Chapter 2 Brie Stotler; Anthony N. Sireci Questions 1. Which one of the following statements is true about the Three-Day Stay Rule of 1990? B. It helps laboratories develop programs that promote highly ethical and lawful conduct, especially in terms of billing. C. It prevents physicians from referring Medicare patients to self-owned laboratories. D. It establishes that all clinical laboratories must be certified by the federal government with programs to ensure quality, appropriate personnel training, and regular proficiency assessment. E. It directs how health care information is managed. 2. Which one of the following statements describes an Advance Beneficiary Notice of Noncoverage? A. A legal notice provided to all uninsured patients before they receive any medical care. B. A legal notice provided to all Medicaid participants. C. A legal notice provided to all uninsured patients receiving outpatient chemotherapy. D. A legal notice provided to Medicare fee-for-service participants delineating services expected to not be covered under their Medicare insurance plan. E. A legal notice to all insured patients delineating services routinely not covered by major insurance carriers. 3. Which one of the following is a mission of the Agency for Healthcare Research and Quality (AHRQ)? A. To compare the effectiveness of new treatments. B. To direct the Department of Health and Human Services (DHHS). C. To oversee the National Institutes of Health (NIH) research programs. D. To be the largest source of funding for biomedical research. E. To provide health care to the elderly and indigent. 4. In method validation studies, Clinical Laboratory Improvement Amendments (CLIA) regulations state that a laboratory must test for analytical specificity. Which one of the following sets of experiments would satisfy this requirement? B. Running samples without the analyte of interest (i.e., Matrix blanks), ensuring that a zero sample actually reads zero. C. Running both positive and negative samples and determining the specificity by taking true negatives and dividing by true negatives plus false positives. D. Running samples spiked with increasing concentrations of an analyte and determining the concentration of the analyte that most specifically discriminates diseased from healthy patients. E. Running samples spiked with the highest measurable concentration of the analyte of interest followed by a Matrix blank. 5. Which one of the following provides the best definition for analytical sensitivity? A. The false-negative rate of the assay. B. The value obtained when the number of true positives is divided by the sum of the true positives and the false negatives. C. The lowest possible concentration of an analyte that is accurately and reproducibly measured by an assay. D. The performance of the assay when measuring an analyte in the presence of an interfering substance. E. The formula that relates instrument response to analyte concentration. 6. The Centers for Disease Control and Prevention (CDC) classifies laboratories by biosafety level. Which one of the following represents the best example of a level 3 laboratory? A. A training laboratory for students. B. A clinical chemistry laboratory. C. A clinical hematology laboratory. D. A laboratory working with highly infectious fungal pathogens. E. A satellite clinical laboratory in an emergency department. 7. Which one of the following is the best definition of “calibration verification”? B. The process of testing known concentrations of an analyte throughout the reportable range and confirming that the assay returns the appropriate value, within an established allowable error. C. The process of comparing the slope and y-intercept of the current calibration curve with the previous curves. D. The process of running previously assayed patient samples with a new lot of calibration material and comparing them with results obtained when the same samples were run using a previous lot of calibration material. E. The process of running samples of known high and low analyte concentrations and plotting them on a Levey-Jennings chart. 8. You are the director of a large hospital laboratory that is accredited by the College of American Pathologists (CAP). Which one of the following indicates how often your laboratory will be subject to a routine inspection by CAP? B. Once every year. C. Once every 2 years. D. Once every 3 years. E. Once every 5 years. 9. A new lot of reagent arrives in the laboratory and after several days of concurrent analysis with the old lot of reagent, a significant positive shift in the value of the high quality control (QC) material using the new reagent lot relative to the same QC material using the old reagent lot is noted. The section supervisor thinks this is a differential matrix interaction of the new lot with the high control and that it will not affect patient results. Which one of the following is the best piece of experimental evidence to support this hypothesis? B. Random patient samples are run using the old and new reagent lots and then compared. The correlation coefficient should be close to 1. C. Calibration standards dissolved in methanol are run across the full analytical range of the instrument using the old and new reagent lots and are then compared. The correlation coefficient should be close to 1. D. The analyte should be extracted from each level of control material using a solid-phase extraction procedure and then run using the old and new reagent lots. The correlation coefficient should be close to 1. E. The assay should be recalibrated; the control material should then be run and compared between both reagent lots. 10. Your laboratory currently runs a full menu of moderate-complexity testing and is accredited by CLIA under a certificate of accreditation. A treating physician asks you to add a test that the Food and Drug Administration (FDA) has classified as a “waived” test. Which one of the following statements is the appropriate course of action with regard to regulatory compliance? A. You must apply for a CLIA certificate of waiver. B. You must apply for a CLIA certificate of registration. C. You must apply for a CLIA certificate of compliance. D. No additional certificates are necessary. E. You cannot run a waived test in a laboratory that performs predominantly moderate-complexity testing. 11. Laboratory testing in the United States is regulated through the CLIA program. The program is overseen by which one of the following regulatory entities? B. The AABB. C. The CAP. D. The Joint Commission. E. The Centers for Medicare and Medicaid Services (CMS). 12. At a minimum, before introducing a new FDA-approved assay into the laboratory, which one of the following groups of performance characteristics must be assayed during a method validation and compared with manufacturer claims, to be compliant with CLIA ’88 regulations? A. Accuracy, precision, reportable range, and confirmation of the manufacturer’s reference range. B. Accuracy, analytical sensitivity and specificity, reportable range, and a full reference range study with a minimum of 120 patient samples. C. Accuracy, clinical sensitivity and specificity, analytical specificity and sensitivity, full reference range study, and reportable range. D. Extraction efficiency, reportable range, full reference range study, QC material stability, and analytical sensitivity and specificity. E. Only accuracy and precision must be confirmed. 13. According to CLIA regulations, which one of the following best indicates how often laboratories must perform QC evaluation (i.e., assay QC samples) for most assays that are performed 24 hours a day? B. Once every 24 hours. C. Every time the assay is performed. D. Once every 48 hours. E. Once every week. 14. You are the director of a newly established laboratory that will perform a mix of waived and nonwaived, moderate-complexity testing. Which one of the following best describes when you can begin testing? B. After you are inspected by your state Department of Health and have received your certificate of compliance. C. After you have been inspected by an approved accreditation agency and have received your certificate of accreditation. D. After your state Department of Health, or an approved accreditation agency, and CMS have both inspected your laboratory. E. Once you have validated your assays to your satisfaction. 15. A laboratory medical director is overseeing the implementation of a new hematology analyzer and is in the process of setting up delta checks for several complete blood cell count parameters. Which one of the following best describes a delta check? A. It is a comparison of reference ranges across laboratories. B. It is a quality measure by which a patient’s previous result is compared with the current one. C. It is a quality measure by which samples are visually checked for hemolysis. D. It is a level of an analyte thought to be life-threatening to the patient, requiring verbal notification to the ordering provider. E. It is a quality measure that occurs when results fall above the analytical limit of an assay. 16. Which one of the following statements best describes the benefit(s) that the Family and Medical Leave Act (FMLA) provides to eligible individuals? A. The FMLA provides employees with up to 12 weeks of paid, job-protected leave per year. B. The FMLA provides employees with up to 16 weeks of paid, job-protected leave for the birth or care of a newborn child. C. The FMLA provides employees with up to 16 weeks of unpaid, job-protected leave for females and 12 weeks for males for the birth or care of a newborn child. D. The FMLA provides employees with up to 12 weeks of unpaid, job-protected leave per year leave for qualifying reasons. E. The FMLA provides employees with continuous health insurance coverage for up to 16 weeks. 17. In the United States, blood banks are regulated by which of the following entities? B. The CAP. C. The CMS. D. The CDC. E. The FDA. 18. Which one of the following statements best describes the role of the FDA in the regulation of laboratory-developed tests (LDTs)? A. LDTs are not medical devices and therefore do not fall under the FDA’s jurisdiction. B. LDTs are considered medical devices, and each laboratory must receive FDA approval before implementing an LDT. C. LDTs are considered medical devices, but the FDA has exercised enforcement discretion on LDTs. D. LDTs are not considered medical devices, but the FDA regulates them. E. LDTs may or may not be considered medical devices, and the FDA considers each assay on a case-by-case basis and decides whether to regulate. 19. During an interview with a potential laboratory technologist, which one of the following questions is appropriate to ask? A. Do you have any disabilities? B. Were you born in the United States? C. What is your native language? D. How old are you? E. Are you capable of performing all of the duties required by this position? 20. The International Organization for Standards (ISO) standard 15189:2007 deals with which one of the following? A. Standards for quality and competence in medical laboratories. B. Standards for pricing and reimbursement in medical laboratories. C. Laboratory services that must be covered under universal health insurance plans. D. Standard requirements for laboratory technologist school curricula. E. Standard requirements for retirement benefit packages. 21. As the director of a laboratory performing high-complexity testing, you are required to ensure that your laboratory is appropriately staffed with all required levels of qualified personnel whose duties and qualifications are clearly defined by CLIA. The laboratory director can assume the responsibility of which one of the following required positions given appropriate training and background? B. Clinical consultant. C. General supervisor. D. Testing personnel. E. The laboratory director can assume any and all of these positions. 22. The Levey-Jennings control chart for an assay performed in your laboratory is shown in Figure 2-1. The chart indicates which type of analytical problem for this assay? A. The assay is accurate but not precise. B. The assay is precise but not accurate. C. The assay is neither precise nor accurate. D. The assay is clinically useless. E. The assay is precise and accurate. 23. Quality control (QC) material is run at the start of each shift (in this case, for one shift per day) for all assays before testing patient samples. The QC results for several days for one of the assays are plotted on a Levey-Jennings chart shown in Figure 2-2. Which one of the following best describes what this Levey-Jennings chart demonstrates? A. Positive trending of QC results. B. Negative trending of QC results. C. Result interference by an unknown substance. D. The use of an inappropriately narrow reference range. E. The need to change the technical range for the assay. 24. Which one of the following best describes how to plot a Levey-Jennings chart? A. The x-axis as “Average value” and the y-axis as “% Difference.” B. The x-axis as the “Gold standard method” and the y-axis as “Test method.” C. The x-axis as “Calibrator level” and the y-axis as “% CV.” D. The x-axis as “Run #” and the y-axis as “Concentration.” E. The x-axis as “Instrument response” and the y-axis as “Expected concentration.” 