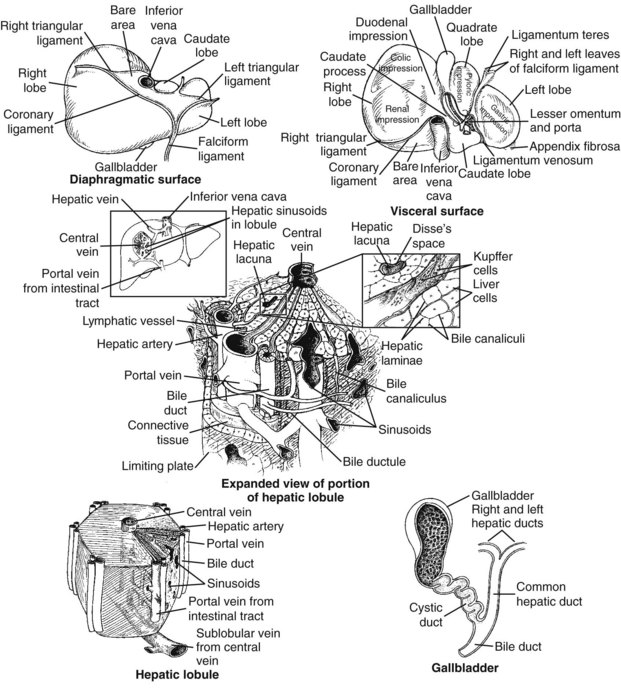
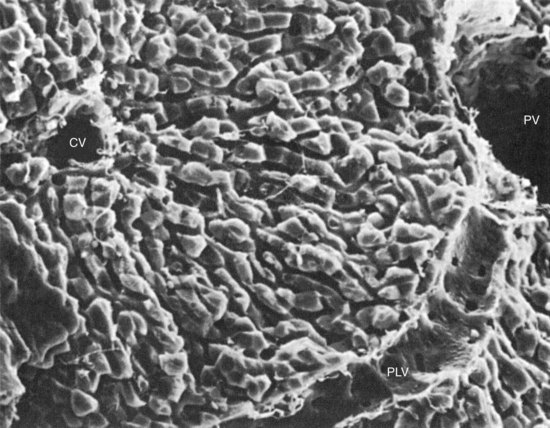
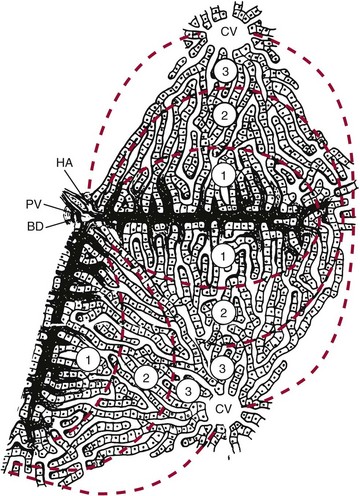
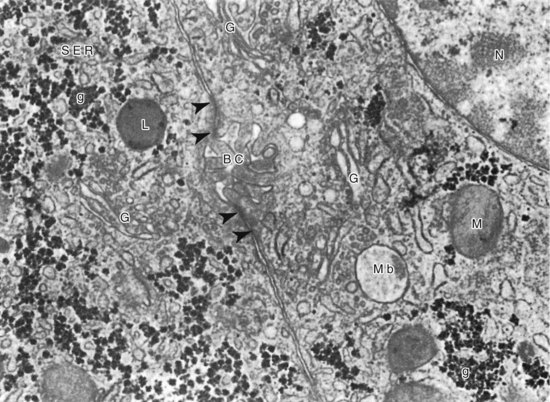
Chapter 50 The liver has a central and critical biochemical role in the (1) metabolism, (2) digestion, (3) detoxification, and (4) elimination of substances from the body. All blood from the intestinal tract initially passes through the liver, where products derived from digestion of food are processed, transformed, and stored. These include amino acids, carbohydrates, fatty acids, cholesterol, lipids, vitamins, and minerals (see Chapters 21, 26, 27, and 31 respetcively). Most major plasma proteins (with the exception of immunoglobulins and von Willebrand factor) are mainly or exclusively synthesized in the liver. The liver responds to multiple hormonal and neural stimuli to regulate the blood glucose concentration. Not only does it extract glucose from blood for use in generating energy, it also stores dietary glucose as glycogen for later use. The liver is also the major site for gluconeogenesis, which is critical for maintaining blood glucose in the fasting state. The liver is central in lipid metabolism; it extracts and processes dietary lipids, and it is the principal site of cholesterol, triglyceride, and lipoprotein synthesis. Another major liver function is the synthesis of bile acids from cholesterol with secretion of these compounds into the bile, facilitating the absorption of dietary fat and fat-soluble vitamins. The liver is also the primary site of metabolism of both endogenous substances and exogenous compounds, such as drugs and toxins. This process, known as biotransformation, converts lipophilic substances to hydrophilic ones for subsequent elimination. The liver is a major site of catabolism of hormones, and thus participates in regulation of plasma hormone concentrations. The liver is also involved in hormone synthesis, producing such hormones as insulin-like growth factor 1, angiotensinogen, hepcidin, thrombopoietin, erythropoietin, and the prohormone 25-OH vitamin D. Many of these hepatic functions may be assessed by laboratory procedures to gain insight into the integrity of the liver. The liver is divided into left and right anatomic lobes by the falciform ligament, an anterior extension of the peritoneal folds that connects the liver to the diaphragm and the anterior abdominal wall (Figure 50-1). Two smaller lobes are found on the posterior surface (caudate lobe) and the inferior surface (quadrate lobe) of the right lobe. Riedel’s lobe, an anatomic extension of the right lobe of the liver, consists of a projection that may feel like a mobile tumor in the right abdomen. Biliary drainage originates at the bile canaliculi; these grooves between adjacent hepatocytes form ductules that merge to create the intrahepatic bile ducts, which ultimately join to form the right and left hepatic bile ducts, which exit from the liver at the porta hepatis and combine to form the common hepatic duct. The hepatic duct is joined by the cystic duct draining the gallbladder to form the common bile duct (see Figure 50-1). The common bile duct then enters the duodenum (usually with the pancreatic duct) at the ampulla of Vater. The duodenal portion of the common bile duct is surrounded by longitudinal and circular muscle fibers that form the sphincter of Oddi. This musculature relaxes when the gallbladder contracts, allowing bile to enter the duodenum; in its normally contracted state, the sphincter prevents reflux of acidic duodenal contents into the bile duct. The gallbladder, located on the undersurface of the right lobe of the liver, is the site for storage and concentration of bile, a complex mixture of bile salts and waste products. In the adult, it averages about 10 cm in length and has a capacity of 30 to 50 mL of bile. Hormonal stimuli initiated by food ingestion cause contraction of the muscular wall of the gallbladder, releasing bile salts into the intestine to facilitate digestion of fat. The functional anatomical unit of the liver is the acinus, adjacent to the portal triad (which consists of a branch of the portal vein, hepatic artery, and bile duct). Each acinus is a diamond-shaped mass of liver parenchyma that is supplied by a terminal branch of the portal vein and of the hepatic artery and is drained by a terminal branch of the bile duct. The blood vessels radiate toward the periphery, forming sinusoids, which perfuse the liver and ultimately drain into the central (terminal) hepatic vein (Figure 50-2). The sinusoids are lined by fenestrated endothelial cells (allowing free filtration of blood) and phagocytic Kupffer cells (see Figure 50-1). The Kupffer cells are derived from blood monocytes. They contain lysosomes with hydrolytic enzymes that break down phagocytized foreign particles, such as bacteria. They also have immunoglobulin and complement receptors and are the main site for clearance of antigen-antibody complexes from blood. Kupffer cells secrete interleukins, tumor necrosis factor, collagenase, prostaglandins, and other factors involved in the inflammatory response. Hepatocytes are the major functioning cells in the liver and are responsible for about 70% of liver mass and perform most of the metabolic and synthetic functions of the liver. Two other cell types are found in small numbers within the liver. The stellate cells (sometimes referred to as Ito cells) are located between the endothelial lining of the sinusoids and the hepatocytes within a small cleft referred to as the space of Disse. In their normal, quiescent state, stellate cells serve as a site of storage for fat-soluble vitamins, particularly vitamin A. When stimulated, stellate cells are morphologically and functionally transformed. They synthesize collagen and are the cells responsible for fibrosis and, eventually, cirrhosis. They also synthesize nitric oxide, which helps to regulate intrahepatic blood flow. Oval cells, found near the portal areas around small bile passages, are believed to be liver stem cells involved in regeneration of hepatocytes and bile ducts after liver injury.268 The blood supply to each acinus consists of three zones (Figure 50-3). Zone 1, the area immediately adjacent to the portal tract, is enriched with lysosomes and mitochondria. The periphery of the acinus, zone 3, is enriched with endoplasmic reticulum, is very active metabolically, and has relatively low oxygen tension. This area is most susceptible to injury, although zone 1 appears to be involved in protecting the liver from external injury and providing a base for hepatic regeneration. Hepatocytes contain a well-developed organelle substructure (Figure 50-4). Mitochondria, which constitute approximately 18% of hepatocyte volume, are the sites of oxidative phosphorylation and energy production. They contain enzymes involved in the citric acid cycle and in β-oxidation of fatty acids. The rough endoplasmic reticulum is the site of synthesis of many proteins, including albumin, coagulation factors, enzymes (e.g., glucose-6-phosphatase), and triglycerides. The smooth endoplasmic reticulum contains microsomes that are involved in bilirubin conjugation, detoxification (cytochrome P450–dependent isoenzymes), steroid