CHAPTER 9
Lidocaine
TUDY HODGMAN, PharmD, FCCM, BCPS
Lidocaine is a local anesthetic that also has Vaughan Williams classification type IB antiarrhythmic properties. It is indicated for ventricular fibrillation in patients who cannot undergo synchronized cardioversion and are hemodynamically stable who do not require electrical cardioversion. It can also be used for both monomorphic and polymorphic ventricular tachycardias. Lidocaine is considered an alternative to amiodarone as a second-line agent in patients with ventricular tachycardia or pulseless electrical activity who are resistant to electric cardioversion and intravenous epinephrine or vasopressin.1 The use of intravenous lidocaine has decreased with the elimination of lidocaine as the standard of practice for prophylaxis of asymptomatic premature ventricular contractions or non-sustained ventricular tachycardia after acute myocardial infarction.
PHARMACOKINETIC PARAMETERS
Lidocaine serum concentrations decrease biexponentially, and intravenous lidocaine follows a two-compartment pharmacokinetic model (see Figure 9-1).2 After an intravenous loading dose, lidocaine distributes into cardiac tissue rapidly, with an alpha t½ of approximately 8 minutes (range 7–30 minutes), achieving maximum serum concentrations within an hour. The cardiac tissue is considered to be part of the central compartment for lidocaine with onset of effects quickly after a loading dose. The beta elimination phase is due to transfer of drug from the larger volume of distribution (Vdss) back into the central compartment (Vdc) with t½ of 87–108 minutes. Therefore, even if a maintenance infusion is started simultaneously to the loading dose, rapid redistribution can lead to subtherapeutic concentrations that may place the patient at risk for life-threatening arrhythmia.3 This rapid distribution phase justifies the repetition of a “loading dose,” generally 50 percent of the initial load, given at 5- to 20-minute intervals to maintain a therapeutic concentration.1,4,5,6
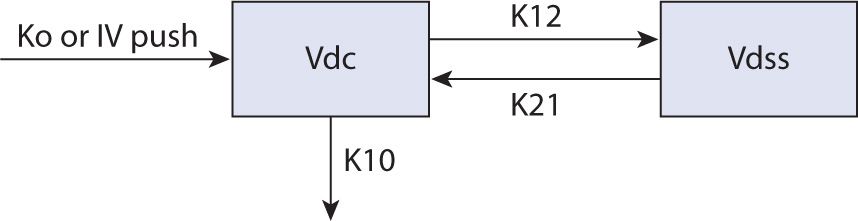
FIGURE 9-1. Two-compartment model.2
THERAPEUTIC CONCENTRATION
Most sources suggest therapeutic concentrations fall in the range of 1.5–5 mcg/mL.1,5 6,7 Unfortunately, lidocaine has a narrow therapeutic index and adverse effects are both dose- and concentration-related. As you approach the upper end of this range, adverse events such as paresthesias, dizziness, drowsiness, and euphoria may appear. If lidocaine concentrations rise above the therapeutic range into toxic concentrations, a host of adverse consequences may be seen, including general adverse events like confusion, dysarthria, muscular twitching or seizures, agitation, psychosis, and even coma.6,7 Cardiovascular adverse events include hypotension, atrioventricular blockade with concurrent hyperkalemia, and circulatory collapse.2,8,9,10,11 However, lidocaine-induced adverse drug events are often missed and attributed to the underlying disease pathology. Routine serum concentration monitoring is not recommended unless the clinician suspects an adverse drug event, the patient experiences recurrent ventricular arrhythmias, or the patient has disease states or conditions known to change the pharmacokinetics of lidocaine.12,13
METABOLISM AND ELIMINATION
Lidocaine is almost exclusively (>95%) eliminated by cytochrome P450 hepatic metabolism. The CYP1A2 and 3A enzyme groups, which are abundant in both the intestinal wall and liver, metabolize efficiently, leading to a large first-pass effect with low oral bioavailability (30%).14 The primary active metabolite is monoethylglycinexylidide (MEGX), which is both renally excreted and further hepatically broken down to glycinexylidide (GX) and other inactive metabolites. Both GX and a portion of MEGX are renally eliminated and have been associated with some of the adverse effects seen with lidocaine.15 Because these metabolites are renally eliminated, patients with significant renal dysfunction can exhibit signs of toxicity despite a concentration within the therapeutic range.4,6 When lidocaine is administered as a prolonged infusion, the metabolites MEGX and GX can compete for hepatic metabolism and lead to accumulation.1,3,4,16–20 Because lidocaine has a low sieving coefficient, it is not removed by hemodialysis or by hemofiltration21; however, no specific dosing guidelines are provided for patients with renal dysfunction either with or without dialysis.