25. In a method validation study, a sample at the proposed lower limit of quantification (LoQ) is run 20 times, 4 times daily for 5 days. Which one of the following answers best describes the performance parameters that can be determined from this experiment? B. Validation of the LoQ, intraday and interday reproducibility, and accuracy at the LoQ. C. Validation of the LoQ and establishment of accuracy at the LoQ. D. Validation of the LoQ and establishment of the reportable range. E. Establishment of reproducibility at the LoQ. 26. A laboratory director is designing a validation experiment for quantifying a drug metabolite in urine by gas chromatography–mass spectrometry. She asks her laboratory technologist to run a blank urine sample spiked with the lowest clinically relevant value of the metabolite 20 times and to average the signal/noise ratio of the analyte peak at this concentration. The laboratory director is trying to establish which one of the following analytical performance parameters? B. Analytical sensitivity C. Clinical specificity. D. Clinical sensitivity. E. The lower limit of linearity. 27. Which one of the following entities administers the Medicaid insurance program? B. The FDA. C. The American Medical Association. D. Individual states. E. Private insurance companies. 28. You are validating a high-complexity assay to measure analyte X. In a method comparison study, you choose 40 samples that span the analytical range of your method, analyze them by the new method, and compare your results with a reference, gold standard method at a reference laboratory. The resulting correlation analysis reveals a coefficient of determination (R2) of 0.99, an intercept of + 0.1, and a slope of 1.34 when the gold standard is plotted on the x-axis. Which one of the following statements best describes your assay? A. Your assay correlates poorly with the reference method. B. Your assay correlates well with the reference method but has a positive proportional bias. C. Your assay correlates well with the reference method but has a negative constant bias. D. Your assay correlates well with the reference method. No other statements can be accurately made. E. Your assay correlates poorly with the reference method and shows a positive proportional bias. 29. Which one of the following government agencies is responsible for developing standardized materials for calibration, weights, and measures? B. The NIH. C. The CMS. D. The National Institute of Standards and Technology (NIST). E. The DHHS. 30. Which one of the following statements best describes a requirement(s) expected of employers by the Occupational Health and Safety Administration (OSHA)? A. Apply for state certification every 2 years and pay the annual membership fee. B. Provide subsidized health insurance for all employees and offer at least two separate insurance plans. C. Display an OSHA poster in the workplace informing employees about the Occupational Safety and Health Act. D. Require that all employees receive an annual physical health exam with urine drug screening. E. Receive state OSHA agency approval before increasing the number of employees. 31. Which one of the following is the best example of a postanalytical error? A. The patient results are manually entered into the wrong patient’s medical record. B. The sample is drawn into an inappropriate tube. C. The sample is run using a reagent lot that has expired. D. The phlebotomist did not fill the tube completely on a sample sent for coagulation testing. E. The autosampling pipette was jostled and is now drawing only 1.3 μL of sample instead of 1.5 μL of sample. 32. Which one of the following statements best describes proficiency testing? A. A quality assessment tool that compares a patient’s previous results with current results. B. A quality assessment tool that involves testing two levels of QC material before testing patient samples. C. A quality assessment tool by which laboratories test samples with unknown results and compare their results. D. A quality assessment tool that involves running calibrators every 6 months to ensure linear assay results. E. A quality assessment test given to all newly hired technologists to ensure appropriate training and competency. 33. Your laboratory receives a proficiency testing (PT) specimen from an approved provider of PT material for a particular analyte. Which one of the following choices best describes how the sample should be handled? B. The PT sample should be split; one aliquot should be run in your laboratory and the other aliquot should be sent to a reference laboratory to ensure concordant results. C. The PT sample should be entered into your laboratory’s workflow and tested and resulted as with any other patient specimen. D. The PT sample should be treated as any other patient sample. If send-out is required as a reflex for this analyte, your laboratory should send it out as per protocol. E. The PT sample should be run multiple times during multiple days and the values averaged to minimize error. 34. Which one of the following is the best definition of “unsuccessful” participation or performance in PT? B. Failure to submit the results of a testing event in the requisite amount of time. C. A clerical error in reporting the results for a testing event. D. Repeated unsatisfactory test scores for an analyte or for the entire testing event in a particular specialty or subspecialty on two consecutive PT events or two of three testing events. E. This is not a term used in assessing performance in PT events. 35. The quality manager of your laboratory recommends that your laboratory register for a PT program for a test that the FDA has classified as “waived.” Which one of the following best describes how you should you respond to this recommendation? B. You should agree. Although PT is not required for waived tests, if a valid PT program exists, participation will help ensure your laboratory’s accuracy in result reporting. C. You should disagree. After looking through the CLIA regulations for PT, you notice the analyte you are measuring is not on the list of regulated analytes for which PT is required. D. You should disagree. The FDA does not require PT for tests it deems waived. E. You should disagree. PT will only highlight your laboratory’s inaccuracy. 36. Which one of the following is most characteristic of root cause analysis (RCA), regardless of the method that is used? A. Identifies a single cause of the error. B. Favors the subjective opinion of experts. C. Results in an executable and quantifiable solution(s). D. Seeks to identify and blame the specific person or group responsible. E. Uses the Ishikawa fishbone diagram. 37. Which one of the following is a specific management infrastructure required for Six Sigma certification? A. Master “Black Belts” and “Green Belts.” B. Merger and acquisitions attorneys. C. Only the chief executive officer commitment is required. D. Six Sigma consultants. E. Support of engineers. 38. Section 1877 of the Social Security Act (42 U.S.C. 1395) prohibits which one of the following? A. Billing Medicare and Medicaid for tests completed in a laboratory without a CLIA license. B. Professional billing for laboratory tests without a physician-documented order that includes the physician’s signature. C. Physician patient referral to an entity for a designated health service from which the physician will financially benefit. D. Performing genetic tests without a signed consent form. E. Performing HIV tests without a signed consent form. 39. A supervisor informs you that the QC values for an assay performed in your laboratory have triggered the 81.5s Westgard rule. Which one of the following types of type of analytical error does this most likely indicate? B. User error. C. Random error. D. Imprecision. E. A bias trend. 40. An area supervisor from your laboratory asks you to review your QC rules. She says that QC is failing too frequently using the 12S Westgard rule and troubleshooting reveals no errors in the method. This has been the case over multiple reagent lots. You would like to maintain the sensitivity of the rule with regard to error detection but would also like to decrease the false-alert rate. Which one of the following is the most logical step? A. Change to using the 13s rule. B. Change to using the 81s rule. C. Adopt a hybrid rule combining the 13s and 22s rules in a multirule scheme. D. Rules are not necessary, and each QC value should be judged individually. E. Ask the supervisor to change lots of QC material; the failure rate is likely due to a faulty batch. 41. Which one of the following is the most common type of CLIA certificate issued? B. Certificate for provider-performed microscopy procedures. C. Certificate of registration. D. Certificate of compliance. E. Certificate of accreditation. 42a. A laboratory is validating a new β-human chorionic gonadotropin assay. Below are the results from a validation study assessing potential carryover. The acceptable limit for carryover was set at 2 times the SD for the low concentration mean. According to Table 2-1, the calculated percent carryover is: B. 2%. C. 15%. D. 10%. E. 5%. Table 2-1 42b. Which one of the following statements best describes whether this amount of carryover acceptable? B. This amount of carryover is acceptable because it is less than 10 mIU/mL. C. This amount of carryover is acceptable because it is less than 15% of the original sample. D. This amount of carryover is not acceptable because it is more than 5% of the original sample. E. This amount of carryover is not acceptable because it is more than 0.2 mIU/mL. 43. According to CLIA regulations, laboratories that hold a certificate of waiver must comply with which one of the following? A. Follow Universal Precautions when testing patient samples. B. Have a designated laboratory director who holds a medical degree. C. Follow the test manufacturer’s instructions. D. Engage in CLIA-directed PT. E. Record all patient test results in an electronic format. 44. Laboratory testing in the United States is regulated through CLIA. The CLIA program is overseen by which one of the following regulatory entities? B. The AABB. C. The CAP. D. The Joint Commission. E. The CMS. 45. CLIA quality system regulations require verification of a manufacturer’s stated performance specifications for which one of the following categories of tests? A. Waived tests approved by the FDA. B. Provider-performed microscopy tests approved by the FDA. C. Moderate-complexity tests approved by the FDA. D. Waived, provider-performed microscopy, and moderate-complexity tests approved by the FDA. E. All FDA-approved tests used for patient care. 46. Which one of the following is categorized as a waived test according to the CLIA classification? B. Fecal occult blood examination. C. Urine sediment examination. D. Fecal leukocyte examination. E. The fern test. 47. According to CLIA regulations, cytology slide preparations must be retained for which one of the following periods of time? B. 2 years. C. 5 years. D. 10 years. E. Indefinitely. 48. According to CLIA regulations, clinical pathology test records must be retained for at least which one of the following periods of time? B. 2 years. C. 5 years. D. 10 years. E. Indefinitely. 49. Which one of the following methods best helps ensure the accuracy of a test? A. Analyze samples from healthy subjects and determine the 95% prediction interval. B. Analyze samples with no measurable analyte spiked with low concentrations of analyte. C. Analyze samples with a high value followed by samples with a low value. D. Analyze a sample 20 times and determine the mean and SD. E. Compare samples between the test method and a reference method. Major points of discussion ■ The physician self-referral ban, or Stark Act, prevents physicians from referring Medicare patients to self-owned laboratories. ■ HIPAA directs how health care information is managed. ■ The OIG compliance guidelines help laboratories develop programs that promote highly ethical and lawful conduct, especially in terms of billing. The OIG is responsible for identifying fraud and abuse in laboratory testing, as regulated by the CMS. 2. A. A legal notice provided to all uninsured patients before they receive any medical care. Major points of discussion ■ An Advance Beneficiary Notice of Noncoverage must be provided by independent laboratories. ■ The Advance Beneficiary Notice of Noncoverage was formerly named the Advance Beneficiary Notice. ■ The law requires a provider to notify a beneficiary in advance if the service will likely be denied as “not reasonable and necessary.” If an Advance Beneficiary Notice of Noncoverage is not given, providers may not bill Medicare or the individual patient for these services. ■ Medicare is a health insurance program for people 65 years or older, people younger than 65 years with certain disabilities, and people of all ages with end-stage renal disease. ■ Medicaid is a joint state-federal–administered health insurance program that provides health coverage for income-qualifying individuals. To participate in Medicaid, federal law requires states to cover certain population groups.3,24 3. A. To compare the effectiveness of new treatments. Major points of discussion ■ Its focus areas are the comparison of effectiveness of new treatments, quality improvement, patient safety, the use of health information technology in patient safety, and determining the cost effectiveness of health care. ■ AHRQ, like its sister agency the NIH, applies more than 80% of its budget for research. However, the NIH is the largest source of funding for biomedical research in the United States. ■ In addition, AHRQ publishes technology assessments to provide information to other agencies. For example, AHRQ provided a summary to the FDA on the scientific evidence on the quality of laboratory-developed tests (i.e., LDTs or “home brew” tests).16 4. A. Running samples of a known analyte concentration spiked with prescription drugs that could potentially interfere with the measurement of the analyte of interest. Major points of discussion ■ Possible interfering substances vary by analyte and analytical method but can include hemolysis, lipemia, icterus, hyperproteinemia, and prescription drug interferences. ■ It is generally impossible to test for all possible interferences; thus, the most commonly encountered interfering substances should be determined and assayed. ■ Analytical specificity is distinct from clinical specificity. ■ Clinical specificity, which is sometimes part of method validation, is determined by taking the number of true negatives identified by the assay and dividing by the sum of the true negatives and false positives. 