synthesis, cholesterol synthesis, and bile acid synthesis. Several microsomal enzymes, including γ-glutamyltransferase, are induced by many drugs and inhibited by others. This is the site of most drug metabolism and many important drug interactions. Bilirubin is the orange-yellow pigment derived from heme, mainly as a product of red blood cell turnover. It is extracted and biotransformed in the liver and excreted in bile and urine. The chemistry, biochemistry, and analytical methodsology for bilirubin and related compounds are discussed in Chapter 32; a brief overview of factors relevant to an understanding of liver disease is included here. Increased direct bilirubin generally results from functional or mechanical impairment in bilirubin excretion from the hepatocyte. Increased conjugated bilirubin is found in most cases of acute hepatitis and cholestasis (stoppage or suppression of the flow of bile); the percentage of direct bilirubin is similar in both types of liver disease.42 Urine bilirubin reflects increased plasma concentrations of conjugated bilirubin. With resolution of liver disease, conjugated bilirubin is rapidly cleared, and biliprotein may become the only form present; urine bilirubin is typically absent in such circumstances. Increased conjugated bilirubin is rarely seen with congenital defects in bilirubin excretion, such as Dubin-Johnson syndrome, and with impaired bilirubin excretion, as occurs in sepsis or other acute illness. Four major bile acids are known. Cholic acid and chenodeoxycholic acid, the primary bile acids, are synthesized in the liver. The sequence of reactions involved in the synthesis of cholic acid from cholesterol is shown in Figure 50-5. To date, nine inborn errors of bile acid synthesis have been identified; these can present with neonatal hepatitis, fat malabsorption, or neurologic defects that can progress to chronic liver disease or liver failure and death.192 The primary bile acids are metabolized (by bacterial 7-α-dehydroxylase) in the intestinal lumen to the secondary bile acids—deoxycholic acid and lithocholic acid. Bile acids (at their carboxylic acid) are conjugated in the liver with the amino acid glycine or taurine. This decreases passive absorption in the biliary tree and proximal small intestine, but permits conservation through active transport in the terminal ileum. Approximately 0.1 to 0.6 g of bile acids is lost in the feces daily. In view of the multiple processes involved in bile acid synthesis, conjugation, and excretion, and in its hepatic and intestinal uptake, several potential sites for primary or secondary disturbances have been identified (Box 50-1). With hepatocyte dysfunction (as occurs in many liver disorders), decreased bile acid synthesis results in low primary bile acid concentrations and a decreased ratio of primary to secondary bile acids in plasma; in addition, decreased extraction from plasma often leads to increased concentrations of bile acids, particularly in the nonfasting state. With cholestatic disorders, decreased delivery of primary bile acids to the intestine with resulting decreased secondary bile acid production causes an increased ratio of primary to secondary bile acids, as well as increased total bile acid concentrations. With intestinal disease (including bypass operations that may be performed to treat obesity), increased fecal loss of bile acids leads to decreased concentrations of both primary and secondary bile acids and, often, a decrease in cholesterol caused by increased need for bile acid synthesis. Although bile acid concentrations are abnormal in many situations, they add little to standard tests of liver function and are rarely used in clinical medicine. The liver has extensive synthetic capacity and plays a major role in the regulation of protein, carbohydrate, and lipid metabolism (see Chapters 21, 26, and 27). A bidirectional flux of precursors and products, such as glucose, amino acids, free fatty acids, and other nutrients, occurs across the hepatocyte membrane. Normal blood glucose concentrations are maintained during short fasts by the breakdown of hepatic glycogen and during prolonged fasts by hepatic gluconeogenesis. The primary sources of carbon atoms for gluconeogenesis are amino acids derived from muscle proteins. To a lesser extent, lactate (produced in skeletal muscle and erythrocytes) and glycerol (obtained from hydrolysis of triglycerides) also serve as substrates for gluconeogenesis. In humans, the oxidation of odd-numbered fatty acids yields propionyl-CoA, which can be converted to glucose. However, the formation of glucose in this manner is not quantitatively significant. Protein, triglyceride, fatty acid, cholesterol, and bile acid synthesis also occurs within the liver. The liver is the primary site of the synthesis of plasma proteins (see Chapter 21). Synthesis occurs in the rough endoplasmic reticulum of hepatocytes, followed by release into the hepatic sinusoids. Although disturbances of protein synthesis occur as a consequence of impaired hepatic function, a variety of other factors may affect plasma protein concentrations. These include (1) decreased availability of amino acids (malnutrition, maldigestion, and malabsorption), (2) catabolic states (hyperthyroidism, Cushing’s syndrome, burns, postsurgery recovery), (3) protein-losing states (nephrotic syndrome and protein-losing enteropathy), (4) actions of cytokines (decrease in transport proteins such as albumin, transferrin, and lipoproteins, but increase in inflammatory response modifiers such as α1-antitrypsin, ceruloplasmin, and α2-macroglobulin), (5) action of hormones (such as growth hormone, cortisol, estrogen, androgens, and thyroid hormones) to increase or decrease production of specific proteins, and (6) congenital deficiency states (Wilson’s disease and α1-antitrypsin deficiency). In addition, the liver has a significant reserve capacity, preventing protein concentrations from decreasing unless liver damage is extensive. In addition, many liver proteins have relatively long half-lives, such as albumin at approximately 3 weeks. For this reason, the sensitivity and specificity of protein concentrations for diagnosis of liver disease are far from ideal. Albumin: Albumin, the most commonly measured serum protein, is synthesized exclusively by the liver. The rate of synthesis varies, depending on hormonal environment, nutritional status, age, and other local factors. In inflammatory conditions, interleukin (IL)-6 inhibits albumin synthesis but induces synthesis of acute-phase response proteins. With liver disease, hypoalbuminemia is noted primarily in cirrhosis, autoimmune hepatitis, and alcoholic hepatitis. The mechanism is multifactorial. In cirrhosis, hepatic synthesis of albumin may be decreased, normal, or increased. Loss of albumin into ascitic fluid seems responsible for the decrease in albumin in many cases. One important consideration in measurement of albumin is the inaccuracy of dye-binding methods in patients with liver disease. Although bromocresol green measurements tend to overestimate albumin concentration at low concentrations,293 bromocresol purple methods give falsely low values in patients with jaundice because of interference of bilirubin at the