Genetic and Developmental Disorders
Linda D. Ward
Key Questions
• How are genes transmitted from parent to offspring?
• How might abnormal meiosis lead to alterations in chromosome number or structure?
• What are the inheritance patterns and general clinical features of some common genetic disorders?
• What is the role of the environment in the development of congenital disorders?

http://evolve.elsevier.com/Copstead/
Geneticists and parents alike have marveled at the development of a recognizable human baby, with eyes and ears, toes and fingers, from its simple beginning as a single cell containing one set of genes. Considering the enormous list of potentially disastrous genetic and environmental influences, the birth of a healthy normal child does indeed seem like a miracle. Although the risk of bearing a child with mental or physical defects is small for most parents, it is real and is often a source of worry during the prenatal period. It has been estimated that most people harbor five to eight defective genes that are recessive and therefore of little consequence until they are transmitted to offspring.1 In addition, there are many known and unknown environmental hazards to which the parent and fetus may be exposed. Disorders that are present at birth are called congenital, whether the cause is genetic, environmental, or both. Some congenital disorders are associated with structural defects attributable to errors in fetal development and are called congenital malformations. It is estimated that about 3% of newborns have a major malformation of cosmetic or functional significance.2,3 Malformations are frequently associated with genetic causes; however, environmental influences (teratogens) also may adversely affect the developing fetus, and in half of cases a clear explanation for a malformation cannot be found. Approximately 30% to 40% of all birth malformations are associated with genetic factors—6% are associated with chromosomal abnormalities, 8% are associated with single-gene disorders, and 20% to 30% are most likely multifactorial.3 Some inherited genetic disorders do not become apparent until later in childhood or adulthood and therefore are not considered to be congenital. In this chapter, the general principles of inheritance; genetic and environmental causes of congenital disorders; and the principles of diagnosis, counseling, and gene therapy are described.
Principles of Inheritance