5. A. The false-negative rate of the assay. Major points of discussion ■ Analytical specificity is best defined as the performance of the assay when measuring an analyte in the presence of an interfering substance. ■ The calibration curve is best defined as the formula that best correlates instrument response to analyte concentration. ■ Clinical sensitivity is defined as the true-positive rate of an assay and is calculated as the ratio of true positives divided by the sum of true positives and false negatives. ■ Clinical specificity is defined as the true-negative rate of an assay and is calculated as the ratio of true negatives dividied by the sum of true negatives and false positives. 6. A. A training laboratory for students. Major points of discussion ■ Biosafety level 2 laboratories are suitable for work involving agents of moderate potential hazard to personnel and the environment. ■ Biosafety level 3 laboratories are suitable for work with indigenous or exotic agents that may cause serious or potentially lethal disease after inhalation. ■ Biosafety level 4 laboratories are suitable for work with dangerous and exotic agents that pose a high individual risk of aerosol-transmitted laboratory infections. These include agents that cause severe to fatal disease in humans for which vaccines or other treatments are not available. 7. A. The process of testing and adjusting an assay so that the instrument measurements best correlate with the known analyte concentrations. Major points of discussion ■ Calibration verification should also be performed after major servicing of the equipment, changing to a new lot of reagents, or when the performance of the assay is questionable. ■ The goal of calibration verification is to ensure that your assay maintains accuracy across its analytical range. ■ Calibration verification is a quality assurance measure. ■ Calibration of an assay is best defined as the process of adjusting an instrument response to maximize the correlation with analyte concentration. This is distinct from calibration verification. ■ A laboratory should establish, a priori, what tolerance limits it will allow for variation within calibration verification. These limits should be based on the clinical accuracy requirement of the assay. ■ Comparing the slope and intercept of a current calibration line with previous lines is an additional piece of quality assurance that is distinct from calibration verification and will identify shifts in calibration in real time.5 8. A. Every 6 months. Major points of discussion ■ A CLIA-accredited laboratory is one that is inspected by accrediting organizations that have been approved by the CMS. ■ An approved accrediting organization can inspect a laboratory in lieu of the CMS. ■ CMS has granted the CAP Laboratory Accreditation Program deeming authority. ■ CAP inspections are guided by detailed checklists created for different areas of the laboratory.25 9. A. Patient samples spanning the analytical range of the assay are run using the old reagent lot and compared with the same samples using the new reagent lot. The correlation coefficient should be close to 1. Major points of discussion ■ Samples should be both QC material and real patient samples (in matrix) that span the analytical range of the assay to prove that the new reagent lot reacts the same as the old lot in matrix and in the “pseudo-matrix” in which many QC materials are prepared. ■ Differences in QC values using two different lots of reagent may be due to different reactions between the new lot and the pseudo-matrix of the QC material. This difference in reaction may extend to patient samples and should be excluded by running patient samples across the analytical range using both lots. ■ New reagent lots should be “verified” in the above manner well before the old lot expires or runs out. This requires good inventory control. ■ It is ultimately the responsibility of the laboratory director to ensure that good QC practices are followed in the laboratory. ■ A priori, the laboratory should establish allowable differences in patient sample values between lots. These tolerance limits should be established based on the clinical accuracy needs of the assay. 10. A. You must apply for a CLIA certificate of waiver. Major points of discussion ■ There are six CMS-approved accreditation agencies: • AABB • American Osteopathic Association • American Society for Histocompatibility and Immunogenetics • COLA • CAP • The Joint Commission7 11. A. The FDA. Major points of discussion ■ CLIA was passed by Congress in 1988. ■ The CLIA program is designed to ensure quality laboratory testing. ■ The CMS oversees the operation of the CLIA program. ■ The CMS recognizes additional accrediting agencies and will issue a certificate of accreditation to laboratories that receive accreditation from the AABB, American Osteopathic Association, American Society for Histocompatibility and Immunogenetics, COLA, CAP, and The Joint Commission. ■ There are five types of CLIA certificates: certificate of waiver, certificate for provider-performed microscopy procedures, certificate of registration, certificate of compliance, and certificate of accreditation.4,7 12. A. Accuracy, precision, reportable range, and confirmation of the manufacturer’s reference range. Major points of discussion ■ Accuracy, reproducibility, reportable range, and reference range are the four performance characteristics of an FDA-approved, moderate-complexity assay that must, at a minimum, be confirmed. ■ Laboratory-developed tests are considered high-complexity tests and are held to a different standard of validation by CLIA regulations. Additional performance parameters must be assessed. ■ The purpose of a method validation is to assess the degree of uncertainty in the measurements made by the assay and to point out assay weaknesses and potential for error. ■ Accuracy is usually validated by running standards of known concentrations and comparing the measured concentration as a percentage of the expected concentration. ■ Precision or reproducibility (inter-run) is generally assessed by running at least two levels of an analyte in quadruplicate over 5 days. Precision is expressed as a % CV ([mean/SD] × 100%). ■ The reportable range is established by running increasing concentrations of the analyte and choosing the range of concentrations in which instrument response increases linearly with analyte concentration. A linear regression procedure is usually used. 13. A. Once every 12 hours. Major points of discussion ■ Laboratories can assay QC samples more frequently if deemed necessary to ensure accurate results. ■ QC samples should be assayed after calibration or maintenance of an analyzer to verify correct method performance. ■ Blood gas measurements have separate, stricter QC requirements. CLIA requires at least one QC level to be assayed every 8 hours for blood gas measurements. ■ In addition to running QC samples every 8 hours for blood gas measurements, CLIA also requires that a control sample be run with every patient sample, unless the analyzer calibrates itself every 30 minutes.6 14. A. After you have applied, paid for, and received a certificate of registration, assuming your state Department of Health has no additional requirements. Major points of discussion ■ A certificate of registration is issued to a laboratory to allow it to conduct nonwaived (moderate- and/or high-complexity) testing until the laboratory is surveyed (inspected) to determine its compliance with CLIA regulations. Only laboratories that apply for a certificate of compliance or a certificate of accreditation will receive a certificate of registration. ■ A certificate of provider-performed microscopy (PPM) is issued to a laboratory in which a physician, midlevel practitioner, or dentist performs specific microscopy procedures during the course of a patient’s visit. A limited list of microscopy procedures is included under this certificate type; these are categorized as moderate-complexity. ■ There are six CMS-approved accreditation organizations: • AABB • American Osteopathic Association • American Society for Histocompatibility and Immunogenetics • COLA • College of American Pathologists • The Joint Commission ■ As defined by CLIA, waived tests are categorized as “simple laboratory examinations and procedures that have an insignificant risk of an erroneous result.” The FDA determines the criteria for “simple” tests as those with a low risk of error and approves manufacturer’s applications for test system waiver.7 15. A. It is a comparison of reference ranges across laboratories. Major points of discussion ■ Delta checks are intended to detect either preanalytic or analytic errors in testing, such as specimen mix-up errors, diluted samples, or analyzer malfunction. ■ Laboratories individually determine (1) which assays will have delta checks and (2) the acceptable thresholds for intraindividual variation. ■ An example of an assay that commonly has a delta check is the mean corpuscular red blood cell volume. ■ Delta checks are an example of patient-derived QC procedures.21,23 16. A. The FMLA provides employees with up to 12 weeks of paid, job-protected leave per year. Major points of discussion ■ Qualifying reasons include the birth of a newborn child; placement of a child for adoption or foster care; care for an immediate family member with a serious health condition; or employee medical leave if unable to work because of a serious health condition. ■ The FMLA applies to all public agencies, elementary and secondary schools, and companies with 50 or more employees. ■ The FMLA is enforced by the U.S. Department of Labor’s Employment Standards Administration, Wage and Hour Division. ■ The FMLA requires health insurance to be maintained during the leave if insurance was provided before the leave was taken. ■ Employees are eligible for leave if they have worked for their employer at least 12 months.15 17. A. The AABB. Major points of discussion ■ The FDA conducts unannounced inspections of all blood banks on a periodic basis. ■ The FDA promulgates criteria for blood donor eligibility. ■ The CMS oversees the CLIA regulatory program. ■ The AABB, CAP, and The Joint Commission have accreditation standards for blood banks and will conduct inspections. 