site of binding.207 Transthyretin: This protein has a short half-life of 24 to 48 hours, making it a sensitive indicator of current synthetic ability. Transthyretin is typically decreased in cirrhosis (among other conditions) as a result of decreased synthesis. It is more commonly used as a measurement of nutritional status. Immunoglobulins: Immunoglobulins are commonly increased in cirrhosis, autoimmune hepatitis, and primary biliary cirrhosis but are normal in most other types of liver disease. Immunoglobulin (Ig)G is increased in autoimmune hepatitis and cirrhosis; IgM is increased in primary biliary cirrhosis. IgA tends to be increased in all types of cirrhosis. None of these findings are specific, and they are seldom used in the diagnosis of liver disease. Ceruloplasmin: This protein is decreased in Wilson’s disease, cirrhosis, and many causes of chronic hepatitis, but may be increased by inflammation, cholestasis, hemochromatosis, pregnancy, and estrogen therapy. It is discussed in greater detail under Wilson’s disease. α1-Antitrypsin: This protein, which is the major serine protease inhibitor (serpin) in plasma, is decreased in homozygous deficiency and cirrhosis and is increased by acute inflammation. It is discussed in greater detail later in this chapter under the heading “α1-Antitrypsin Deficiency.” α-Fetoprotein: This protein, a normal component of fetal blood, falls to adult concentrations by 1 year of age. Mild increases are seen in patients with acute and chronic hepatitis and indicate hepatocellular regeneration. It is present at higher concentrations in hepatocellular carcinoma (HCC) and is discussed in greater detail later and in Chapters 21 and 24. The coagulation proteins that are synthesized in the liver are listed in Table 50-1. These proteins interact to produce a fibrin clot (see Chapter 59). Inhibitors of the coagulation system, including antithrombin, protein C, and protein S, are also synthesized in the liver. Some of the coagulation factors (II, VII, IX, and X) require vitamin K for post-translational carboxylation within the hepatocyte. Proteins C and S are also carboxylated by a vitamin K–dependent enzyme. Activated protein C in plasma inhibits coagulation by inactivating factors V and VIII. Parenchymal liver disease of sufficient severity to impair protein synthesis or obstructive liver disease sufficient to impair intestinal absorption of vitamin K is therefore a potential cause of bleeding disorders. Because of the great functional reserve of the liver, failure of hemostasis usually does not occur except in severe or long-standing liver disease. TABLE 50-1 *Protein synthesized in liver. Prothrombin time (PT) measures the activity of fibrinogen (factor I), prothrombin (factor II), and factors V, VII, and X. Because all of these factors are made in the liver and several are vitamin K dependent, a prolonged PT often indicates the presence of significant liver disease. In cholestasis, vitamin K deficiency may cause an increase in PT. In this case, the coagulation abnormality is corrected within a few days by parenteral injection of 10 mg of vitamin K. In contrast, if PT is prolonged because of hepatocellular disease, factor synthesis is decreased and administration of vitamin K does not typically correct the problem. PT is also prolonged in some patients with liver disease because of the presence of dysfibrinogenemia, an abnormal form of fibrinogen that does not clot normally, and that may predispose to thrombosis.188 INR has been found to standardize interpretation of PT measurements between laboratories in those taking warfarin. Unfortunately, INR does not have the same relationship to impairment of clotting in individuals with liver disease.216,235 The apparent explanation lies in the mechanism of clotting factor deficiency in liver disease and warfarin administration. Although liver disease inhibits synthesis of clotting factors, warfarin impairs vitamin K–dependent carboxylation, impairing the ability of the factors to bind calcium. These noncarboxylated clotting factors (termed proteins induced by vitamin K antagonists, or PIVKAs) appear to act as inhibitors of coagulation; thus when lower amounts of tissue factor are present, clotting times are more prolonged.235 In contrast, in liver disease, factor deficiency is due to impaired factor synthesis, and no PIVKAs are present (except in HCC, as discussed later in this chapter). This leads to lesser increases in PT in individuals with liver disease and underestimation of the degree of clotting impairment when reagents with low ISI are used. Studies have shown that calculation of a different ISI using plasma samples from patients with liver disease can standardize PT results with different reagents,33,454 but to date, such “liver ISI” information is not readily available to laboratories. The liver plays a key role in the metabolism of lipids and lipoproteins (see Chapter 27). On a daily basis, approximately 33% of the fatty acids originating from adipose tissue enter the liver, where they undergo esterification into triglycerides or are oxidized. Oxidation is favored in the fasting state and esterification is favored in the nonfasting state. Excessive esterification results in “fatty liver,” a disorder in which excess triglycerides are deposited in large vacuoles that displace other cellular components. Most cholesterol is endogenously synthesized in the liver. It and cholesterol of dietary origin enter the hepatic pool, where they are converted to bile acids, incorporated into lipoproteins, or used in the synthesis of liver cell membranes. The relative rates of secretion of bile acids, cholesterol, and lecithin are important factors in the pathogenesis of cholesterol gallstones. Patients with end-stage liver disease may have low concentrations of urea in plasma (see Chapter 48). The rate of urea excretion in urine is lower than in healthy individuals. In addition, plasma concentrations are elevated for the urea precursors—ammonia and amino acids. Lower specific activities of enzymes involved in urea synthesis are also seen. These findings suggest that patients with liver disease have an impaired ability to metabolize protein nitrogen and to synthesize urea. The rate of hepatic urea synthesis also depends on exogenous intake of nitrogen and on endogenous protein catabolism. The major source of circulating ammonia is the GI tract. Plasma ammonia concentration in the hepatic portal vein is typically fivefold to tenfold higher than that in the systemic circulation. It is derived from the action of bacterial proteases, ureases, and amine oxidases on the contents of the colon and from the hydrolysis of glutamine in both the small and large intestines. Under normal circumstances, most of the portal vein ammonia load is metabolized to urea in hepatocytes in the Krebs-Henseleit (urea) cycle during the first pass through the liver; this process includes intramitochondrial and cytosolic enzyme-catalyzed steps (Figure 50-6). Animal and human studies have shown that an elevated concentration of ammonia (hyperammonemia) exerts toxic effects on the central nervous system. Several causes, both inherited and acquired, of hyperammonemia are known. Inherited deficiencies of urea cycle