“Whom does the baby look like?” is frequently asked of new parents. It is common knowledge that traits tend to run in families, but Gregor Mendel, a nineteenth-century monk turned geneticist, was the first to notice that traits were transmitted in a predictable way from parent to offspring.4 Height, weight, skin color, eye color, and hair color are some of the physical traits that characterize an individual; other inherited traits contribute to risk for disease. Phenotype refers to the physical and biochemical attributes of an individual that are outwardly apparent. These traits are a result of the expression of the individual’s unique genetic makeup, or genotype. In humans, genes are organized into 46 different chromosomes that become visible under the microscope only during cell division (see Chapter 5).
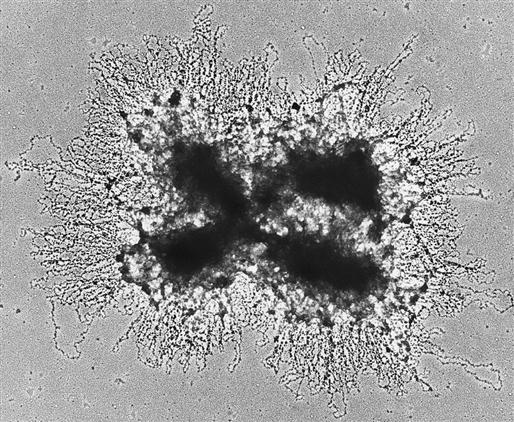
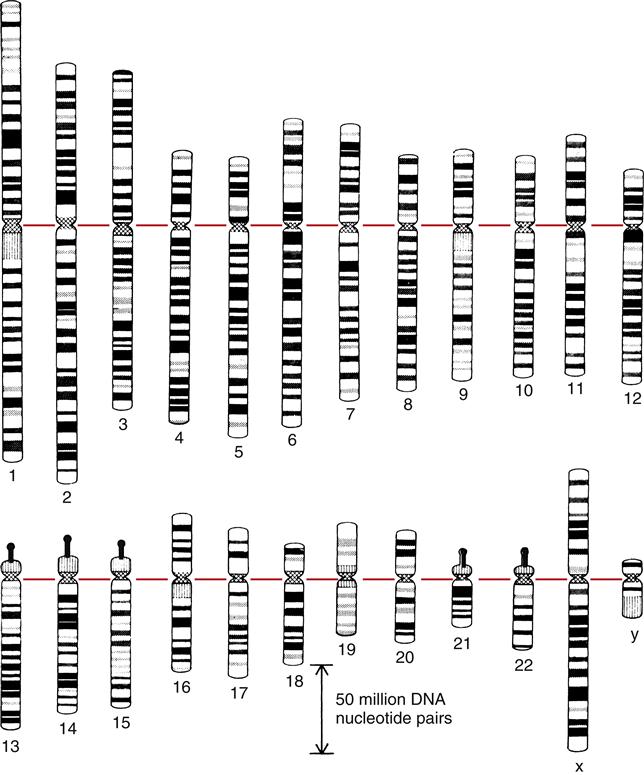
Before cell division, chromosomes look like X’s of varying sizes and shapes. The X-shaped chromosome is really made up of two identical linear chromosome units, called chromatids, which separate during meiosis. The point at the middle of the X at which the two sister chromatids are united is the centromere (Figure 6-1). Human chromosomes are diploid; they occur as pairs. One member of the pair comes from the mother, and one member comes from the father. Under the microscope the members of a pair appear to be identical (homologous), although they are different in DNA sequence. Chromosomes are characterized on the basis of total size, length of the arms of the X, and characteristic banding patterns when exposed to certain stains (karyotype) (Figure 6-2).
Of the 23 pairs of chromosomes, 22 are homologous and are called autosomes. The remaining pair, the sex chromosomes, differs in males and females. Females receive an X chromosome from each parent, whereas males receive an X chromosome from their mother and a Y chromosome from their father. Thus, the genotype is a result of the union of 23 maternal and 23 paternal chromosomes at conception. Sexual reproduction allows the mixing of genomes from two different individuals to produce offspring that differ genetically from one another and from their parents. This source of genetic variability is advantageous to the species because it allows for adaptation and evolution in a changing environment.
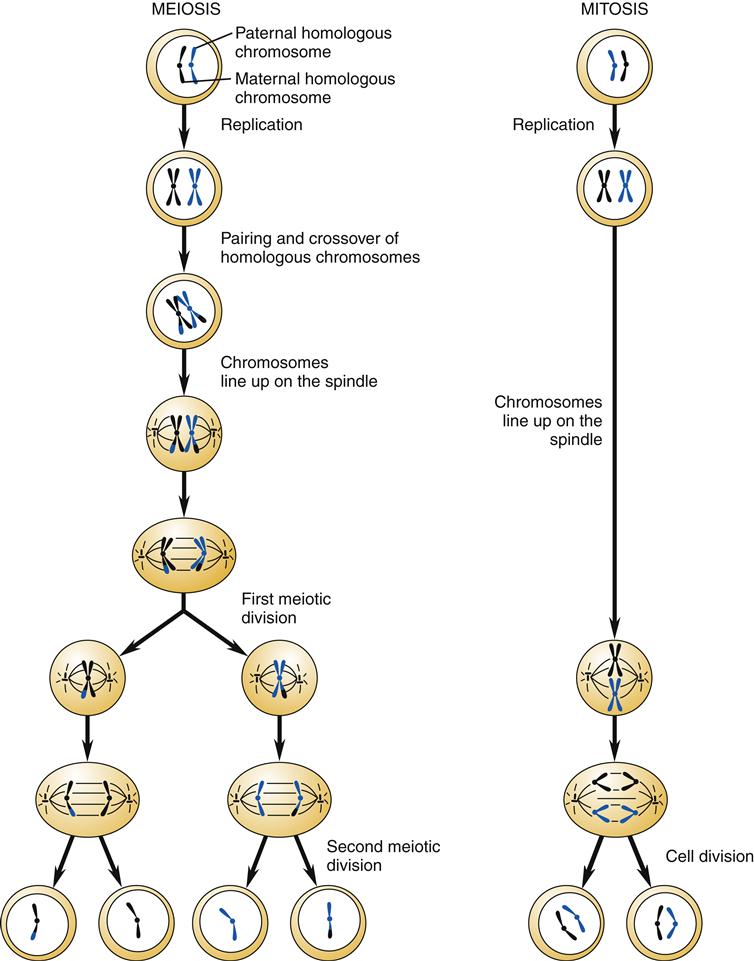
For the 2 germ cells (i.e., egg and sperm) to combine to form a cell with the normal complement of 46 chromosomes (23 pairs), each germ cell must contribute half of the total. Meiosis refers to a special form of cell division that results in germ cells that are haploid; they have half of the normal number of chromosomes. In contrast to mitosis (see Chapter 3), meiosis involves two divisions of chromosomal DNA. A comparison of meiotic and mitotic cell division is shown in Figure 6-3.