18. A. LDTs are not medical devices and therefore do not fall under the FDA’s jurisdiction. Major points of discussion ■ The definition of an LDT is a test designed in a laboratory for specific use only in that laboratory. ■ The FDA exercises enforcement discretion with regard to LDTs to enable implementation of the newest diagnostics in clinical laboratories in a meaningful time period. ■ The FDA’s proposed jurisdiction over LDTs is the subject of debate, particularly from the CMS, which considers LDTs a service offered by the laboratory to ordering physicians without any reagents or devices crossing state borders. Therefore, they argue that the FDA has no authority in regulating LDTs. ■ Although LDTs are not actively regulated by the FDA, CLIA has a strict set of performance criteria that must be established in method validation before releasing patient results. These include, but are not limited to, linearity, reproducibility, analytical sensitivity/specificity, reference range, and recovery/accuracy. ■ LDT validation studies should be tailored to address issues specific to the technology being used. For example, ionization suppression experiments should always be included in methods using electrospray ionization–mass spectrometry, despite this not being specifically listed by CLIA regulations.14,17 19. A. Do you have any disabilities? Major points of discussion • A person may be disabled if he or she has a history of a disability (such as cancer that is in remission). • A person may be disabled if he or she is believed to have a physical or mental impairment that is not transitory (lasting or expected to last 6 months or less) and minor (even if he or she does not have such an impairment). ■ The Age Discrimination in Employment Act of 1967 protects people who are age 40 years or older from discrimination because of age. The law also makes it illegal to retaliate against a person because the person complained about discrimination, filed a charge of discrimination, or participated in an employment discrimination investigation or lawsuit. ■ Title VII of the Civil Rights Law of 1964 makes it illegal to retaliate against a person because the person complained about discrimination, filed a charge of discrimination, or participated in an employment discrimination investigation or lawsuit. The law also requires that employers reasonably accommodate applicants’ and employees’ sincerely held religious practices, unless doing so would impose an undue hardship on the operation of the employer’s business. ■ Title I of the Americans with Disabilities Act of 1990 makes it illegal to discriminate against a qualified person with a disability, in the private sector or in state and local governments. The law also makes it illegal to retaliate against a person because the person complained about discrimination, filed a charge of discrimination, or participated in an employment discrimination investigation or lawsuit. The law also requires that employers reasonably accommodate the known physical or mental limitations of an otherwise qualified individual with a disability who is an applicant or employee, unless doing so would impose an undue hardship on the operation of the employer’s business. ■ The Genetic Information Nondiscrimination Act of 2009 makes it illegal to discriminate against employees or applicants because of genetic information. Genetic information includes information about an individual’s genetic tests and the genetic tests of an individual’s family members, as well as information about any disease, disorder, or condition of an individual’s family members (i.e., an individual’s family medical history). The law also makes it illegal to retaliate against a person because the person complained about discrimination, filed a charge of discrimination, or participated in an employment discrimination investigation or lawsuit. 20. A. Standards for quality and competence in medical laboratories. Major points of discussion ■ ISO is a network of the national standards institutes of 163 countries. ■ International Standards for technical regulations on products, production methods, and services help ensure a certain level of performance and safety worldwide. ■ Adherence to ISO standards is voluntary. ISO has no legal authority to enforce its standards unless countries have incorporated them into their national laws. ■ ISO 15189:2007 (“Medical laboratories—Particular requirements for quality and competence”) is designed specifically for clinical and medical laboratories and includes quality management standards, technical competence standards, and test reliability standards.20 21. A. Technical supervisor. Major points of discussion ■ If the laboratory performs a mix of moderate- and high-complexity testing, only those employees working on high-complexity testing must meet the above requirements. ■ These four positions can be filled by one person (i.e., the laboratory director) as long and he or she meets the qualifications and is able to meet the established responsibilities defined in Subpart M of CLIA. ■ It is the laboratory director’s responsibility to ensure that appropriate personnel work in the laboratory. ■ The personnel requirements for a laboratory are determined by the level of complexity of the assays run in the laboratory. 22. A. The assay is accurate but not precise. Major points of discussion ■ A Levey-Jennings control chart plots QC results around their expected mean value. ■ Acceptable limits for the deviation of the QC results from the expected value are determined by the laboratory. ■ This is often achieved through the use of the Westgard rules, which are QC interpretive rules for statistical monitoring of assay accuracy and precision. ■ Accuracy can be assessed by measuring an analyte in reference material and comparing the result with the known certified concentration. ■ Precision is the agreement between result values obtained by repeat measurements of a given quantity of the analyte.18,30 23. A. Positive trending of QC results. Major points of discussion ■ For all tests classified as CLIA nonwaived tests, a minimum of two levels of controls must be run on each day that patient testing is performed. ■ Levey-Jennings charts show QC results over time and plot how far each point lies from the mean expected value. ■ Just by chance, QC values should fall outside 2 SDs from the mean 5% of the time. ■ There are many Levey-Jennings rules that are used to flag a QC violation. For example, if a QC value falls 2 SDs beyond the mean, then it would be denoted a 12 s violation. If it falls 3 SDs beyond the mean, it is denoted 13 s. ■ An action plan should be in place to evaluate QC violations.6 24. A. The x-axis as “Average value” and the y-axis as “% Difference.” Major points of discussion ■ Levey-Jennings charts are a graphic way of plotting QC data to monitor test quality. ■ The graphic representation allows for quick trend spotting and identification of rule violations. ■ The typical Levey-Jennings chart has the QC Run # on the x-axis and the QC result on the y-axis. ■ At least two levels of QC are typically run on an assay, and different levels should be graphed on separate Levey-Jennings chart. ■ It is often useful to denote the expected/average value of the QC material and then ± 2 SDs on the Levey-Jennings chart to help spot rule violations. 25. A. Validation of LoQ. Major points of discussion ■ By FDA method validation guidelines, the CV at the LoQ should be less than 15%. ■ By FDA method validation guidelines, the accuracy at the LoQ should be greater than 80%. ■ An appropriate LoQ is a value below the lowest, clinically relevant value of the analyte. ■ If no proposed LoQ is provided in the literature or from experience, the LoQ of the assay can be determined by either: • Establishing the analyte value for which the signal/noise ratio is more than 10, or • Running a matrix blank 20 times and adding 5 SDs to the average value obtained in this experiment.19 26. A. Analytical specificity. Major points of discussion ■ There are several ways to determine this value. One way is to average the ratio of analyte signal to background noise. For the limit of quantification, the signal/noise ratio is typically more than 10. For lower limits of detection, the signal/noise ratio is typically more than 3. ■ Another approach to establish a lower limit of quantification is by running a matrix sample without analyte (i.e., matrix blank) 20 times. To the average concentration of the blank, 4 to 5 times the SD of the multiple runs is added. This sum is now the lower limit of quantification. ■ Clinical sensitivity is the true-positive rate of an assay, which differs from analytical sensitivity. ■ Clinical specificity is the true-negative rate of an assay and differs from the analytical specificity. For laboratory-developed assays, CLIA regulations require that analytical specificity be established during test validation. There is no specific regulation for clinical specificity.19 27. A. The CDC. Major points of discussion ■ Medicaid is jointly funded by the federal government and state governments. ■ To participate in Medicaid, states are required to offer the health insurance coverage to certain population groups. ■ States can expand the health insurance coverage to additional population groups if they deem this appropriate. ■ States receive federal matching funds to provide the benefits.25 28. A. Your assay correlates poorly with the reference method. Major points of discussion ■ Classically, the reference method is plotted on the x-axis and your laboratory’s method on the y-axis. ■ If the plot is classically drawn, a slope greater than 1 implies a positive and proportional bias of your method relative to the reference method. ■ If plotted classically, a y-intercept term greater than 0 implies a constant positive bias. ■ Both constant and proportional biases may coexist in a method comparison study. ■ An R2 value approaching 1 indicates that the linear regression line drawn between the two methods is an excellent fit. However, high coefficient of determination does not rule out bias in your method. 29. A. The FDA. Major point of discussion ■ The DHHS oversees the CMS, FDA, and OIG. 30. A. Apply for state certification every 2 years and pay the annual membership fee. Major points of discussion ■ The OSHA, which enforces the Occupational Safety and Health Act, is part of the U.S. Department of Labor. ■ OSHA standards are rules that employers must follow to protect employees from hazards. ■ These standards limit hazardous chemical exposure, require certain safety practices and equipment, require monitoring hazards, and require keeping records of workplace injuries. ■ All employers are required to display a poster prepared by the Department of Labor informing employees of their protections under the Occupational Safety and Health Act. ■ OSHA regulates occupational exposure to hazardous chemicals in laboratories.26 31. A. The patient results are manually entered into the wrong patient’s medical record. Major points of discussion ■ The analytical phase of testing is defined by those procedures dealing with sample manipulation, reagent preparation, calibration, measurement, and instrument readout. ■ The postanalytical phase involves recording test results, interpretation of results, and transmission of reports. ■ Errors can occur in any and all phases of the total testing process. ■ Manual result entering and reporting increase the risk of postanalytical errors, such as the one described in this question.30 32. A. A quality assessment tool that compares a patient’s previous results with current results. Major points of discussion ■ Proficiency samples should be tested in the same manner as patient samples. ■ Proficiency samples cannot be sent to reference laboratories for result correlation. ■ Laboratories should not discuss test results with other laboratories prior to the deadline for submission. ■ PT ensures accuracy and reliability of laboratory testing. ■ Delta checks compare current result values with previous result values for a given patient and flag samples that have exceeded predefined acceptable limits for variation. ■ Delta checks are intended to detect either preanalytical or analytical errors in testing, such as specimen mix-up errors, diluted samples, or analyzer malfunction.10 33. A. The PT samples should be specifically marked and personally walked through the testing procedure by the laboratory director or his or her designee. Major points of discussion ■ The purpose of PT is to ensure the accuracy and reliability of your laboratory with regard to other laboratories performing the same or comparable assays. ■ To maintain a CLIA certificate, a laboratory must participate in a CLIA-approved PT program for all regulated moderate- and high-complexity tests performed under the CLIA certificate. ■ If a moderate- or high-complexity analyte does not fall under the “regulated” category established by CLIA, the laboratory must establish a semiannual program by which accuracy and reliability are checked.10 34. A. Failure to attain the minimum satisfactory score for an analyte, test, subspecialty, or specialty for a testing event. Major points of discussion ■ An unsatisfactory performance in PT means failure to attain the minimum satisfactory score for an analyte, test, subspecialty, or specialty for a testing event. ■ Clerical errors in entering PT results are counted as incorrect results and, therefore, as unsatisfactory performance for the analyte in question. ■ Failure to report the results of PT within the allotted time frame will cause the laboratory to receive a score of 0 for that event. ■ If a laboratory receives a nonpassing score for an analyte, the laboratory must investigate the cause of the error and consider, implement, and document corrective actions.10 35. A. You should agree. To maintain CLIA certification, all assays performed in your laboratory must be evaluated in a CLIA-approved PT program. Major points of discussion ■ FDA-waived tests need not participate in PT programs. ■ The purpose of PT programs is to ensure quality, accuracy, and reliability of your laboratory results. ■ CMS provides a list of approved PT programs in which your laboratory can enroll to meet PT regulatory criteria. ■ PT participation is required for each CLIA certificate, not for each test site. Therefore, if you have multiple testing locations under one CLIA certificate, you need to register only for one PT program for any one regulated analyte. ■ Importantly, even if a moderate- or high-complexity test is not listed as a regulated analyte in Subpart I, CLIA requires that the accuracy of the test be confirmed at least twice annually.10 36. A. Identifies a single cause of the error. Major points of discussion ■ An RCA assumes that errors are best reduced by identifying and preventing the root cause(s) of an error. ■ There are many methods and tools used in performing an RCA: • Methods: Event and causal factor mapping, tree diagrams, etc. • Tools: Ishikawa fishbone diagram, change analysis, Pareto analysis, etc ■ All methods for performing RCAs have limitations. For example, a Pareto analysis requires data to be collected to track the frequency of certain errors. If the database is inaccurate or incomplete, the RCA will not result in an appropriate solution. ■ All RCAs share common characteristics regardless of the method or tools that are used: 1. The adverse event or the problem is clearly identified. 