enzymes are the major cause of hyperammonemia in infants.55 The two major inherited disorders are those involving the metabolism of the dibasic amino acids lysine and ornithine and those involving the metabolism of organic acids, such as propionic acid, methylmalonic acid, isovaleric acid, and others (see Chapter 58). The fasting venous plasma ammonia concentration is useful in the differential diagnosis of encephalopathy when it is unclear whether encephalopathy is of hepatic origin.126 It is especially helpful in diagnosing Reye’s syndrome and the inherited disorders of urea metabolism, as well as increased ammonia concentrations due to drugs such as salicylates or valproate. In acute liver injury, ammonia concentrations greater than 200 µmol/L are associated with cerebral edema and a poor prognosis,83 and it has been suggested that ammonia concentrations should be used as part of the evaluation of prognosis in acute liver failure.37 However, plasma ammonia is not useful in patients with known chronic liver disease.26 Although ammonia concentrations are higher as the degree of encephalopathy worsens, significant overlap between concentrations is seen in different stages of encephalopathy, and about 70% of those with cirrhosis without encephalopathy have elevated ammonia concentrations.350 Ammonia concentrations may actually better reflect the presence of shunting blood around the portal veins than the degree of liver dysfunction.441 Because the liver is a major processor of dietary and endogenous carbohydrates, liver disease affects carbohydrate metabolism in a variety of ways (see Chapter 26). However, none of the conventional modes of evaluating carbohydrate metabolism have value in the diagnosis of liver disease. Because the liver is the major site of both glycogen storage and gluconeogenesis, hypoglycemia is a common complication in certain liver diseases, particularly Reye’s syndrome, fulminant hepatic failure, advanced cirrhosis, and hepatocellular carcinoma. Xenobiotics are chemical substances that are foreign to the biological system. Biochemically, they are cleared and/or metabolized by the liver; some have been used as tests of liver function. Rates of metabolism of these compounds are sometimes referred to as quantitative liver function tests, to distinguish them from the more commonly used term, liver function tests, often used to refer to measurements of liver-associated enzymes. As liver disease progresses, quantitative liver function test results gradually worsen, and their measurement adds slightly to that obtained by widely used tests such as bilirubin, albumin, and INR measurement.130 Even when these tests are used, significant overlap of values is noted in persons with cirrhosis and less severe degrees of liver scarring, limiting their utility. A variety of drugs that are metabolized by the liver have been used to study the action of various P450 enzymes. Aminopyrine is demethylated to form carbon dioxide and aminoantipyrine. With the use of 13C- or 14C -labeled aminopyrine, the resulting isotopically labeled CO2 is measured in breath as a reflection of functioning liver mass. Decreases in metabolism are common in persons with cirrhosis,162 but metabolism is also affected by other factors such as cigarette smoking and use of drugs such as oral contraceptives; significant intraindividual variation in results has been noted.63 Overall diagnostic sensitivity is similar to that of other more routine laboratory tests.340 Caffeine is rapidly and nearly completely absorbed from the GI tract and then undergoes N-demethylation by the hepatic mixed-function oxidase system. Caffeine clearance is altered during hepatic injury, being prolonged in both chronic hepatitis and cirrhosis.484 A single dose of caffeine (3.5 mg/kg to a maximum dose of 200 mg, dissolved in water, fruit juice, or milk for oral administration) is administered. This caffeine dose is equivalent to that found in one cup of brewed coffee or in one can of commercial soft drink. Blood (or salivary samples) obtained before and at timed intervals after caffeine ingestion can be analyzed by reversed-phase HPLC or immunoassay. A close correlation is found between plasma and salivary caffeine concentrations. Caffeine half-life is approximately 5.5 hours in healthy adults and 3 hours in healthy children, with clearance of approximately 2 mL/min/kg in healthy adults and 10 mL/min/kg in healthy children. Caffeine clearance correlates with the aminopyrine breath test and has similar limitations, although it is less subject to effects of variables, such as smoking and oral contraceptive use.63 Lidocaine undergoes N-de-ethylation in the liver by cytochrome P450 to form monoethylglycinexylidide (MEGX); the rate of appearance of MEGX in plasma reflects hepatic lidocaine clearance. Because lidocaine is highly extracted, its clearance is flow dependent. Thus alterations in hepatic blood flow also influence lidocaine elimination.502 Lidocaine (1 mg/kg) is given by intravenous bolus; plasma is obtained at baseline and at 15 minutes for MEGX concentration (time of plateau concentration in healthy individuals). MEGX is most commonly measured using immunoassay. Lidocaine clearance has been used to assess liver transplant function, but its use is limited by the effect of hypoperfusion (as occurs in sepsis or volume depletion).155 Jaundice (or icterus) is a physical sign characterized by a yellow appearance of the skin, mucous membranes, and sclera caused by bilirubin deposition. It is the most specific clinical manifestation of hepatic dysfunction, but is not present in many individuals with liver disease (especially chronic liver disease) and may occur in states of bilirubin overproduction (such as hemolysis). Jaundice is seen most easily in the sclera of the eyes, where yellow contrasts sharply with the usual bright white color. Jaundice is usually apparent clinically when the plasma bilirubin concentration reaches 2 to 3 mg/dL (34 to 51 µmol/L), although higher concentrations may be required when fluorescent lighting is used. When bilirubin clearance from the liver to the intestinal tract is impaired (as in acute hepatitis and bile duct obstruction), acholic (gray-colored) stools may be noted. Bilirubin is the source of stercobilin, which produces the brown color of normal stool. Increases in plasma conjugated bilirubin lead to tea-colored urine, because conjugated bilirubin is water soluble. Jaundice may also be due to disorders of bilirubin metabolism. Bilirubin metabolism is discussed more fully in Chapter 32. Classification of jaundice, based on the site of altered bilirubin metabolism, is shown in Box 32-1 of that chapter. The portal circulation handles all venous outflow of the GI tract, the spleen, the pancreas, and the gallbladder (Figure 50-7). The portal vein is formed by the union of the splenic vein and the superior mesenteric vein. Portal flow is normally 1000 to 1200 mL/min with pressure of 5 to 7 mm Hg. Portal hypertension occurs when portal flow is obstructed anywhere along its course. Causes of obstruction leading to portal hypertension are classified by site: (1) presinusoidal, (2) sinusoidal, and (3) postsinusoidal. Presinusoidal portal hypertension is most commonly caused