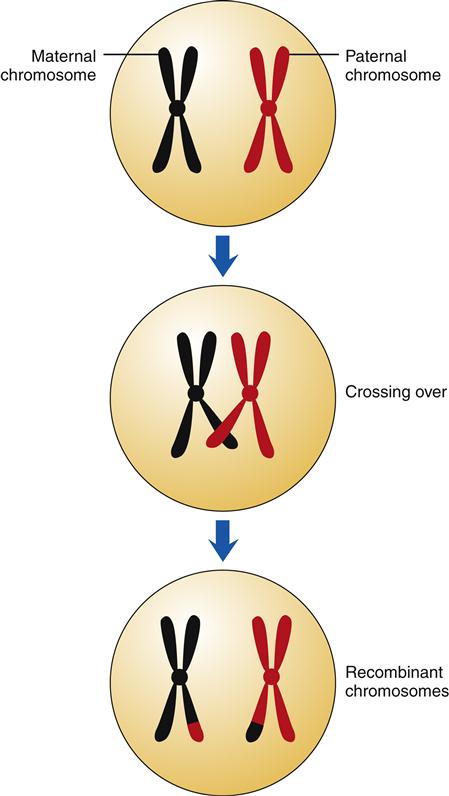
During the first phase of meiosis, pairs of homologous chromosomes with duplicated sister chromatids come in close contact. Portions of the homologous chromosomes are exchanged in a process called crossing over (Figure 6-4). This results in a mixing of the maternal and paternal genes of the cell to form a new combination of genes within the chromosomes. Genetic recombination is very precise, such that genes are exchanged intact and not interrupted in the middle. On average, each homologous pair of chromosomes has one to three crossover events occurring during the first meiotic division.5 The first cellular division of meiosis results in two cells, each with 46 chromosomes. These two cells undergo a second division in which the sister chromatids are pulled apart (similar to normal mitosis), resulting in four cells, each having only 23 chromosomes. Each of the germ cells has a different combination of genes that, when passed on through sexual reproduction, will form a new, genetically unique individual.
The genes that code for a particular gene product, such as an enzyme, are located at a particular position (locus) on the chromosome. Genes come in several forms, called alleles. A person has two alleles for each gene, one received from each parent. If both alleles are identical, the individual is said to be homozygous for that gene. If two different alleles are present, the individual is heterozygous.
