clinical manifestations
Pneumoconioses (anthracosis, asbestosis, silicosis) generally produce no symptoms in the early stages. Physical evidence of the disease occurs when the pulmonary circulation is impaired because of increased pulmonary vascular resistance or development of a pulmonary infection. Workers may remain symptom free for up to 10 to 20 years with chronic exposure.1,2,14 Once again, symptom manifestation is dependent on the predisposing factors. As pneumoconioses progress, patients present with a progressive, productive cough and dyspnea, especially with exercise. In addition, patients may complain of progressive weakness and fatigue. Clubbing of fingers may also be present. Late clinical features include chronic hypoxemia, cor pulmonale, and respiratory failure.
Diagnosis
The reliability of pulmonary changes noted on chest radiographs varies with the severity of the disease. When the patient is symptom free, no changes may be noted. However, as the pneumoconioses progress, micronodular mottling and haziness become apparent.1 In addition, nodules, fibroses, and calcifications resulting from dust particle deposition are noted. Pneumoconioses usually produce one of three radiographic findings: nodular, reticular, or linear. However, because of the insidious progression of occupational lung diseases, radiographs negative for lung disease do not exclude the presence of the disease process. Changes in pulmonary function tests demonstrate predominantly restrictive impairment (see Figure 22-19) with a component of obstructive functional impairment, depending on the severity and type of dust inhalation.1
Finally, hypoxemia is evident from arterial blood gas measurements in the late disease stages. Falling PaO2 levels are accompanied by decreased PaCO2 levels as the body initially compensates for the hypoxemia with an increased respiratory drive. However, as the disease progresses, both hypoxemia and hypercapnia are evident.
Treatment
Preventive measures are the key to limiting the onset and severity of occupational lung diseases. Adherence to federal standards for exposure to dust and particulate matter, as well as continuing education of workers and employers, could dramatically impact the incidence of respiratory diseases. The use of respirators and water sprays for miners to decrease airborne particles in mines are two prevention techniques. Early evaluation of a work environment predisposed to occupational lung diseases is where “treatment” must begin.14 Two primary goals in the management of active occupational lung diseases are to prevent further parenchymal damage and to relieve signs and symptoms, when possible. Ideally, if the problematic dust can be identified, the individual should be removed from the environment. However, if a job change is unrealistic, every possible measure must be implemented to prevent further inhalation of dust particles. Included in this treatment is the evaluation of current health maintenance behaviors. Treatment consists of corticosteroids, inhaled bronchodilators, oxygen therapy, and respiratory treatments (intermittent positive-pressure ventilation, postural drainage, and deep breathing exercises). The effectiveness and utilization of these therapies depend on the patient’s condition and the stage of disease. Rarely are those pathologic conditions reversed with medical treatment. The primary goal is to halt symptom progression.
Atelectatic Disorders
Acute (Adult) Respiratory Distress Syndrome
Etiology
Acute (adult) respiratory distress syndrome is characterized by damage to the alveolar-capillary membrane. In the United States there are more than 150,000 cases per year. Mortality statistics range from 30% to 63%.1,2,15,16 Clinically, ARDS is associated with a decline in the PaO2 that is refractory (does not respond) to supplemental oxygen therapy. Damage to the alveolar-capillary membrane causes widespread protein-rich alveolar infiltrates (visible on chest radiographs) and severe dyspnea. Patients who recover from the acute injury can expect to return to relatively normal lung function.1,15 Follow-up studies (9 months to 4 years) in ARDS survivors show a mild restrictive pulmonary function accompanied by cough, dyspnea, and excess sputum production.2,4 Some individuals continue to have abnormalities in diffusing capacity, oxygenation, and lung mechanics.2,4 ARDS is associated with severe trauma, sepsis (greater than 40% of cases), aspiration of gastric acid (greater than 30% of cases), fat emboli syndrome, and shock from any cause (Box 23-1). The precise mechanism of lung injury is not known, but the common denominator appears to be increased permeability of the pulmonary vasculature and flooding of the alveoli with proteinaceous fluid, leading to the development of protein-rich pulmonary edema (noncardiogenic pulmonary edema). The acute lung injury triggers the immune system to activate the complement system and to initiate neutrophil sequestration in the lung (Figure 23-2).