2. No blame is placed on a single individual or group because adverse events are usually due to a chain of errors. 3. Specific causes and effects are identified; typically it identifies a system of errors rather than a single cause. 4. These causes and effects are supported by evidence. 5. Results in a clearly defined, executable, quantifiable solution(s) that can be monitored quantitatively. 37. A. Master “Black Belts” and “Green Belts.” Major points of discussion ■ Six Sigma seeks to minimize the number of defects by eliminating the cause(s) of the defects. ■ This management style requires the full commitment of upper management, as well as all employees involved in the manufacturing process. ■ Six Sigma is characterized by champions called “Black Belts” and “Green Belts,” who lead initiatives to identify errors and develop executable solutions. ■ Six Sigma has been applied to health care for cost savings and quality improvement. However, some experts feel that the method may not be as effective as it has been in the manufacturing industry.22 38. A. Billing Medicare and Medicaid for tests completed in a laboratory without a CLIA license. Major points of discussion ■ The following are considered designated health services: clinical laboratory services, physical and occupational therapy services, outpatient speech-language pathology services, radiology services, radiation therapy services, durable medical equipment and supplies, parenteral/enteral nutrients and supplies, orthotics and prosthetic devices and supplies, home health services, outpatient prescription drugs, and inpatient and outpatient hospital services. ■ There are three CLIA test categories: waived, moderate-complexity, and high-complexity testing. ■ The most commonly issued CLIA certificate is a waived certificate. ■ All laboratories must have a CLIA license to test patient samples legally.9 39. A. Volume error. Major points of discussion ■ Common causes for a bias trend in QC results include a need for recalibration of the analyzer or an error in reconstituting the QC material. ■ A wide scatter of QC values suggests imprecision or poor reproducibility of results. ■ Westgard rules are QC interpretive rules used by laboratories for statistical monitoring of the accuracy and precision of their assays. ■ The Levey-Jennings control chart is used to plot QC data results.23,31 40. A. Change to using the 13s rule. Major points of discussion ■ Westgard developed power functions in 1979 to express the probability that a particular QC interpretive rule would detect systemic error biases at a variety of magnitudes. ■ A 12s rule (i.e., one value > 2 SDs above or below the mean), although very sensitive for detecting systematic error biases, even small ones, also has a fairly high false-alert rate. ■ To improve the efficiency of QC interpretative rules, a combination of rules applied simultaneously may be considered (i.e., multirules). ■ A combination such as the 13s/22s rule would trigger investigation if either rule is broken. This combines the sensitivity and specificity of each individual rule.29 41. A. Certificate of waiver. Major points of discussion ■ The number of laboratories issued a certificate of waiver has grown to more than 50% of the approximate 225,000 laboratories enrolled as of 2011. ■ CLIA is user-fee funded; the biennial certificate fee is based on the type of certificate and the test volume of the performing entity. ■ Provider-performed microscopy procedures are considered moderately complex. ■ The certificate of compliance and certificate of accreditation are issued to laboratories that perform nonwaived (i.e., moderate- and/or high-complexity) testing. These two certificates also confer the ability to perform waived and provider-performed microscopy tests.4,5 42a. A. 12%. 42b. A. This amount of carryover is acceptable because the result of the potentially contaminated sample falls in the range of 5.0 to 5.4 mIU/mL. Major points of discussion ■ Carryover of an analyte could potentially cause a false elevation in patient results and, therefore, should be assessed when validating a new test system, if applicable to the methodology. ■ Accuracy can be assessed by measuring an analyte in reference material with a known concentration and comparing the result with the certified value. ■ Precision is the agreement between result values obtained by repeat measurements of a quantity. ■ The limit of detection is the lowest concentration of an analyte in a sample that can be reliably detected.1,13,28 43. A. Follow Universal Precautions when testing patient samples. Major points of discussion ■ The available CLIA certificates are the certificate of waiver, certificate for provider-performed microscopy procedures, certificate of registration, certificate of compliance, and certificate of accreditation. ■ The CMS-approved laboratory accreditation organizations include the AABB, COLA, American Society for Histocompatibility and Immunogenetics, CAP, and The Joint Commission. ■ Waived tests are defined as “simple laboratory examinations and procedures that have an insignificant risk of an erroneous result.” ■ The FDA is responsible for determining the level of test complexity for approved testing platforms.8 44. A. The FDA. Major points of discussion ■ CLIA was passed by Congress in 1988. ■ The CLIA program is designed to ensure laboratory testing quality. ■ The CMS oversees the operation of the CLIA program. ■ The CMS recognizes additional accrediting agencies and will issue a certificate of accreditation to laboratories that receive accreditation from the AABB, American Osteopathic Association, American Society for Histocompatibility and Immunogenetics, COLA, CAP, and/or The Joint Commission. ■ There are five types of CLIA certificates: certificate of waiver, certificate for provider-performed microscopy procedures, certificate of registration, certificate of compliance, and certificate of accreditation.4,7 45. A. Waived tests approved by the FDA. Major points of discussion ■ CLIA Quality System Regulations were implemented in 2003. ■ Measuring accuracy determines whether the test results are correct. This can be tested by analyzing samples with known concentrations. ■ Precision ensures that comparable results are obtained when the same sample is tested repeatedly. ■ The reportable range of a test is the result range that is accurate. Values above or below this range should not be reported. ■ Records of performance verification of the test system must be retained while the test is in use and for 2 years after discontinuing the test.12,13 46. A. Pinworm examination. Major points of discussion ■ The FDA determines the classification category for all CLIA-regulated tests. ■ CLIA-regulated tests fall into one of three categories: waived, moderate complexity, or high complexity. ■ Provider-performed microscopy procedures are considered moderate-complexity tests and include vaginal, cervical, or skin wet mounts; potassium hydroxide preparations; pinworm examinations; fern tests; postcoital qualitative examinations of vaginal or cervical mucus; microscopic urinalysis; fecal leukocyte examination; semen analysis for presence and/or motility of sperm; and nasal smears for eosinophils. ■ A certificate for provider-performed microscopy procedures is available from CLIA and confers the permission to perform any waived test, or any moderate-complexity test, as long as it is a provider-performed microscopy procedure.4,7,11 47. A. 1 year. Major points of discussion ■ Histopathology slide preparations must be retained for 10 years after the date of examination. ■ The CAP requires blood bank QC records be kept for a minimum of 5 years. ■ The CAP requires retention of QC records for the general laboratory for 2 years. ■ Flow cytometry plots and histograms must be kept for 10 years.2,27 48. A. 1 year. Major points of discussion ■ The CAP requires all peripheral blood smears/body fluid smears to be retained for 7 days. ■ The CAP requires all stained slides for microbiology (i.e., Gram stains) be kept for 7 days.2,27 49. A. Analyze samples from healthy subjects and determine the 95% prediction interval. Major points of discussion ■ Accuracy can also be assessed by comparing the test results from one method with the results from a reference method, using the same samples. ■ The analytical range of an assay refers to the interval between the highest and lowest concentrations of the analyte in which precision, accuracy, and linearity have been demonstrated to be acceptable. ■ The limit of detection is the lowest concentration of an analyte that can be reliably detected.13,18
Laboratory Management
Quality Management, Regulations, and Quality Control
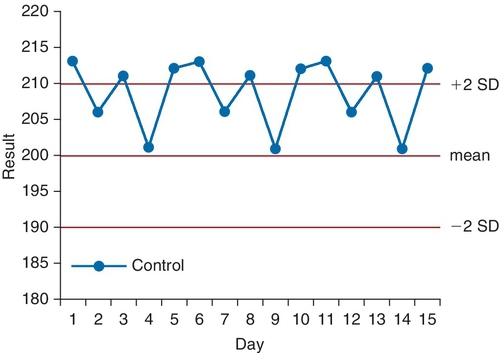
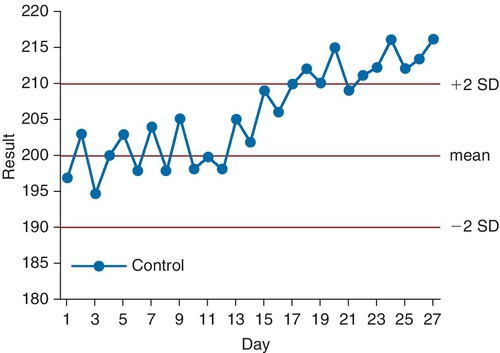
Assay Result
Standard Deviation (SD)
Low control sample, mean concentration
5 mIU/mL
0.2 mIU/mL
High control sample concentration
50,000 mIU/mL
10 mIU/mL
Potentially contaminated sample (low control sample repeated), concentration
5.1 mIU/mL
B. A legal notice provided to all Medicaid participants.
C. A legal notice provided to all uninsured patients receiving outpatient chemotherapy.
D. A legal notice provided to Medicare fee-for-service participants delineating services expected to not be covered under their Medicare insurance plan.
Rationale: An Advance Beneficiary Notice of Noncoverage is a notice provided by independent laboratories, health care practitioners, and medical suppliers to Medicare participants for services when payment is expected to be denied if the patient receives the service/supply.
E. A legal notice to all insured patients delineating services routinely not covered by major insurance carriers.
Rationale for A, B, C, and E: An advance beneficiary notice is required for people who have Medicare insurance (not uninsured patients).
Rationale: The mission of the AHRQ is to improve the safety and quality of patient care. One way to meet this goal is to support research that compares the effectiveness of new treatments.
B. To direct the Department of Health and Human Services (DHHS).
Rationale: AHRQ is 1 of 12 agencies that are directed by the DHHS.
C. To oversee the National Institutes of Health (NIH) research programs.
Rationale: AHRQ provides research funding along with its sister agency, the NIH.
D. To be the largest source of funding for biomedical research.
Rationale: The NIH is the largest source of funding for biomedical research. AHRQ provides research funding for its initiatives, but not on the order of magnitude provided by the NIH.
E. To provide health care to the elderly and indigent.
Rationale: The DHHS is responsible for Medicare and Medicaid.
Rationale: Analytical specificity refers to the assay’s ability to (1) measure the analyte in question specifically and (2) not measure other interfering substances that may affect the result.
B. Running samples without the analyte of interest (i.e., Matrix blanks), ensuring that a zero sample actually reads zero.
Rationale: This is one method for establishing the analytical sensitivity or, equivalently, the lower limit of detection/quantification.
C. Running both positive and negative samples and determining the specificity by taking true negatives and dividing by true negatives plus false positives.
Rationale: This is the method used for determining clinical specificity, or the true-negative rate.
D. Running samples spiked with increasing concentrations of an analyte and determining the concentration of the analyte that most specifically discriminates diseased from healthy patients.
Rationale: This experiment could be used to establish the appropriate cutoff for a test.
E. Running samples spiked with the highest measurable concentration of the analyte of interest followed by a Matrix blank.
Rationale: This experiment could be used to establish carryover within the analytical system.
Rationale: This would be determined by the subtracting the clinical sensitivity from 1.
B. The value obtained when the number of true positives is divided by the sum of the true positives and the false negatives.
Rationale: This is the clinical sensitivity.
C. The lowest possible concentration of an analyte that is accurately and reproducibly measured by an assay.
Rationale: See Major Points of Discussion.
D. The performance of the assay when measuring an analyte in the presence of an interfering substance.
Rationale: This is the analytical specificity.
E. The formula that relates instrument response to analyte concentration.
Rationale: This is the calibration curve.
Rationale: A training laboratory for student education typically deals only with samples that are not usually infectious and is classified as a level 1 laboratory.
B. A clinical chemistry laboratory.
Rationale: A clinical chemistry laboratory is classified as a level 2 laboratory.
C. A clinical hematology laboratory.
Rationale: A clinical hematology laboratory is classified as a level 2 laboratory.
D. A laboratory working with highly infectious fungal pathogens.
Rationale: A biosafety level 3 laboratory works with microbiological agents that may cause fatal disease through inhalation. A laboratory that works with highly infectious fungal pathogens meets this definition.