by portal vein thrombosis or schistosomiasis but may also occur with increased portal flow, such as occurs with Felty syndrome (a combination of chronic rheumatoid arthritis, splenomegaly, leukopenia, pigmented spots on the lower extremities, and sometimes other evidence of hypersplenism, such as anemia and thrombocytopenia). Sinusoidal hypertension is most commonly caused by cirrhosis but may occur transiently with acute and chronic hepatitis or acute fatty liver. The most important cause of postsinusoidal hypertension is hepatic vein occlusion or Budd-Chiari syndrome,149A in which sudden obstruction or occlusion of the hepatic veins (associated with myeloproliferative disorders in half of cases) causes hepatomegaly, abdominal pain, severe ascites, mild jaundice, and eventually portal hypertension and liver failure.364 The most common cause of postsinusoidal hypertension is cardiac disease, most commonly congestive heart failure. Chronic congestive heart failure is usually associated with portal hypertension and ascites, and may even lead to increased activities of aminotransferases.323 Other causes include abscesses, membranous obstruction of the vena cava, and veno-occlusive disease (as may be seen in patients following bone marrow transplantation). Although increased portal resistance is the major factor causing portal hypertension, it is often accompanied by decreased resistance to blood flow through other blood vessels, which enhances blood flow through the portal veins. When portal pressure increases, the portal venous system becomes dilated and forms collateral connections to the systemic venous flow (Figure 50-8), leading to portosystemic shunting. Initially, this is clinically silent, but as portal hypertension worsens, it compromises many of the metabolic functions of the liver. One such abnormality is altered estrogen metabolism, which increases the ratio of estrogen to testosterone. Clinical consequences include spider telangiectasias and palmar erythema, gynecomastia in men, and abnormal vaginal bleeding and irregular menstrual periods in women. Impaired protein metabolic functions cause the accumulation of ammonia and abnormal neurotransmitters, ultimately leading to hepatic encephalopathy.60A Because most nutrients arrive through the portal vein, synthetic functions are also impaired, leading to hypoalbuminemia (contributing to ascites), decreased clotting factors (predisposing to bleeding), and reduced thrombolytic factors, such as antithrombin (predisposing to venous thrombosis). The most life-threatening consequence of portosystemic shunting is the development of varices (enlarged and tortuous veins), which can occur throughout the GI tract but are most common in the esophagus and stomach. Bleeding from varices is one of the leading causes of morbidity and mortality in patients with cirrhosis. Varices are present at the time of diagnosis of cirrhosis in about 40% of patients and occur in an additional 6% per year.156 In general, the risk of bleeding is low until portal vein pressure exceeds 12 mm Hg. The major consequences of varices are rupture and bleeding, usually presenting as hematemesis. Treatment of portal hypertension and varices is directed at obliterating the dilated blood vessels or reducing portal pressure. Pressure can be reduced by pharmacologic agents, such as nonselective β-adrenergic blockers, but if this is not effective, invasive procedures can be used, most commonly by putting rubber bands around large varices (“banding”) or, if not successful, by placing a stent through the jugular and hepatic veins to connect to the portal vein, called transjugular intrahepatic portosystemic shunt (TIPS).46 Because portal flow is already significantly reduced before shunting, minimal change in liver function is usually seen, but the incidence of hepatic encephalopathy following shunts is markedly increased. Ascites has many causes, and it is important to differentiate ascites secondary to portal hypertension from ascites due to other causes. This is done by analyzing ascitic fluid. The feature that best distinguishes portal hypertension is an increase in the serum-ascites albumin gradient (sometimes abbreviated as SAAG). A gradient greater than 1.1 g/dL (11 g/L) is characteristic of ascites caused by portal hypertension,5 but a high SAAG can also be seen in congestive heart failure or nephrotic syndrome. Ascites due to portal hypertension is managed by creating a negative sodium balance or by relieving portal hypertension. A negative sodium balance is obtained by reducing sodium intake and enhancing sodium excretion with the use of diuretics. In cirrhosis, activation of the renin-aldosterone axis (caused by a variety of factors) necessitates use of agents that act at the distal nephron as the primary diuretic used, but these can be combined with other diuretics that act more proximally. In patients who require more urgent treatment for relief of symptoms, or who do not respond adequately to diuretics, ascitic fluid may be removed with a catheter placed percutaneously through the abdominal wall. More than 10 L of fluid may be drained to relieve patient discomfort or respiratory compromise. Repeated or large-volume paracentesis also requires plasma volume expansion to prevent renal failure; albumin is effective as an expander.315 Ascites predisposes to spontaneous bacterial peritonitis, defined as bacteremia (typically gram negative), in the absence of mechanical disruption of the bowel.234 The condition usually presents in an individual with known cirrhosis who develops abdominal pain, fever, or leukocytosis. The diagnosis is established by examination of the ascitic fluid; greater than 250 neutrophils per microliter, or more than 500 in the absence of a positive blood culture, is considered diagnostic. In contrast, secondary peritonitis is usually associated with higher neutrophil counts, along with high protein and low glucose in ascitic fluid. Several studies have suggested that dipsticks that detect the presence of leukocyte esterase could be used to identify increased leukocytes and to diagnose spontaneous bacterial peritonitis; however, a recent review found poor sensitivity and frequent false-negative results in asymptomatic patients.334,387 Unless cell counts are not available (e.g., in an office setting in a remote site), use of dipsticks for leukocyte esterase is not recommended. Hepatic encephalopathy is a metabolic disorder characterized by a wide spectrum of neuropsychiatric dysfunction.60A,86 It may occur as an acute syndrome in patients with acute hepatic failure, or as a chronic, relapsing syndrome associated with cirrhosis. As implied by the synonym, chronic hepatic encephalopathy occurs in the setting of portosystemic shunting, usually as a result of cirrhosis. The pathophysiology of hepatic encephalopathy is not completely understood but includes an increased sensitivity to dietary proteins. Ammonia concentrations are always increased with acute encephalopathy476 and are usually increased with chronic encephalopathy. A reduction in plasma ammonia is often associated with symptomatic improvement. However, because plasma ammonia concentrations do not correlate with the severity of the encephalopathy, it has