With normal circulatory function, lidocaine has 100 percent bioavailability after intravenous injection. Intramuscular administration is generally avoided because it may interfere with assessment of creatine kinase enzyme concentrations used in the evaluation of acute myocardial infarction and also the need for rapid onset. Lidocaine is a drug with an extraction ratio of about 70 percent, placing it in the category of high extraction ratio where clearance is approximated by liver blood flow, approximately 10 ml/kg/min.9 Therefore, diseases that affect liver blood flow are likely to significantly affect lidocaine clearance. In either CHF or cirrhosis, clearance decreases by about 40 percent to 6 ml/kg/min, whereas in major trauma or critical illness clearance is approximately 6.8 ml/kg/min.6,17,22,23
Unlike the general usefulness of using a serum creatinine to estimate the degree of renal insufficiency, no serum hepatic marker correlates to significant changes in hepatic dysfunction. Therefore, since hepatic disease leads to variable protein binding and elimination, some have used an objective measurement of hepatic function, the Child-Pugh classification (see Table 9-1).24
| TABLE 9-1 | Child-Pugh Classification24 |

A Child-Pugh score of ≥8 would suggest poor hepatic function that would necessitate decreased dose.14 Although lidocaine has a high extraction ratio, in decompensated cirrhotic patients, the clearance is not related to blood flow, but likely decreases relative to the amount of circulating hepatic enzymes that are produced. Additionally many of the patients with hepatic dysfunction will also be treated with a nonselective beta blocker that is known to decrease hepatic enzyme activity as well.14 Patients with known hepatic dysfunction would best have serum lidocaine concentrations utilized to prevent toxicity in this high-risk population.
Changes in plasma protein binding are also likely to affect high-extraction-ratio drugs like lidocaine.6 Normally, plasma protein binding is about 70 percent, with a small portion (30%) to albumin and the rest bound to alpha 1-acid glycoprotein (AAG).20 AAG is an acute phase reactant secreted in high quantities during stress situations (e.g., acute MI, CHF, or trauma that leads to even lower free concentrations of lidocaine as the bound fraction increases). The pharmacokinetics of AAG are also known to be effected by other disease processes such as rheumatoid arthritis, cancer, morbid obesity, nephritic syndrome, or drugs (estrogen).20,25,26 After a myocardial infarction, AAG concentrations may increase for the first 72 hours leading to a decrease in the unbound percentage of lidocaine from 30 percent to 20 percent. This process may be exhibited by decreased lidocaine clearance, placing these patients at risk for adverse effects. It has been suggested that monitoring of unbound drug may be necessary for the most accurate assessment of drug effect.27
The terminal half-life of lidocaine with normal hepatic function is 1–2 hours and increases to more than 5 hours in patients with hepatic dysfunction.1,4,9 In order to estimate steady state, three to five half-lives should pass (8–24 hours) before assessing steady-state serum concentrations. To avoid the delay in achieving a therapeutic serum concentration and allowing the potential for breakthrough arrhythmias, it is suggested a loading dose be given prior to instituting the maintenance dose. The maintenance dose should then be started immediately to avoid subtherapeutic serum concentrations until steady state is reached. One to two repeat bolus doses can be given after 5- to 20-minute intervals to accommodate the initial distribution phase, because the maintenance dose causes only a slow rise in serum concentrations.1,15 The half-life of lidocaine can increase to approximately 5 hours with liver disease (cirrhosis, hepatitis) due to the lack of hepatic enzyme activity. With prolonged infusions, greater than 24 hours, the terminal t½ increases.3,18,19,28 Obesity—defined as total body weight (TBW) >130 percent of lean body weight (LBW)—is not known to specifically affect terminal t½.
The volume of central compartment (Vdc) is not easily measurable, therefore, most clinicians use a population average of 0.5 L/kg for this parameter with a total body volume of distribution (Vdss) of 1.5–2 L/kg.4 The volume of distribution increases minimally with hepatic dysfunction to approximately 0.6 L/kg with Vdss of 2.3 L/kg due to decreased protein stores (albumin and AAG).9 Vdc decreases with acute heart failure to approximately 0.3 L/kg due to increases in AAG, with a Vdss of 0.88 L/kg.4,9 Trauma patients or the critically ill have a Vdc of 0.25 and Vdss of 0.75 L/kg.22,23 Renal failure does not change the volume of distribution. Obesity (TBW >130% LBW) is not associated with a larger Vdc; therefore, doses should be based upon LBW. However, because Vdss does increase with weight, controversy exists as to which weight is best to utilize for computing the total bolus (i.e., the number of bolus doses given).29 Of note, the volume of distribution changes should not lead to changes in individual loading doses administered, but rather changes the total “loading dose” administered as several intermittent boluses (e.g., decreasing or increasing the number of repeat boluses at a dose of 0.5–0.75 mg/kg).
MONITORING
Because lidocaine is an antiarrhythmic agent, the electrocardiogram should be monitored for its effect on the presenting dysrhythmia. The standard goal of therapy is suppression of dysrhythmia with concurrent avoidance of adverse effects. Commonly lidocaine is only employed for a short duration while other therapeutic interventions are done, as there is not a therapeutic class oral agent to transition to. Rarely lidocaine is continued for recalcitrant ventricular dysrhythmias while assessing other long-term antiarrhythmic options. Monitoring of serum lidocaine concentrations is often not necessary as it used short term. However, in the case when a prolonged course is necessary for recalcitrant dysrhythmia or evaluation of drug-related adverse events, serum concentrations should be assessed to avoid lidocaine toxicity or increase morbidity.30,31 Some clinicians suggest stopping lidocaine infusions after 6–24 hours to assess the need for continued therapy.
DRUG INTERACTIONS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