E. A satellite clinical laboratory in an emergency department.
Rationale: A satellite clinical laboratory in an emergency department usually performs only routine tests, including chemistry and hematology assays; therefore, it is classified as a level 2 laboratory.
Rationale: This is the definition of “assay calibration.” This should be done more frequently than every 6 months.
B. The process of testing known concentrations of an analyte throughout the reportable range and confirming that the assay returns the appropriate value, within an established allowable error.
Rationale: This is the correct definition of “calibration verification.”
C. The process of comparing the slope and y-intercept of the current calibration curve with the previous curves.
Rationale: This is good practice. When recalibrating an assay, tracking the slope and intercept helps identify calibration shifts. However, this is not the definition of “calibration verification.”
D. The process of running previously assayed patient samples with a new lot of calibration material and comparing them with results obtained when the same samples were run using a previous lot of calibration material.
Rationale: This is good laboratory practice when changing lots of calibration reagent. The patient samples should be chosen to span the analytical range of the assay. However, this is not the definition of “calibration verification.”
E. The process of running samples of known high and low analyte concentrations and plotting them on a Levey-Jennings chart.
Rationale: This describes a quality control (QC) performance procedure and not calibration verification.
B. Once every year.
Rationale: These are too frequent.
C. Once every 2 years.
Rationale: This is the normal frequency for inspections.
D. Once every 3 years.
Rationale: Regular CAP inspections occur more frequently than every 3 years.
E. Once every 5 years.
Rationale: Regular CAP inspections occur more frequently than every 5 years.
Rationale: The key here is that the patient samples are preselected to span the analytical range of the assay.
B. Random patient samples are run using the old and new reagent lots and then compared. The correlation coefficient should be close to 1.
Rationale: Random patient samples would not be ideal because the majority of the analyte values will cluster within the normal range. To prove no effect on patient samples, one should select a panel of patient samples that span the analytical range of the assay.
C. Calibration standards dissolved in methanol are run across the full analytical range of the instrument using old and new reagent lots and are then compared. The correlation coefficient should be close to 1.
Rationale: Running standards out of their matrix would not prove that patient samples (in matrix) will quantify correctly using a new lot of reagent.
D. The analyte should be extracted from each level of control material using a solid-phase extraction procedure and then run using the old and new reagent lots. The correlation coefficient should be close to 1.
Rationale: This is a valid approach to prove that the matrix in the high-QC material is, in fact, causing the discrepancy. However, it does nothing to prove that patient samples, which are always in matrix, will react the same between different reagent lots.
E. The assay should be recalibrated; the control material should then be run and compared between both reagent lots.
Rationale: This could help, particularly if the QC run using the old reagent lot were quantified relative to a different calibration curve than the QC run using the new reagent lot. However, it does nothing to prove that patient samples, which are always in matrix, will react the same between different reagent lots.
Rationale: This is issued to a laboratory that performs only waived testing. This certificate is not needed if the laboratory already operates under a certificate that allows for higher complexity testing.
B. You must apply for a CLIA certificate of registration.
Rationale: This certificate is issued to a laboratory to allow it to conduct nonwaived (i.e., moderate- and/or high-complexity) testing until the laboratory is surveyed (by the state Department of Health or an accreditation agency) to determine its compliance with CLIA regulations. Only laboratories that apply for a certificate of compliance or a certificate of accreditation will receive a certificate of registration. This is not needed for waived testing.
C. You must apply for a CLIA certificate of compliance.
Rationale: This certificate is issued to a laboratory once the state Department of Health conducts an inspection and determines that the laboratory is compliant with all applicable CLIA requirements. This type of certificate is issued to a laboratory that performs nonwaived testing and is not needed to add a waived test.
D. No additional certificates are necessary.
E. You cannot run a waived test in a laboratory that performs predominantly moderate-complexity testing.
Rationale: You can perform a waived test under either a certificate of compliance or a certificate of accreditation, both of which allow the laboratory to conduct moderate- or high-complexity tests.
Rationale: The FDA does not directly oversee the CLIA program. However, the FDA is responsible for classifying laboratory test systems as waived, moderate-complexity, or high-complexity.
B. The AABB.
Rationale: The AABB publishes standard guidelines for blood banks, cell therapy, and transfusion services, not for general clinical laboratories.
C. The CAP.
Rationale: The CAP does not oversee the CLIA program. CAP does publish laboratory standards and offers a laboratory accreditation program, which is a voluntary program separate from CLIA.
D. The Joint Commission.
Rationale: The Joint Commission does not oversee CLIA. The Joint Commission does publish laboratory standards and offers an accreditation program for laboratories, which is a voluntary program separate from CLIA.
E. The Centers for Medicare and Medicaid Services (CMS).
Rationale: CMS oversees the enforcement of CLIA regulations.
Rationale: These are the four performance characteristics that CLIA regulations require a laboratory to confirm prior to performing an FDA-approved moderate-complexity assay. The reference range study does not have to be a full 120-patient study; rather, it is meant to confirm that manufacturer’s ranges are appropriate for your patient population.
B. Accuracy, analytical sensitivity and specificity, reportable range, and a full reference range study with a minimum of 120 patient samples.
Rationale: A full reference range study requires 120 individuals in each demographic subset. This is not required to confirm the manufacturer’s reference ranges. Generally, 20 to 40 healthy individuals will suffice for a reference range confirmation.
C. Accuracy, clinical sensitivity and specificity, analytical specificity and sensitivity, full reference range study, and reportable range.
Rationale: These are some of the more advanced performance parameters that laboratories introducing laboratory-developed tests would assess.
D. Extraction efficiency, reportable range, full reference range study, QC material stability, and analytical sensitivity and specificity.
Rationale: These are more detailed and assay-specific performance characteristics that some laboratories will establish when validating laboratory-developed tests.
E. Only accuracy and precision must be confirmed.
Rationale: There are four performance characteristics defined by CLIA. Accuracy and precision are two of them. The other two are reportable range and reference range.
B. Once every 24 hours.
Rationale: QC samples must be assayed once every 24 hours, or more frequently if recommended by the manufacturer.
C. Every time the assay is performed.
Rationale for A and C: These are too frequent.
D. Once every 48 hours.
Rationale: QC must be run more often than every 48 hours if the test is offered continuously (i.e., 24 hours a day).
E. Once every week.
Rationale: QC samples must be assayed more often than once a week for tests that are offered 24 hours day.
Rationale: The certificate of registration is an intermediate step that allows your laboratory to begin testing until the laboratory is inspected by either your state Department of Health or a deemed accreditation agency.
B. After you are inspected by your state Department of Health and have received your certificate of compliance.
C. After you have been inspected by an approved accreditation agency and have received your certificate of accreditation.
Rationale: Neither of these certificates is required to begin testing, unless otherwise specified by your state Department of Health.
D. After your state Department of Health, or an approved accreditation agency, and CMS have both inspected your laboratory.
Rationale: CMS inspects only a small number of laboratories on a random basis. This is not required to begin testing.
E. Once you have validated your assays to your satisfaction.
Rationale: You must apply for a CLIA license to provide patient testing results.
Rationale: Delta checks do not involve reference range comparisons.
B. It is a quality measure by which a patient’s previous result is compared with the current one.
Rationale: Delta checks compare current result values with previous result values.
C. It is a quality measure by which samples are visually checked for hemolysis.
Rationale: A delta check is not a visual assessment for hemolysis.
D. It is a level of an analyte thought to be life-threatening to the patient, requiring verbal notification to the ordering provider.
Rationale: This is a critical value.
E. It is a quality measure that occurs when results fall above the analytical limit of an assay.
Rationale: A delta check does not assess the analytical limit of an assay.
B. The FMLA provides employees with up to 16 weeks of paid, job-protected leave for the birth or care of a newborn child.
Rationale: The FMLA does not enforce paid leave.
C. The FMLA provides employees with up to 16 weeks of unpaid, job-protected leave for females and 12 weeks for males for the birth or care of a newborn child.
Rationale: The FMLA does not have gender-specific standards.
D. The FMLA provides employees with up to 12 weeks of unpaid, job-protected leave per year leave for qualifying reasons.
Rationale: Employers must grant up to12 weeks of unpaid leave for qualifying reasons.
E. The FMLA provides employees with continuous health insurance coverage for up to 16 weeks.
Rationale: The FMLA provides health insurance coverage for only 12 weeks.
B. The CAP.
Rationale: The AABB and CAP have accreditation standards for blood banks, but they do not set the federal regulations.
C. The CMS.
D. The CDC.
Rationale: The CMS and CDC do not regulate blood banks.
E. The FDA.
Rationale: The FDA oversees the regulations, and their compliance, for all blood banks.
Rationale: The FDA considers laboratory assays to be medical devices.
B. LDTs are considered medical devices, and each laboratory must receive FDA approval before implementing an LDT.
Rationale: Although LDTs are considered medical devices, the FDA exercises enforcement discretion on this issue to encourage rapid implementation of the newest, most-effective diagnostics into laboratories to improve public health.
C. LDTs are considered medical devices, but the FDA has exercised enforcement discretion on LDTs.
Rationale: Although LDTs are, indeed, laboratory devices, the FDA exercises enforcement discretion on this issue allowing laboratories to implement their assays after an in-house, CLIA-appropriate validation.
D. LDTs are not considered medical devices, but the FDA regulates them.
Rationale: The FDA does not regulate LDTs.
E. LDTs may or may not be considered medical devices, and the FDA considers each assay on a case-by-case basis and decides whether to regulate.
Rationale: The FDA is clear on its stance on the status of LDTs as medical devices over which it could exercise regulatory control, but chooses not to, in the interest of enhancing public health.
Rationale: Title 1 of the Americans with Disabilities Act makes it unlawful for an employer to discriminate against a qualified applicant based on disability.
B. Were you born in the United States?
C. What is your native language?
Rationale: Title VII of the Civil Rights Act of 1964 makes it illegal to discriminate against someone on the basis of race, color, religion, national origin, or gender.
D. How old are you?
Rationale: The Age Discrimination in Employment Act (ADEA) forbids age discrimination only against people who are age 40 years or older.
E. Are you capable of performing all of the duties required by this position?
Rationale: This is the best way to gather information about whether the potential employee is mentally and physically capable of performing the job. There is no need to ask about specific disabilities, age, or language fluency.
Rationale: ISO 15189:2007 is the “Medical laboratories—Particular requirements for quality and competence” standards.
B. Standards for pricing and reimbursement in medical laboratories.
Rationale: ISO 15189:2007 does not regulate laboratory test pricing.
C. Laboratory services that must be covered under universal health insurance plans.
Rationale: ISO 15189:2007 does not regulate health insurance.
D. Standard requirements for laboratory technologist school curricula.
Rationale: ISO 15189:2007 does not regulate laboratory technologist training.
E. Standard requirements for retirement benefit packages.
Rationale: ISO 15189:2007 does not regulate employee work benefits.