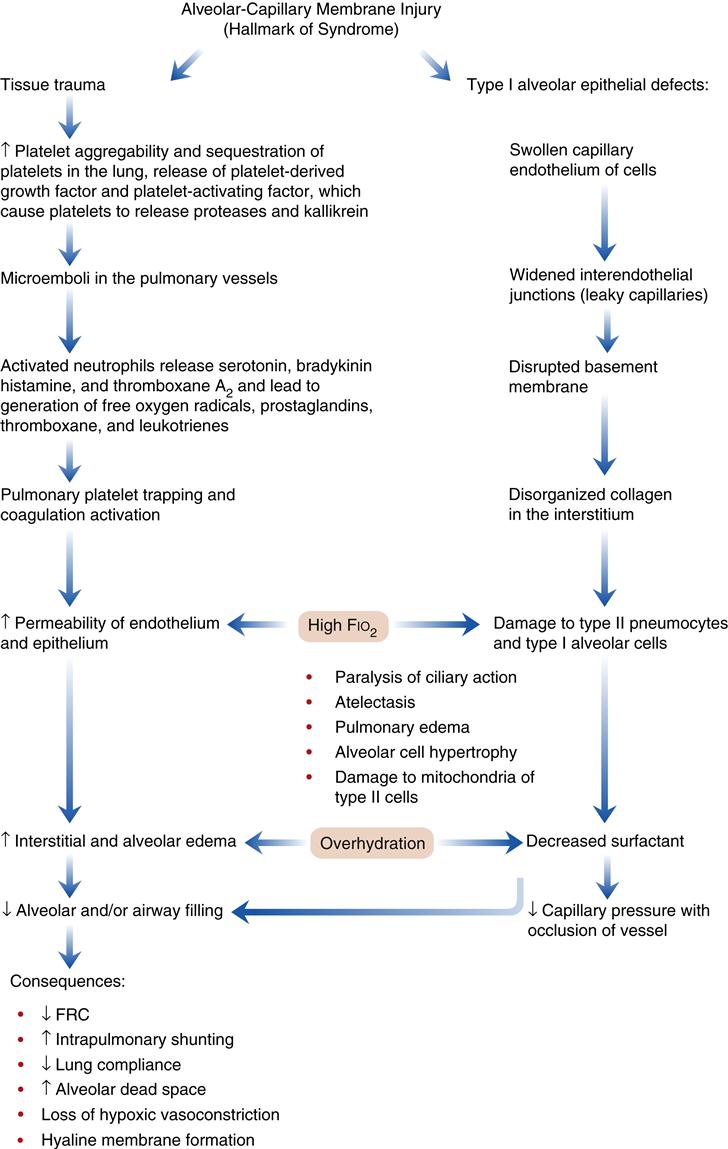
Pathogenesis
The pathogenetic sequence of events in ARDS is shown in Figure 23-2. The initial injury to the alveolar-capillary membrane may be caused by direct damage, as seen in aspiration of acidic gastric contents, or by indirect damage, as occurs in shock from any cause. Therapeutic interventions (high oxygen and overhydration) may act to compound the effects of the initial lung injury. The resulting injury leads to an increase in alveolar-capillary permeability, which results in interstitial and alveolar edema. The four characteristic pathophysiologic abnormalities of ARDS involve: (1) injury to the alveoli from a wide variety of disorders, (2) changes in alveolar diameter, (3) injury to the pulmonary circulation, and (4) disruptions in oxygen transport and utilization.15,16 Common findings in this type of injury include (1) severe hypoxemia caused by intrapulmonary shunting of blood; (2) a decrease in lung compliance; (3) a decrease in FRC; (4) diffuse, fluffy alveolar infiltrates on the chest radiograph; and (5) noncardiogenic pulmonary edema.1,9,15,16
The mechanism by which the FRC is decreased appears to be the result of stiff, noncompliant lungs associated with the presence of alveolar edema and exudate, which exaggerate surface tension forces.1,2,15,16 Early alveolar closure and continued closure lead to atelectasis and loss of lung volume.1 The decrease in lung compliance, often severe in ARDS, is reflected in the high ventilatory pressures required to deliver an adequate volume of gas. It is thought that this decrease in lung compliance is due to loss or inactivation of surfactant with subsequent increased recoil pressure of the lungs.1,2,15,16 In addition, proteinaceous fluid fills the alveoli and impairs ventilation. Figure 23-3 shows alveolar damage attributable to ARDS. Alveoli contain dense proteinaceous debris, desquamated cells, and hyaline membranes. The decrease in PaO2 is a result of perfusion of large numbers of alveoli that are poorly ventilated (areas of low ventilation-perfusion ratio) or not ventilated (areas of shunt).
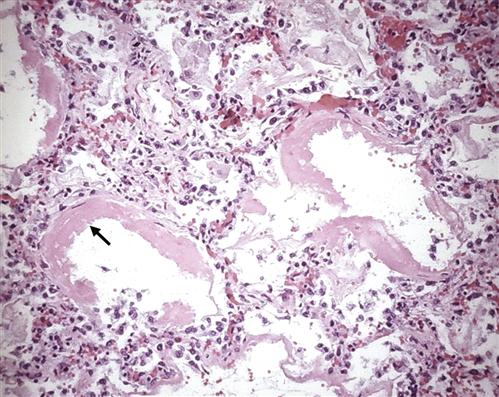
Some of the alveoli are collapsed; others are distended. Many contain dense proteinaceous debris, desquamated cells, and hyaline membranes (arrow). (From Kumar V et al, editors: Robbin’s basic pathology, ed 8, Philadelphia, 2007, Saunders, p 483.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