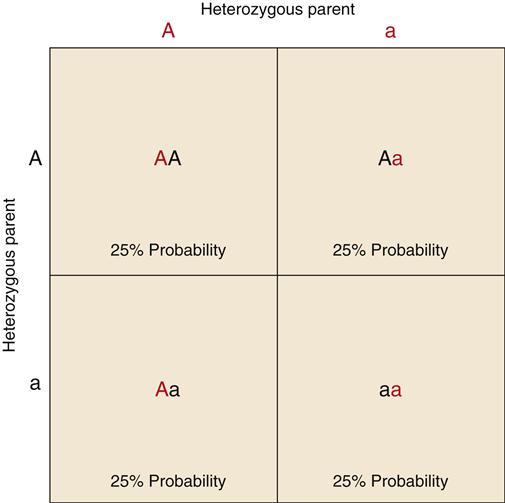
Some traits involve only one gene locus and are called single-gene (or monogenic) traits. The transmission of single-gene traits from parent to offspring follows predictable patterns that can be demonstrated using the Punnett square (Figure 6-5). In a Punnett square, alleles for a gene are represented by capital and lowercase letters. A capital letter is used for a dominant allele, and a lowercase letter represents a recessive allele. As the term implies, a dominant allele will mask a recessive allele, making the associated trait apparent. A recessive trait is apparent only if both alleles for the trait are recessive (homozygous). Dominant genes often code for functional enzymes or structural proteins, and recessive genes code for nonfunctional ones. The Punnett square is based on the mendelian principle that all genes are inherited independently from each other in a random manner. Thus, if both parents are heterozygous for a dominant trait (Aa), the offspring will have a 25% probability of being AA, a 50% probability of being Aa, and a 25% probability of being aa. Persons having the AA and Aa genotypes will express the trait in a similar manner. The trait will be absent in the aa genotype. Many genetic diseases are carried on the recessive allele and are manifested only by the homozygous (aa) genotype (e.g., cystic fibrosis, phenylketonuria). Persons who are heterozygous for the disease (Aa) are said to be carriers because they are able to pass the defective recessive gene to their offspring even though they do not exhibit the trait.
Some alleles are not clearly dominant or recessive and result in a blending or codominant expression of the trait. Blood type, for example, has three distinct alleles: A, B, and O. The A and B alleles may both be expressed together, resulting in the AB blood type. Most traits result from the interaction of several gene loci and are called polygenic. Polygenic traits are heritable, but predicting their occurrence is more difficult than with single-gene traits. Polygenic traits are often affected by environmental factors, in which case they are called multifactorial. Examples of multifactorial traits are height, weight, and blood pressure. These traits are influenced by multiple genes as well as environmental factors (such as dietary intake, which influences the ultimate expression of those genes). Most common diseases, such as heart disease, asthma, diabetes, and cancer, are multifactorial as well.
DNA Mutation and Repair
The term mutation refers to a permanent change in DNA structure. Genetic mutation is a rare event despite the daily exposure of cells to numerous mutagenic influences. Radiation, chemicals, viruses, and even some products of normal cellular metabolism are all potential mutagens. Most mutations, however, occur spontaneously as a result of copying errors during DNA replication.3 Regardless of the cause, only a few of these changes in DNA result in permanent alterations (mutations). The stability of the genes, and thus the low mutation rate, depends on efficient DNA repair mechanisms.
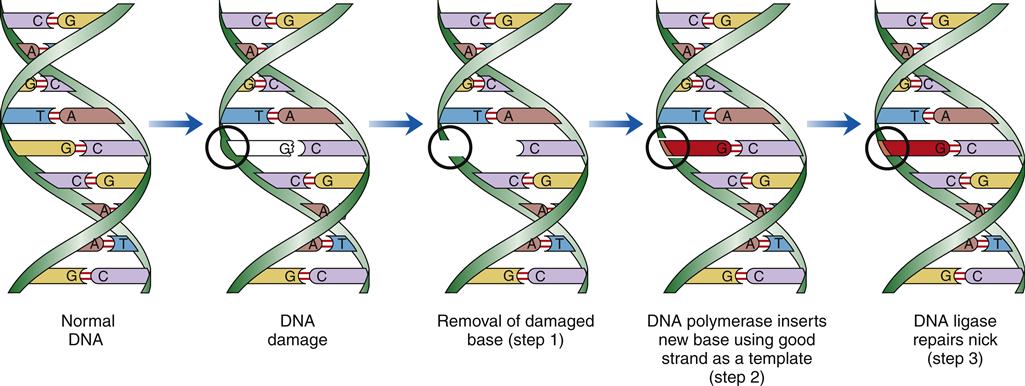
There are a variety of cellular DNA repair mechanisms. Most require the presence of a normal complementary DNA template to correctly repair the damaged strand of DNA. Single-stranded breaks or loss of bases from only one DNA strand are readily repaired. Double-stranded breaks, involving both strands of complementary DNA, may result in permanent loss of genetic information at the break point when the broken strands are reunited. Different types of DNA damage are detected and repaired by different enzyme systems. The steps in one type of DNA repair are shown in Figure 6-6.