Rationale: A high-complexity laboratory requires a technical supervisor whose training requirements and responsibility are defined in CLIA Subpart M Section 493.1449 Standard; Technical supervisor qualifications.
B. Clinical consultant.
Rationale: A high-complexity laboratory requires a clinical consultant whose training requirements and responsibility are defined in CLIA Subpart M Section 493.1455 Standard; Clinical consultant qualifications.
C. General supervisor.
Rationale: A high-complexity laboratory requires a general supervisor whose training requirements and responsibility are defined in CLIA Subpart M Section 493.1461 Standard: General supervisor qualifications.
D. Testing personnel.
Rationale: A high-complexity laboratory requires a testing personnel whose training requirements and responsibility are defined in CLIA Subpart M Section 493.1489 Standard; Testing personnel qualifications.
E. The laboratory director can assume any and all of these positions.
Rationale: Although all of the positions described in answers A through D are required by CLIA to operate a high-complexity laboratory, the regulations do not require that separate individuals fill each role. Indeed, as long as the laboratory director meets the qualification standards in Subpart M for each position, he or she may fill any and all of the positions.
Rationale: Accuracy refers to how closely an assay result value matches the true value of the analyte. This chart would show scatter around the expected value.
B. The assay is precise but not accurate.
Rationale: Precision refers to the reproducibility of the results.
C. The assay is neither precise nor accurate.
Rationale: The QC results are scattered, representing a large SD, and there is a bias trend away from the expected mean value. This suggests imprecision and inaccuracy.
D. The assay is clinically useless.
Rationale: Levey-Jennings control charts do not assess the clinical utility of laboratory tests but, rather, assess if the assay is performing as expected.
E. The assay is precise and accurate.
Rationale: A precise and accurate assay would have an observed mean value close to the expected mean value and a small SD.
Rationale: These QC results trend at approximately 2 SDs above the mean value.
B. Negative trending of QC results.
Rationale: These QC results trend in a positive direction, above the mean value, not in a negative direction.
C. Result interference by an unknown substance.
Rationale: Levey-Jennings charts are not used to analyze interference.
D. The use of an inappropriately narrow reference range.
Rationale: Levey-Jennings charts are not used to analyze reference ranges.
E. The need to change the technical range for the assay.
Rationale: Levey-Jennings charts are not used to determine the technical range of an assay.
Rationale: This is typical for a Bland-Altman or bias plot.
B. The x-axis as the “Gold standard method” and the y-axis as “Test method.”
Rationale: This is an example of a method comparison plot.
C. The x-axis as “Calibrator level” and the y-axis as “% CV.”
Rationale: This is a nice way of visualizing trends in reproducibility with increasing analyte concentration.
D. The x-axis as “Run #” and the y-axis as “Concentration.”
Rationale: This is the classic Levey-Jennings chart. Often, the expected, average value for the control material and the ± 2 SD limits are also noted as dashed or dotted lines on the plot.
E. The x-axis as “Instrument response” and the y-axis as “Expected concentration.”
Rationale: This is a calibration curve.
Rationale: If the reproducibility (CV < 15%) and accuracy (> 80%) meet a prespecified value, the LoQ is validated. However, there is additional information available from these experiments.
B. Validation of the LoQ, intraday and interday reproducibility, and accuracy at the LoQ.
Rationale: The within-day and between-day reproducibility are determined by running the same sample multiple times per day for multiple days. The values of these 20 runs can then be averaged to obtain a value for the sample that can be compared with the “known” value of the sample to establish accuracy at the LoQ. As long as these two measures fall within preestablished criteria, the proposed LoQ has been verified.
C. Validation of the LoQ and establishment of accuracy at the LoQ.
Rationale: The intraday and interday reproducibility is also determined by this experiment.
D. Validation of the LoQ and establishment of the reportable range.
Rationale: The reportable range is generally established by running samples across the hypothesized range and assessing linearity, usually by linear regression methods.
E. Establishment of reproducibility at the LoQ.
Rationale: Reproducibility is established at the LoQ, but additional information is also available.
Rationale: Analytical specificity refers to an assay’s ability to discriminate the analyte of interest from possible interferences.
B. Analytical sensitivity.
Rationale: Analytical sensitivity defines the lowest value of an analyte that can reproducibly and accurately be measured by an assay. The higher the sensitivity, the lower is the possible concentration of analyte that it can measure.
C. Clinical specificity.
Rationale: Clinical specificity is defined as the true-negative rate—that is, how often the assay categorizes samples from patients without disease as “healthy.”
D. Clinical sensitivity.
Rationale: Clinical sensitivity is defined as the true-positive rate—that is, how often the assay categorizes samples from patients with disease as “diseased.”
E. The lower limit of linearity.
Rationale: Although the lower limit of linearity may be the same as the lower limit of detection/quantification, more often, it is a lower value. Lower limits of linearity are usually determined by running serial dilutions of a standard and then analyzing the results using linear regression.
Rationale: The CDC primarily focuses on infectious disease epidemiology and research.
B. The FDA.
Rationale: The FDA primarily regulates food safety, dietary supplements, medications, vaccines, blood transfusions, medical devices, and veterinary products.
C. The American Medical Association.
Rationale: The American Medical Association is a physician trade association.
D. Individual states.
Rationale: Individual states establish and administer their own Medicaid programs.
E. Private insurance companies.
Rationale: Medicaid is not a form of private health insurance.
Rationale: The R2 value is 0.99, which indicates excellent correlation.
B. Your assay correlates well with the reference method but has a positive proportional bias.
Rationale: The slope greater than 1 implies that for every unit increase in concentration detected in the gold standard method, your method detects an increase of 1.34 concentration units. This is proportional and positive bias. The R2 value of 0.99 implies excellent correlation.
C. Your assay correlates well with the reference method but has a negative constant bias.
Rationale: A negative constant bias would be reflected in a negative intercept term.
D. Your assay correlates well with the reference method. No other statements can be accurately made.
Rationale: Although your assay does correlate well with the reference method, the slope and y-intercept provide additional data that can and must be interpreted in this method correlation experiment.
E. Your assay correlates poorly with the reference method and shows a positive proportional bias.
Rationale: The two assays correlate very well with one another.
Rationale: The FDA is a part of the DHHS and regulates the manufacture of biologics, medical devices, and test kits.
B. The NIH.
Rationale: The NIH is an agency of the DHHS, the main aim of which is the performance and funding of biomedical research.
C. The CMS.
Rationale: CMS sets quality standards and reimbursement rates that apply to laboratories. These rates are often then adopted by other third-party payers.
D. The National Institute of Standards and Technology (NIST).
Rationale: The NIST is a branch of the U.S. Department of Commerce. Among other things, the NIST develops and distributes standardized calibration materials for a finite list of analytes.
E. The DHHS.
Rationale: This federal agency oversees the CMS, FDA, and OIG. It is not involved in developing standards for calibration.
Rationale: There is no application process for OSHA.
B. Provide subsidized health insurance for all employees and offer at least two separate insurance plans.
Rationale: OSHA does not mandate that employees provide health insurance.
C. Display an OSHA poster in the workplace informing employees about the Occupational Safety and Health Act.
Rationale: Employers are required to display a poster prepared by the Department of Labor containing this information.
D. Require that all employees receive an annual physical health exam with urine drug screening.
Rationale: OSHA does not mandate health exams or drug testing.
E. Receive state OSHA agency approval before increasing the number of employees.
Rationale: OSHA does not regulate staffing numbers.
Rationale: This is an example of postanalytical error.
B. The sample is drawn into an inappropriate tube.
C. The sample is run using a reagent lot that has expired.
D. The phlebotomist did not fill the tube completely on a sample sent for coagulation testing.
Rationale for B and D: These are examples of preanalytical errors.
E. The autosampling pipette was jostled and is now drawing only 1.3 μL of sample instead of 1.5 μL of sample.
Rationale for C and E: These are examples of analytical errors.
Rationale: This is a delta check.
B. A quality assessment tool that involves testing two levels of QC material before testing patient samples.
Rationale: This describes standard QC procedures that ensure real-time test accuracy.
C. A quality assessment tool by which laboratories test samples with unknown results and compare their results.
Rationale: PT is an external assessment tool used to grade laboratory testing quality.
D. A quality assessment tool that involves running calibrators every 6 months to ensure linear assay results.
Rationale: PT does not involve recalibration of analyzers.
E. A quality assessment test given to all newly hired technologists to ensure appropriate training and competency.
Rationale: PT occurs on a regular basis and is not restricted to newly hired individuals.
Rationale: PT samples should be entered into your laboratory’s normal workflow and run by regular testing personnel (i.e., treated like a patient sample).
B. The PT sample should be split: one aliquot should be run in your laboratory and the other aliquot should be sent to a reference laboratory to ensure concordant results.
Rationale: PT samples should never be sent out to a reference laboratory or to any other laboratory.
C. The PT sample should be entered into your laboratory’s workflow and tested and resulted as with any other patient specimen.
Rationale: This is the correct answer.
D. The PT sample should be treated as any other patient sample. If send-out is required as a reflex for this analyte, your laboratory should send it out as per protocol.
Rationale: If your laboratory protocol requires a reflex test, which is offered only at a reference laboratory as a send-out, the PT sample should never be sent out for analysis. Instead, check the box on the proficiency test result form that indicates “would send out” or “confirmatory testing not performed in house.”
E. The PT sample should be run multiple times over multiple days and the values averaged to minimize error.
Rationale: PT samples should be evaluated as if they were routine patient samples. Unless your laboratory averages multiple runs to obtain a patient value, this is an incorrect answer.
Rationale: This is the definition of an unsatisfactory PT performance.
B. Failure to submit the results of a testing event in the requisite amount of time.
Rationale: Although this is counted as a score of 0 for the analyte for that testing event and is, therefore, an unsatisfactory performance and/or an unsuccessful performance, this is not the correct definition.
C. A clerical error in reporting the results for a testing event.
Rationale: Although this is counted as an incorrect result and is, therefore, an unsatisfactory PT performance for the analyte in question and, depending on previous performance, may also be an unsuccessful performance, this is not the correct answer.
D. Repeated unsatisfactory test scores for an analyte or for the entire testing event in a particular specialty or subspecialty on two consecutive PT events or two of three testing events.
Rationale: This is the definition of unsuccessful participation/performance in PT.
E. This is not a term used in assessing performance in PT events.
Rationale: This is indeed a term used to express multiple unsatisfactory performances for an analyte or within a specialty/subspecialty.
Rationale: PT program registration is required only for a defined number of moderate- and high-complexity tests specified in CLIA ’88 Subpart I (Proficiency Testing of Nonwaived Testing).
B. You should agree. Although PT is not required for waived tests, if a valid PT program exists, participation will help ensure your laboratory’s accuracy in result reporting.
Rationale: This answer reflects the best laboratory medicine practice. Although PT is not required for waived tests, the purpose of PT is to ensure quality and accuracy. If a PT program is available and your laboratory can feasibly participate, it should.