been suggested that other factors are involved.186 It is recognized that a variety of neurotransmitter systems are dysfunctional in hepatic encephalopathy, but the exact cause of the changes is not known. One important contributor is the endogenous benzodiazepine agonist system. The diagnosis of chronic hepatic encephalopathy is usually made on clinical grounds. Plasma ammonia concentrations are rarely helpful for diagnosis or for monitoring the patient’s disorder; normal ammonia concentrations are helpful in excluding hepatic encephalopathy as a cause of cerebral dysfunction when the clinical picture is not clear. As discussed earlier, ammonia is more helpful in acute encephalopathy in proving a hepatic cause, and is of some prognostic importance in acute liver failure.37 Elevated ammonia concentrations in that situation suggest acute hepatic failure or Reye’s syndrome. Hepatorenal syndrome (HRS) refers to decreased renal function secondary to hepatic disease; portal hypertension is a common factor in all cases of HRS that develop in chronic liver disease, but HRS may also occur in acute liver failure. Although formerly thought to be a rapidly progressing, terminal event in a person with end-stage liver disease, it is now recognized that HRS falls into two major varieties.322 Type 2 HRS is more common; it represents a slowly progressive or stable decline in renal function that is due to peripheral vasodilation and renal vasoconstriction. Type 1, or classic, HRS represents rapidly declining renal function, which is usually seen in a person with preexisting type 2 HRS. Type 1 HRS usually develops in the setting of an acute decrease in blood pressure, often due to spontaneous bacterial peritonitis or variceal bleeding. A common feature in both forms of HRS is activation of the renin-angiotensin-aldosterone axis caused by intravascular volume depletion.26 As with other forms of prerenal azotemia (elevated concentrations of urea, creatinine, and other compounds rich in nitrogen.), HRS in the untreated patient is generally associated with increased antidiuretic hormone and with profound thirst. This leads to the development of (1) hyponatremia, (2) hypokalemia, (3) metabolic alkalosis, (4) low urine sodium, (5) high urine potassium excretion, and (6) high urine osmolality. BUN, creatinine, and creatinine clearance are not reliable indicators of renal function in HRS.77 Urea is produced by the liver and is often decreased in advanced liver disease; it is also increased after upper GI bleeding—a common cause of worsening renal function in HRS. Creatinine production by muscle is reduced in cirrhosis, causing falsely low serum creatinine and creatinine clearance. Although cystatin-C has better correlation with measured glomerular filtration rate (GFR),110,462 it has not been widely adopted for monitoring persons with cirrhosis, and one study has suggested that it may be misleading following liver transplantation.44 Despite its limitations, the most widely accepted criterion for diagnosis of HRS is an increase in serum creatinine or a reduction in estimated GFR. Because no specific clinical or laboratory features of HRS have been identified, diagnosis depends on (1) the presence of severe liver disease, (2) a rise in creatinine to greater than 1.5 mg/dL (25.6 µmol/L), (3) no evidence of other renal disease by urinalysis and clinical history, and (4) lack of improvement in renal function with treatments that increase intravascular volume (such as stopping diuretics, or administration of fluids and/or albumin).322 The latter two criteria are important because laboratory findings are similar to those of volume depletion, with low urine output, low urine sodium concentration, and increased urine osmolality. Treatment of HRS is best accomplished by increasing systemic vascular resistance, as with the vasopressin analog terlipressin, or by reducing portal venous pressure, which is most commonly accomplished with TIPS. Both approaches have shown promise in improving renal function in HRS.291 Because of the central role of the liver in drug metabolism and disposition, alterations in drug metabolism may occur in patients with liver disease. In general, this is reflected in delayed metabolism. Only patients with evidence of liver failure, such as encephalopathy, coagulopathy, or ascites, need alterations in dosing. In general, patients with liver disease are not more susceptible to drug-induced hepatotoxicity. However, those with alcoholic liver disease who continue to consume alcohol are susceptible to liver injury from acetaminophen, even at therapeutic doses.411 The intake and disposition of nutrients in patients with chronic liver disease are altered, which subjects them to nutritional imbalance. Severe metabolic and nutritional derangements have been observed in the cirrhotic patient, including alterations in glucose metabolism caused by insulin resistance, and hypokalemia caused by secondary hyperaldosteronism. In addition, hypoalbuminemia is frequently present because of decreased production and sinusoidal leakage of albumin in patients with portal hypertension. Also, in patients with chronic cholestasis, impaired delivery of bile salts to the duodenum results in malabsorption of lipids and fat-soluble vitamins, leading to deficiencies in vitamins A, D, E, and K (see Chapters 31 and 52). Vitamin A deficiency may cause night blindness, but rarely progresses to serious visual impairment. Vitamin D deficiency causes osteopenia and, in severe cases, osteomalacia. In fact, osteopenic bone disease may be one of the most crippling results of chronic cholestatic liver disease, such as primary biliary cirrhosis.189 Vitamin E deficiency is of little clinical significance. Vitamin K deficiency leads to hypoprothrombinemia, with easy bruising and bleeding. As was discussed earlier and in Chapter 59, the liver manufactures most of the soluble clotting factors (the major exceptions being factor VIII and von Willebrand factor), as well as a number of inhibitors of clotting (proteins C and S, antithrombin). The liver also clears activated clotting factors from the circulation. Bile acids are necessary for vitamin K absorption and are needed to produce the active forms of several clotting factors, as well as proteins C and S. Disorders of fibrinogen also occur in liver disease. For example, dysfibrinogenemia may be seen in both acute and chronic liver disease and leads to prolongation of the partial thromboplastin time.150 Patients with autoimmune hepatitis may have anticardiolipin antibodies and antibodies to platelets. The liver is the major source of thrombopoietin, which is needed to produce platelets. Portal hypertension results in splenomegaly, which often leads to thrombocytopenia. In addition, persons with liver disease often have evidence of platelet-associated antibodies,218 although their contribution to low platelet counts in liver disease is questionable.164 Although these facts suggest that hemostatic problems are common in patients with liver disease, discordance is often noted between degree of abnormality of laboratory tests of coagulation and clinical evidence of bleeding.260,446,455 Even in patients bleeding from esophageal varices and having prolonged clotting