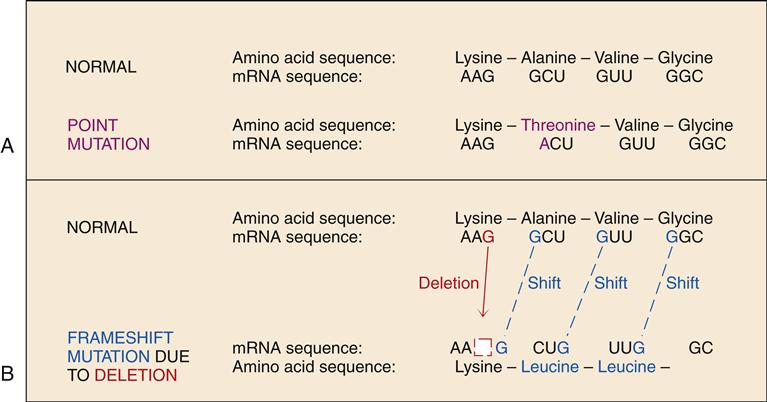
Genetic mutations are generally of two types: a point mutation, which involves a single base pair substitution, or a frameshift mutation, which often changes the genetic code dramatically. A sequence of three DNA bases (codon) is required to code for each amino acid. A point mutation in the gene may cause the affected codon to signify an abnormal amino acid. The inclusion of the abnormal amino acid in the sequence of the protein may or may not be of clinical significance. Sickle cell anemia and α1-antiprotease deficiency are examples of point mutation disorders in which a single amino acid substitution causes significant dysfunction. A frameshift mutation is due to the addition or deletion of one or more bases, which changes the “reading frame” of the DNA sequence. The DNA sequence is normally “read” in groups of three bases, with no spaces between codons. All of the codon triplets will be changed in the DNA downstream from a frameshift mutation, resulting in a protein with a greatly altered amino acid sequence (Figure 6-7). Numerous genetic disorders are due to mutations in a gene that codes for a particular protein. These are termed single-gene or mendelian disorders. Mutations may also alter chromosome structure through loss, gain, or translocation of chromosome segments. These processes are discussed later in the chapter.
Genetic Disorders
Genetic disorders may be apparent at birth or may not be clinically evident until much later in life. The majority of genetic disorders are inherited from the affected individual’s parents; however, new (de novo) mutations sometimes occur during gamete formation or arise during fetal development. Genetic disorders encountered clinically are only a small percentage of those that occur and represent the less extreme aberrations that permit live birth.
Disorders that are genetic in origin traditionally have been divided into three groups: (1) chromosomal aberrations, (2) mendelian single-gene disorders, and (3) polygenic or multifactorial disorders. A fourth group encompasses a number of single-gene defects that do not follow classic mendelian patterns of inheritance. This group includes triplet repeat (trinucleotide) mutations, mitochondrial gene mutations, and mutations influenced by genomic imprinting. General principles of transmission and selected examples are included for each of the four groups.