C. You should disagree. After looking through the CLIA regulations for PT, you notice the analyte you are measuring is not on the list of regulated analytes for which PT is required.
Rationale: This test is not subject to PT requirements because it is a waived test, not because it does not appear on the list of CLIA regulated analytes. “Regulated” analytes (defined under subpart I of CLIA regulations) are, by definition, tested using moderate- or high-complexity methods.
D. You should disagree. The FDA does not require PT for tests it deems waived.
Rationale: The FDA does not regulate PT; this is handled by CMS through CLIA.
E. You should disagree. PT will only highlight your laboratory’s inaccuracy.
Rationale: The purpose of PT is to help a laboratory identify areas in which it needs improvement in terms of analytical accuracy.
Rationale: A single cause of error is rarely identified. It is usually a system of errors that contributes to an adverse event.
B. Favors the subjective opinion of experts.
Rationale: An RCA should be supported by evidence.
C. Results in an executable and quantifiable solution(s).
Rationale: The solution(s) from an RCA should be executable and should be monitored by a quantitative method.
D. Seeks to identify and blame the specific person or group responsible.
Rationale: An RCA should not seek to place blame on any individual or group. Rather, it usually identifies multiple systemic errors that contribute to an event.
E. Uses the Ishikawa fishbone diagram.
Rationale: A fishbone diagram is a specific tool that can be, but is not always, used to identify specific reasons an error occurred.
Rationale: Master “Black Belts” and “Green Belts” champion Six Sigma initiatives.
B. Merger and acquisitions attorneys.
Rationale: Attorneys may be needed for certain projects but are not required for implementation of Six Sigma.
C. Only the chief executive officer commitment is required.
Rationale: A commitment from all employees in the organization is required for Six Sigma to be effective.
D. Six Sigma consultants.
Rationale: Six Sigma consultants may be useful in initiatives but are not required.
E. Support of engineers.
Rationale: All employees of an organization must commit to Six Sigma, but not all organizations have engineers.
B. Professional billing for laboratory tests without a physician-documented order that includes the physician’s signature.
Rationale: Section 1877 of the Social Security Act does not address laboratory CLIA licensing or physician laboratory test ordering.
C. Physician patient referral to an entity for a designated health service from which the physician will financially benefit.
Rationale: This is commonly known as the Stark Law. This law prohibits a physician from making referrals for designated health services payable by Medicare to an entity with which he or she or an immediate family member has a financial relationship, unless a specific exception applies.
D. Performing genetic tests without a signed consent form.
E. Performing HIV tests without a signed consent form.
Rationale: Section 1877 of the Social Security Act does not address legal issues surrounding patient consent.
Rationale: It is unlikely that a volume error would produce this pattern of QC results.
B. User error.
Rationale: It is unlikely that user error would produce this pattern of QC results.
C. Random error.
Rationale: Eight sequential QC results falling 1.5 SDs above or below the mean expected value does not suggest random error but rather a systemic problem.
D. Imprecision.
Rationale: Imprecision would be suggested by a wide range of QC results for the same level of QC material.
E. A bias trend.
Rationale: Eight sequential results that exceed 1.5 SDs of the expected mean value suggest a bias trend in the results.
Rationale: This would definitely decrease your false-alert rate, but it would also decrease your sensitivity for error detection.
B. Change to using the 81s rule.
Rationale: This would decrease false-alert rates but would lack the real-time sensitivity for error detection. This rule also is made to detect systematic errors that may develop subtly over time.
C. Adopt a hybrid rule combining the 13s and 22s rules in a multirule scheme.
Rationale: This combines the sensitivity of a 22s rule and the false-alert rate of the 13s rule. Combination rules (i.e., multirules) are often used to combine the sensitivity and specificity of individual rules.
D. Rules are not necessary, and each QC value should be judged individually.
Rationale: Every laboratory should have clearly stated QC interpretation rules that it follows daily.
E. Ask the supervisor to change lots of QC material; the failure rate is likely due to a faulty batch.
Rationale: The supervisor’s complaint was not lot specific but more of a trend she noticed in QC over time.
Rationale: The most commonly issued type of CLIA certificate is the certificate of waiver. This allows providers to perform only waived tests and is effective for 2 years after the date of issue.
B. Certificate for provider-performed microscopy procedures.
Rationale: This is the second most commonly issued certificate. It is issued to providers that want to perform only approved microscopy procedures but no other moderately complex tests.
C. Certificate of registration.
D. Certificate of compliance.
Rationale: These two certificates are issued to laboratories that perform moderate- or high-complexity testing after inspection that determines their compliance with CLIA requirements.
E. Certificate of accreditation.
Rationale: This certificate is issued to laboratories accredited by an organization other than CLIA, such as the CAP.
Rationale: 12% carryover of this sample would correspond to a concentration of 0.6 mIU/mL.
B. 2%.
Rationale: Percent carryover equals [(Results from potentially contaminated low sample – Original results from the uncontaminated low sample)/(Original results from the uncontaminated sample)] multiplied by 100. In this example, it is (5.1 –5.0)/5.0 × 100 = 2%.
C. 15%.
Rationale: 15% carryover of this sample would correspond to a concentration of 0.75 mIU/mL.
D. 10%.
Rationale: 10% carryover of this sample would correspond to a concentration of 0.5 mIU/mL.
E. 5%.
Rationale: 5% carryover of this sample would correspond to a concentration of 0.25 mIU/mL.
Rationale: The acceptable limit for the carryover study was determined to be 2 times the SD of the original low control result. The SD is 0.2 mIU/mL; therefore, the acceptable range is 5.0 to 5.4 mIU/mL. This corresponds to no more than an 8% increase over the original result of 5.0 mIU/mL.
B. This amount of carryover is acceptable because it is less than 10 mIU/mL.
Rationale: 10 mIU/mL is above the acceptable amount of carryover. The acceptable amount of carryover in this example was determined to be no greater than 0.4 mIU/mL. This is twice the SD of the low control result mean. This corresponds to an acceptable result value up to 0.4 mIU/mL above the “true” result.
C. This amount of carryover is acceptable because it is less than 15% of the original sample.
Rationale: 15% of the original sample corresponds to 0.75 mIU/mL (15% of 5.0), which is more than the acceptable amount of carryover.
D. This amount of carryover is not acceptable because it is more than 5% of the original sample.
Rationale: The amount of carryover is not greater than 5% of the original sample (0.1 mIU/mL of carryover corresponds to 2% of 5.0 mIU/mL).
E. This amount of carryover is not acceptable because it is more than 0.2 mIU/mL.
Rationale: This limit is too low.
Rationale: Universal Precautions are not addressed in the CLIA requirements for the certificate of waiver.
B. Have a designated laboratory director who holds a medical degree.
Rationale: There is no special training or education required to apply for a certificate of waiver.
C. Follow the test manufacturer’s instructions.
Rationale: The CLIA requirements for waived testing include enrolling in the CLIA program; paying the certificate fee every 2 years; following the manufacturer’s instructions on how to perform the test; notifying the appropriate state agency of any changes in ownership, name, address, or directorship; and permitting inspections by the CMS.
D. Engage in CLIA-directed PT.
Rationale: PT is not required for waived tests.
E. Record all patient test results in an electronic format.
Rationale: Electronic test reporting is not required by CLIA.
Rationale: The FDA does not directly oversee the CLIA program; however, the FDA is responsible for classifying laboratory test systems as waived, moderate complexity, or high complexity.
B. The AABB.
Rationale: The AABB publishes standard guidelines for blood banks, not general clinical laboratories.
C. The CAP.
Rationale: The CAP does not oversee the CLIA program. CAP does publish laboratory standards and offers a laboratory accreditation program, which is a voluntary program and is separate from CLIA.
D. The Joint Commission.
Rationale: The Joint Commission does not oversee CLIA. The Joint Commission does publish laboratory standards and offers an accreditation program for laboratories, which is a voluntary program and separate from CLIA.
E. The CMS.
Rationale: The CMS oversees the enforcement of the CLIA regulations.
Rationale: CLIA requires entities performing waived tests to follow the manufacturer’s instructions. Performance verification is not required.
B. Provider-performed microscopy tests approved by the FDA.
Rationale: Performance verification is not required for provider-performed microscopy procedures.
C. Moderate-complexity tests approved by the FDA.
Rationale: The Quality System Regulations became effective in 2003. These regulations require laboratories to verify the manufacturer’s stated performance for accuracy, precision, reportable range, and reference ranges for moderate-complexity tests (other than provider-performed microscopy) that the laboratory wishes to perform. This helps ensure that the test performs as the manufacturer intended.
D. Waived, provider-performed microscopy, and moderate-complexity tests approved by the FDA.
Rationale: Performance verification is not required for waived or provider-performed microscopy tests.
E. All FDA-approved tests used for patient care.
Rationale: Performance verification is not required for tests classified as waived.
Rationale: This is a provider-performed microscopy test.
B. Fecal occult blood examination.
Rationale: Fecal occult blood examination is a waived test. Other common waived tests include urine pregnancy tests by visual color comparison, blood glucose tests that use glucose monitoring devices cleared by the FDA for home use, and dipstick or tablet reagent urinalysis. All of the other choices are categorized as provider-performed microscopy procedures. These include vaginal, cervical, or skin wet mounts; potassium hydroxide preparations; pinworm examinations; fern tests; postcoital qualitative examinations of vaginal or cervical mucus; microscopic urinalysis; fecal leukocyte examination; semen analysis for presence and/or motility of sperm; and nasal smears for eosinophils.
C. Urine sediment examination.
D. Fecal leukocyte examination.
E. The fern test.
Rationale: These are provider-performed microscopy tests.
B. 2 years.
C. 5 years.
D. 10 years.
E. Indefinitely.
Rationale: According to the Code of Federal Regulations, Title 42, section 493.1105 Standard: Retention requirements, laboratories must retain cytology slide preparations for at least 5 years from the date of examination.
B. 2 years.
C. 5 years.
Rationale: Cytopathology slide preparations must be retained for 5 years from the date of examination.
D. 10 years.
Rationale: Histopathology slide preparations must be retained for 10 years after the date of examination.
E. Indefinitely.
Rationale for A, B, and E: According to the Code of Federal Regulations, Title 42, section 493.1105 Standard: Retention requirements, laboratories must retain or be able to retrieve a copy of the original test report (including final, preliminary, and corrected reports) for at least 2 years after the date of reporting.
Rationale: This determines the reference range.
B. Analyze samples with no measurable analyte spiked with low concentrations of analyte.
Rationale: This determines the analytical sensitivity of the test—that is, the lower limit of detection.
C. Analyze samples with a high value followed by samples with a low value.
Rationale: This assesses for analyte carryover.
D. Analyze a sample 20 times and determine the mean and SD.
Rationale: This assesses the precision of the test.
E. Compare samples between the test method and a reference method.
Rationale: Accuracy refers to how close a measured value is to the actual concentration of the analyte in the sample.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