times, administration of blood components (including activated factor VII) was not associated with any clinical difference in degree of bleeding or need for blood transfusions.455 Because hepatic function is often normal in many patients with liver disease, the plasma activities of several cytosolic, mitochondrial, and membrane-associated enzymes are measured, because they are increased in many forms of liver disease. Because plasma enzyme measurements are discussed in greater detail in Chapter 22, only those factors relevant to an understanding of liver disease will be summarized here. One area of significant concern is the reference limits for liver-associated enzymes, particularly for alanine aminotransferase (ALT, EC 2.6.1.2). In most laboratories, reference intervals are based on samples of the “apparently healthy” population. For ALT, that upper reference limit is often around 40 to 45 U/L in males, but many laboratories have upper reference limits of 65 to 70 U/L.332 These differences are greater than can be explained by analytical differences between methods.332 Although ALT values are approximately 40% lower in females (a difference found even in children128), not all laboratories have different reference limits for the two genders. Population-based reference intervals may not, however, be adequate for identifying persons with liver disease, or for recognizing persons who may be at risk for metabolic syndrome or cardiovascular disease. Because many chronic liver disorders (e.g., hepatitis C, alcoholic liver disease, nonalcoholic fatty liver disease) are prevalent in the population, such reference intervals may include many persons with liver disease. A widely cited Italian study, which excluded persons with known or likely liver disease, suggested lowering reference limits for ALT to about 30 U/L in males and 19 U/L in females.370 A similar study in the author’s laboratory (data unpublished), excluding persons with positive anti–hepatitis C virus (HCV), hepatitis B surface antigen (HBsAg), or obesity, found upper reference limits of 33 U/L in males and 19 U/L in females. A study among dialysis patients [who had 25% lower aspartate aminotransferase (AST) and ALT compared with healthy controls] in Taiwan found that the optimal cutoff value for detecting viral hepatitis was 17 U/L.201 In Korea, risk of development of liver steatosis increased with serum ALT activity, even among those within its usual reference limits,69 as was true with risk of death from cardiovascular disease.230 A study using the Framingham offspring in the United States found that risk of metabolic syndrome and cardiovascular disease increased significantly, starting at ALT, above the lowest quartile of the normal reference limits.166 The National Academy of Clinical Biochemistry and American Association for the Study of Liver Diseases guidelines on liver-related tests recommend that health-related reference limits should be developed for alanine aminotransferase.118 These data provide some preliminary information toward that end. For cytoplasmic enzymes, the relative amount of enzyme in the liver relative to plasma is an important determinant of diagnostic sensitivity. The activity of AST within hepatocytes is about twice that of ALT, although plasma activities are similar. In contrast, hepatocyte activities of LD are much lower (relative to plasma) than those of the other two enzymes, and plasma activities of LD are several times higher than those of AST and ALT. This means that a smaller increase in LD is seen with liver injury than occurs with AST and ALT. The relative amount of enzyme in tissue is not necessarily the same in disease; in cirrhosis and malnutrition, and with alcohol abuse, greater decreases are seen in cytoplasmic ALT than in cytoplasmic AST.272 In addition, other mechanisms may be responsible for this difference in enzyme activity. The development of immunoassays for measuring ALT has led to the observation of discordance between enzymatic activity and mass in several types of liver disease.227 In chronic hepatitis and in healthy individuals, ALT activity and mass change in parallel. In acute hepatitis, activity is increased to a much greater degree than is mass; the opposite pattern is seen in cirrhosis and hepatocellular carcinoma. Additional studies are necessary to confirm this finding, but these results suggest that as yet, poorly understood factors affect enzyme activity. Several mechanisms appear to be involved in release of enzymes from hepatocytes. Cell injury, the simplest mechanism, appears to allow leakage of cytoplasmic enzymes from cells, but minimal release of other types of enzymes. Thus necroinflammatory disease leads to release of AST and ALT, and to a lesser extent LD, but not of mitochondrial isoenzyme of AST nor ALP or GGT. Alcohol appears to induce expression of mitochondrial AST on the surface of hepatocytes.501 The mechanism of release of membrane-bound enzymes such as GGT and ALP into the circulation is less well understood. Synthesis of GGT and ALP appears to be increased in diseased human liver.318 How this enhanced synthesis of tissue-bound enzymes translates into increased activity in plasma is not clear. However, fragments of hepatocyte membrane rich in GGT and ALP activity have been detected in the plasma of patients with cholestasis; this process may be a result of membrane fragmentation by bile acids. Furthermore, bile acids, which are detergents, could solubilize and release GGT and ALP from plasma membranes. In vitro studies of membranes treated with bile acids show that this possibility exists.398 The liver has a limited number of ways of responding to injury.371A Acute injury to the liver may be asymptomatic, but often presents as jaundice. The major acute liver diseases are (1) acute hepatitis and (2) cholestasis. Chronic liver injury generally takes the clinical form of chronic hepatitis; its long-term complications include cirrhosis and HCC. The discussion of liver disease will focus mainly on these patterns and on a few diseases that differ from this general pattern. Cell death occurs by necrosis (death of cell) or apoptosis (programmed cell death) or both. The target cell determines the pattern of injury, with hepatocyte injury leading to hepatocellular disease and biliary cell injury leading to cholestasis. All cellular injury induces fibrosis as an adaptive or healing response, with the duration of injury and genetic factors determining whether cirrhosis and ultimately carcinoma occur (Figure 50-9). Cellular necrosis occurs as the result of an injurious environment and has been referred to as “murder.” It is characterized by cellular swelling with loss of membrane integrity. Toxic injury from compounds such as carbon tetrachloride, aspirin, and acetaminophen (Figure 50-10) occurs for the most part by necrosis. Apoptosis occurs as the result of accelerated programmed death in which the cell participates in its own demise and thus commits “suicide.” It is characterized by cell shrinkage, with nuclear chromatin condensation and fragmentation forming apoptotic bodies. Regardless of the cause, cell death typically leads to leakage of cytoplasmic enzymes. Most forms of hepatitis are associated with apoptosis.