Chromosomal Abnormalities
Chromosomal defects are generally due to an abnormal number of chromosomes or alterations in the structure of one or more chromosomes. Errors in the separation of chromosomes during meiosis may result in abnormal numbers of chromosomes. These defects, as well as large structural defects (involving more than 5 to 10 million base pairs), can be detected using traditional genetic testing methods such as karyotyping.6 Chromosomal abnormalities occur commonly but are most often lethal; as many as 50% of spontaneous pregnancy losses (miscarriages) involve a chromosomal abnormality.1 Based on traditional chromosome analysis, 0.5% to 1% of newborn infants have been found to have some form of chromosomal abnormality.1,3 In recent years, the development of molecular testing methods has allowed detection of much smaller structural defects, increasing the detection rate of chromosomal abnormalities.6
Aberrant Number of Chromosomes
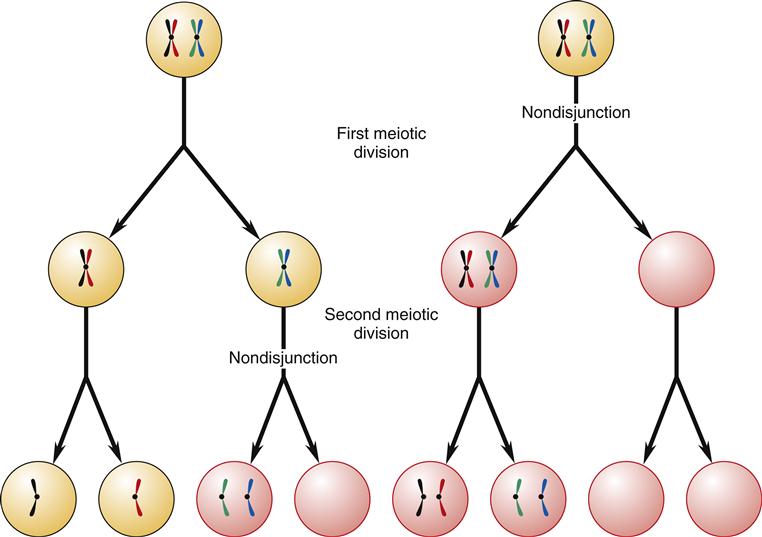
The union of sperm and egg results in a fertilized egg (zygote) with the full complement of 46 chromosomes: 22 pairs of autosomes and 2 sex chromosomes (euploid). Aneuploidy refers to an abnormal number of chromosomes—in humans, either more or less than 46. Aneuploidy is most commonly caused by nondisjunction.3 Nondisjunction means that paired homologous chromosomes fail to separate normally during either the first or the second meiotic division (Figure 6-8). The resulting germ cells then have an abnormal number of chromosomes: one germ cell will have 22 chromosomes and the other will have 24 chromosomes. When the abnormal germ cell combines with a normal germ cell containing 23 chromosomes, the resulting zygote will either be deficient by one chromosome (45) or have an extra chromosome (47). In anaphase lag, one chromosome lags behind and is therefore left out of the newly formed cell nucleus. This results in one daughter cell with the normal number of chromosomes and one with a deficiency of one chromosome, a condition called monosomy. Polysomy refers to the condition of having too many chromosomes.
The causes of aneuploidy are poorly understood; however, advanced maternal age, abnormalities in parental chromosome structure, and abnormalities in crossing over are known to increase the risk of abnormalities in chromosome number in humans.7 Aneuploidy occurs rather frequently during human gametogenesis; however, zygotes with abnormal chromosome numbers are usually nonviable. Approximately one third of human miscarriages involve an aneuploid fetus.8 Although monosomy involving the autosomes is not usually compatible with life, autosomal polysomy may result in a viable fetus when a single extra copy of a chromosome carrying relatively few genes is present. Severe disability, however, nearly always results (e.g., trisomy 21—Down syndrome). Disorders involving extra or missing sex chromosomes are more common and less debilitating.
Abnormal Chromosome Structure
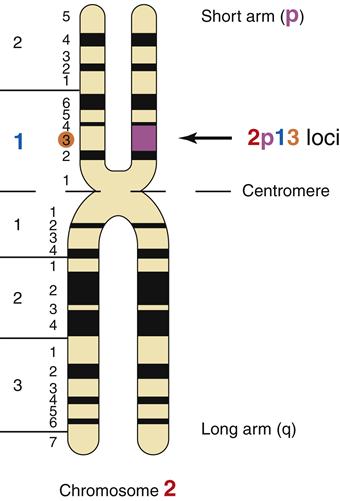
Alterations in chromosome structure are usually due to breakage and loss or rearrangement of pieces of the chromosomes during meiosis or mitosis. During meiosis, the homologous chromosomes normally pair up and exchange genetic alleles in a process called crossing over. Normal crossing over involves precise gene exchange between homologues only, with no net gain or loss of DNA. When the normal process of crossing over goes awry, portions of chromosomes may be lost, attached upside down, or attached to the wrong chromosome. Mitosis also presents opportunities for chromosomal breakage and rearrangement. The severity of the chromosomal rearrangement ranges from insignificant to lethal, depending on the number and importance of the gene loci involved. Gene locations can be described by their position on the long arm (q arm) or the short arm (p arm) of the chromatid. For example, the gene locus 2p13 is located on the short arm of chromosome 2 at region 1, band 3 (Figure 6-9). The common types of chromosomal rearrangements are translocations, inversions, deletions, and duplications (Figure 6-10).
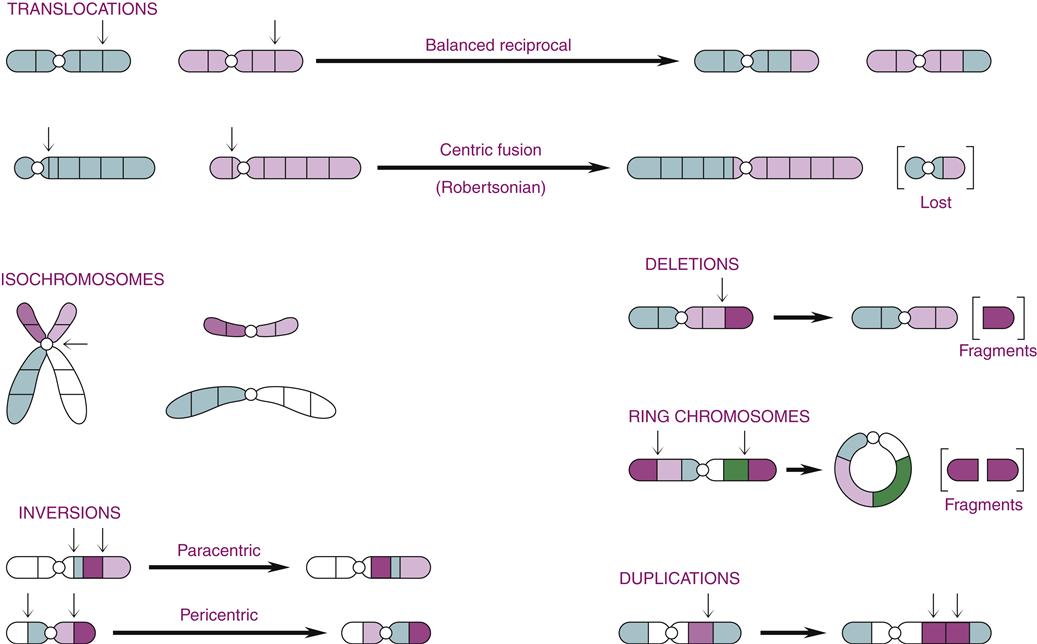
Chromosomal translocations result from the exchange of pieces of DNA between nonhomologous chromosomes. If no genetic material is lost, as in a reciprocal translocation, the individual may have no symptoms or disorder. However, an individual with a reciprocal translocation is at increased risk of producing abnormal gametes. The exchange of a long chromatid arm for a short one results in the formation of one very large chromosome and one very small chromosome (see Figure 6-10). This is called a robertsonian translocation and is responsible for a rare hereditary form of Down syndrome, discussed later in the chapter. Isochromosomes occur when the sister chromatids separate incorrectly at the centromere such that the two identical short arms remain together, as do the two long arms.
Inversion refers to the removal and upside-down reinsertion of a section of chromosome (see Figure 6-10). Like balanced translocations, inversions involve no net loss or gain of genetic material and are often without consequence to the individual. Difficulties result, however, when homologous chromosomes attempt to pair up during meiosis. The chromosome with an inverted section may not pair up properly, resulting in duplications or loss of genes at the time of crossing over. Thus, the offspring of an individual harboring an inversion may be affected.
Loss of chromosomal material is called deletion. Deletions result from a break in the arm of a single chromosome, resulting in a fragment of DNA with no centromere. The piece is then lost at the next cell division. Chromosomal deletions have been associated with some forms of cancer, including retinoblastoma (see Chapter 7). Deletions at both ends of a chromatid may cause the free ends to attach to one another, forming a ring chromosome.
In contrast to a deletion, where genes are lost, duplication results in extra copies of a portion of DNA. The consequences of duplications are generally less severe than those from loss of genetic material.
Examples of Autosomal Chromosome Disorders
Trisomy 21 (Down Syndrome)
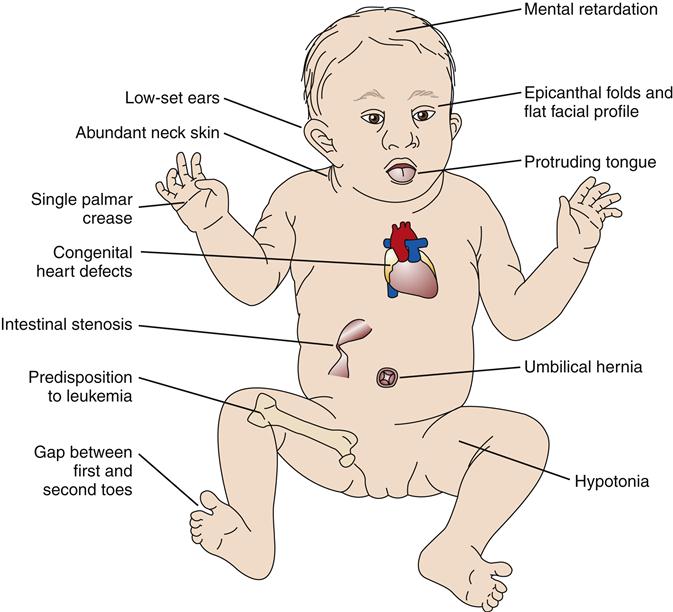
Trisomy 21 is a chromosomal disorder in which individuals have an extra copy of chromosome 21. It is the most common of the chromosomal disorders and a leading cause of mental disability, occurring in about 1 in 700 live births.3 The incidence varies among populations and over time, however, and reflects trends in maternal age, prenatal diagnosis, and termination of affected pregnancies.9 The syndrome, first described by Langdon Down in 1866,10 includes intellectual disability, protruding tongue, low-set ears, epicanthal folds, poor muscle tone, and short stature (Figure 6-11). Children with Down syndrome often are afflicted with congenital heart deformities and an increased susceptibility to respiratory tract infections and leukemia. Precise causes of these signs and symptoms are unknown, although the gene-dosage hypothesis relates them to overexpression of certain genes contained on chromosome 21.11
In 95% of cases, the extra chromosome 21 is thought to be of maternal origin, and the incidence of trisomy 21 is clearly associated with advanced maternal age.3 Table 6-1 demonstrates a rise in the incidence of Down syndrome from maternal age 20 to 50 years. The reason for increased susceptibility of the ovum to nondisjunction with age remains unknown.
TABLE 6-1
FREQUENCY OF TRISOMY 21 (DOWN SYNDROME) IN RELATION TO MATERNAL AGE
| AGE OF MOTHER AT BIRTH (YEAR) | FREQUENCY OF TRISOMY 21 AT BIRTH |
| 20 | 1/1470 |
| 25 | 1/1333 |
| 30 | 1/935 |
| 35 | 1/353 |
| 37 | 1/200 |
| 39 | 1/112 |
| 41 | 1/68 |
| 43 | 1/46 |
| 45 | 1/36 |
| 50 | 1/26 |
Data from Morris JK et al: Comparison of models of maternal age-specific risk for Down syndrome live births, Prenat Diagn 23:252-258, 2003.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