Liver Disease
Anatomy Of The Liver
Gross Anatomy
Microscopic Anatomy
Ultrastructure of the Hepatocyte
Biochemical Functions Of The Liver
Hepatic Excretory Function
Bilirubin
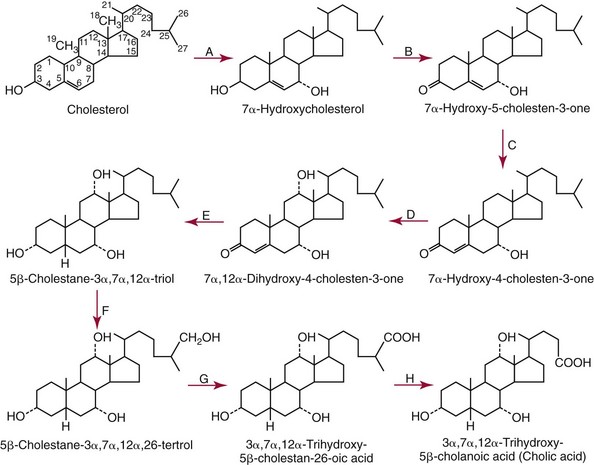
Bile Acids
Chemistry
Clinical Significance of Bile Acids
Hepatic Synthetic Function
Protein Synthesis
Plasma Proteins
Coagulation Proteins
Number or Acronym
Name
I
Fibrinogen*
II
Prothrombin*†
III
Tissue factor
IV
Calcium (Ca2+)
V
Proaccelerin*
VI
–
VII
Proconvertin*†
VIII
Antihemophilic factor
IX
Christmas factor*†
X
Stuart-Prower factor†
XI
Plasma thromboplastin antecedent*
XII
Hageman factor*
XIII
Fibrin-stabilizing factor*
PK
Prekallikrein (Fletcher factor)*
HMWK
High molecular weight kininogen*

Lipid and Lipoprotein Synthesis
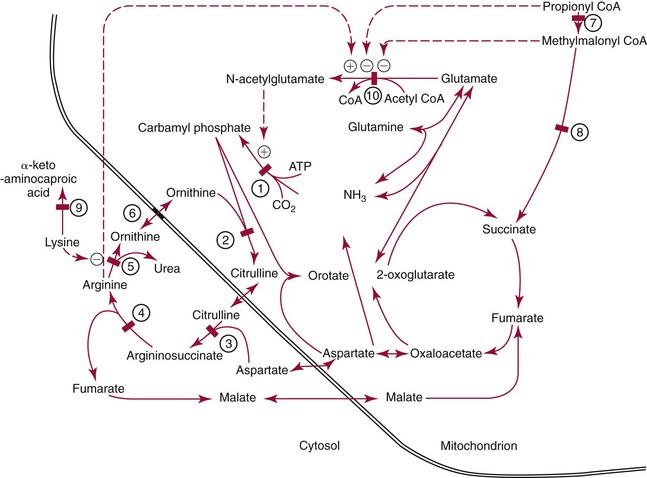
Urea Synthesis
Hepatic Metabolic Function
Ammonia Metabolism
Biochemistry and Physiology
Clinical Significance
Carbohydrate Metabolism
Xenobiotic Metabolism and Excretion
Drug Clearance Tests
Clinical Manifestations Of Liver Disease
Jaundice
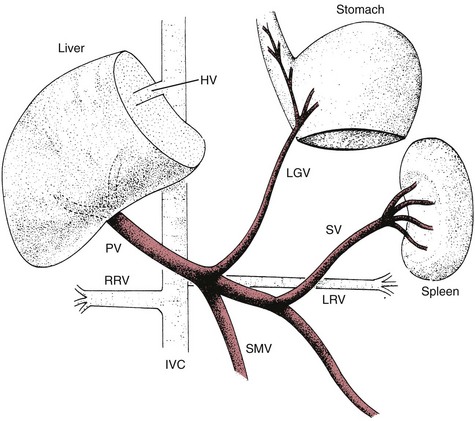
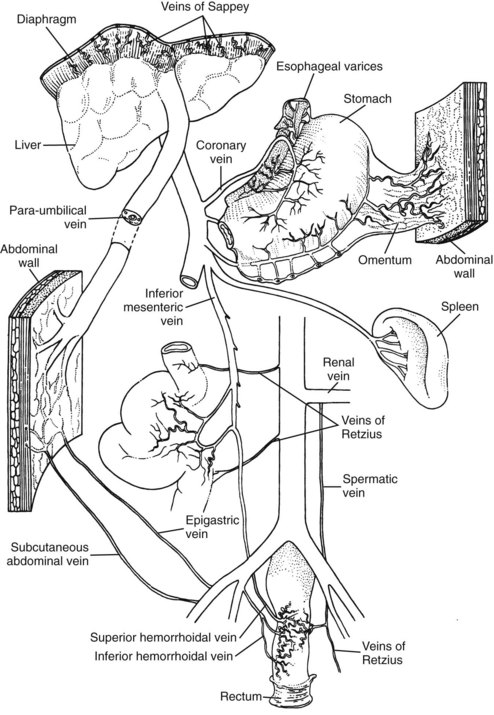
Portal Hypertension
Bleeding Esophageal Varices
Ascites
Spontaneous Bacterial Peritonitis
Hepatic (Portosystemic) Encephalopathy
Hepatorenal Syndrome
Altered Drug Metabolism
Nutritional and Metabolic Abnormalities
Disordered Hemostasis in Liver Disease
Enzymes Released from Diseased Liver Tissue
Reference Limits for Alanine Aminotransferase
Factors Affecting Plasma Enzyme Quantities
Relative Activity in Liver and Plasma
Mechanisms of Release
Diseases Of The Liver
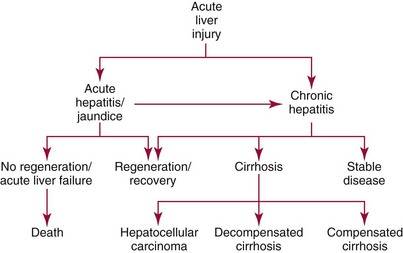
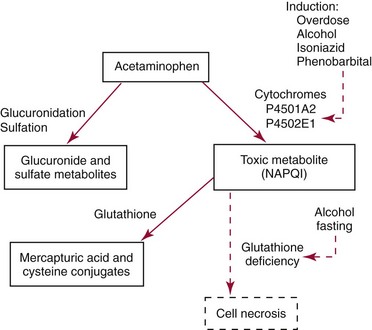
Mechanisms and Patterns of Injury
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine