Intrarenal Disorders
Robin Beeman and Roberta J. Emerson
Key Questions
• How are renal tumors differentiated, detected, and managed?
• How do autosomal dominant and autosomal recessive forms of polycystic kidney disease differ?
• What risk factors and clinical findings are associated with pyelonephritis?
• How are the various forms of glomerulonephritis differentiated?
• What laboratory and clinical findings suggest a diagnosis of nephrotic syndrome?

http://evolve.elsevier.com/Copstead/
Functional kidneys are necessary for the removal of waste products from the blood and the maintenance of fluid, electrolyte, and acid-base balance despite wide variations in intake and losses. Systemic disorders that alter the delivery of blood flow to the kidney may adversely affect the kidney’s ability to perform its filtering and homeostatic functions. In addition, many disorders occur primarily within the kidney and have the potential to result in chronic kidney disease or end-stage renal disease (ESRD). In general, these disorders can be categorized as (1) congenital, (2) neoplastic, (3) infectious, (4) obstructive, and (5) glomerular.
Common Manifestations of Kidney Disease
Pain
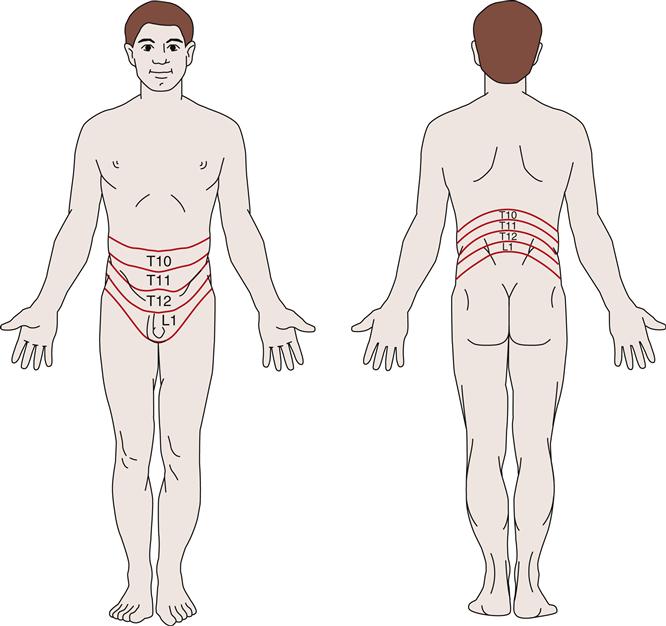
Thorough pain assessment is an essential component of the history and physical examination of any patient. The results can be useful in localizing the etiology of the pain, but assessment is also challenging because pain perceived as coming from the abdomen can originate from many varied organs and tissues within the abdomen or extraabdominally (see Chapter 47). Pain associated with the urinary tract may originate from the lower urinary tract (ureters, bladder, or urethra) or the kidney itself. Renal or kidney pain is also referred to as nephralgia (-algia is from the Greek algos, meaning pain). Extensive damage to a kidney can occur without nephralgia because most of the kidney lacks pain receptors. However, the renal capsule is innervated by nociceptors, and when a disease process causes it to be distended, inflamed, or punctured, a dull to sharp pain is felt. Distention or inflammation produces a dull, constant pain. This may be the result of intrarenal fluid accumulation such as occurs with inflammation, infected or bleeding cysts, hemorrhage from blunt trauma, or neoplastic expansion. In addition, whenever the renal capsule is penetrated (e.g., during biopsy or trauma), a dull pain or intense pressure may be felt. The renal pelvis and the rest of the urinary tract are innervated by many pain receptors. Obstruction of the intrarenal collecting system causes pain if the obstruction leads to distention of the renal pelvis or capsule. Large calculi, however, can develop insidiously in the renal pelvis or calices and may be painless until they start to move into the ureteral junction. Ischemia caused by the occlusion of renal blood vessels (e.g., from an embolus, atherosclerotic disease, or neoplasm) results in a constant dull or sharp pain.
Pain associated with intrarenal disorders affecting the capsule is classically assessed by palpation or light percussion over the costovertebral angle posteriorly and is recorded as CVA tenderness or flank pain. Sympathetic nerves transmit information from renal and ureteral nociceptors to the spinal cord between the T10 and L1 levels. Because these sympathetic nerves enter the spinal cord at this level, the pain can be felt throughout the corresponding T10-L1 dermatomes. A dermatome is an area of skin innervated by a specific spinal cord segment (Figure 27-1). Visceral and cutaneous afferent fibers enter the spinal cord in close proximity and converge on some of the same neurons at the spinal, thalamic, and cortical levels of the central nervous system. When visceral pain fibers are stimulated, concurrent stimulation of cutaneous fibers occurs and the visceral pain is perceived as though it had originated in the skin. Nerve fibers from the renal plexus communicate with the spermatic plexus, and because of this association, scrotal pain in males and labial pain in females may accompany renal pain.
Abnormal Urinalysis Findings
Urinalysis is an essential laboratory test for all suspected problems of the genitourinary system1 (see Chapter 26). After history taking and a physical examination, urinalysis generally serves as a starting point for the differential diagnosis. First, urine is examined grossly, encompassing both the solvent and the solutes. The color, odor, and turbidity of the urine offer the first clues. Dark, strong-smelling urine may be an indicator of decreased renal function. Cloudy, pungent urine generally indicates an infectious process, the turbidity being a result of leukocytes in the urine. Dipstick tests and microscopic analysis provide a great deal of additional information. Microscopic examination entails the assessment of the urine sediment, the portion that remains after the urine specimen is centrifuged.1 Table 27-1 lists major kidney disorders that are associated with abnormalities identified by urine dipstick testing. Table 27-2 provides abnormal microscopic urinalysis results indicative of kidney disorders.
TABLE 27-1
URINE DIPSTICK FINDINGS ASSOCIATED WITH KIDNEY DISORDERS
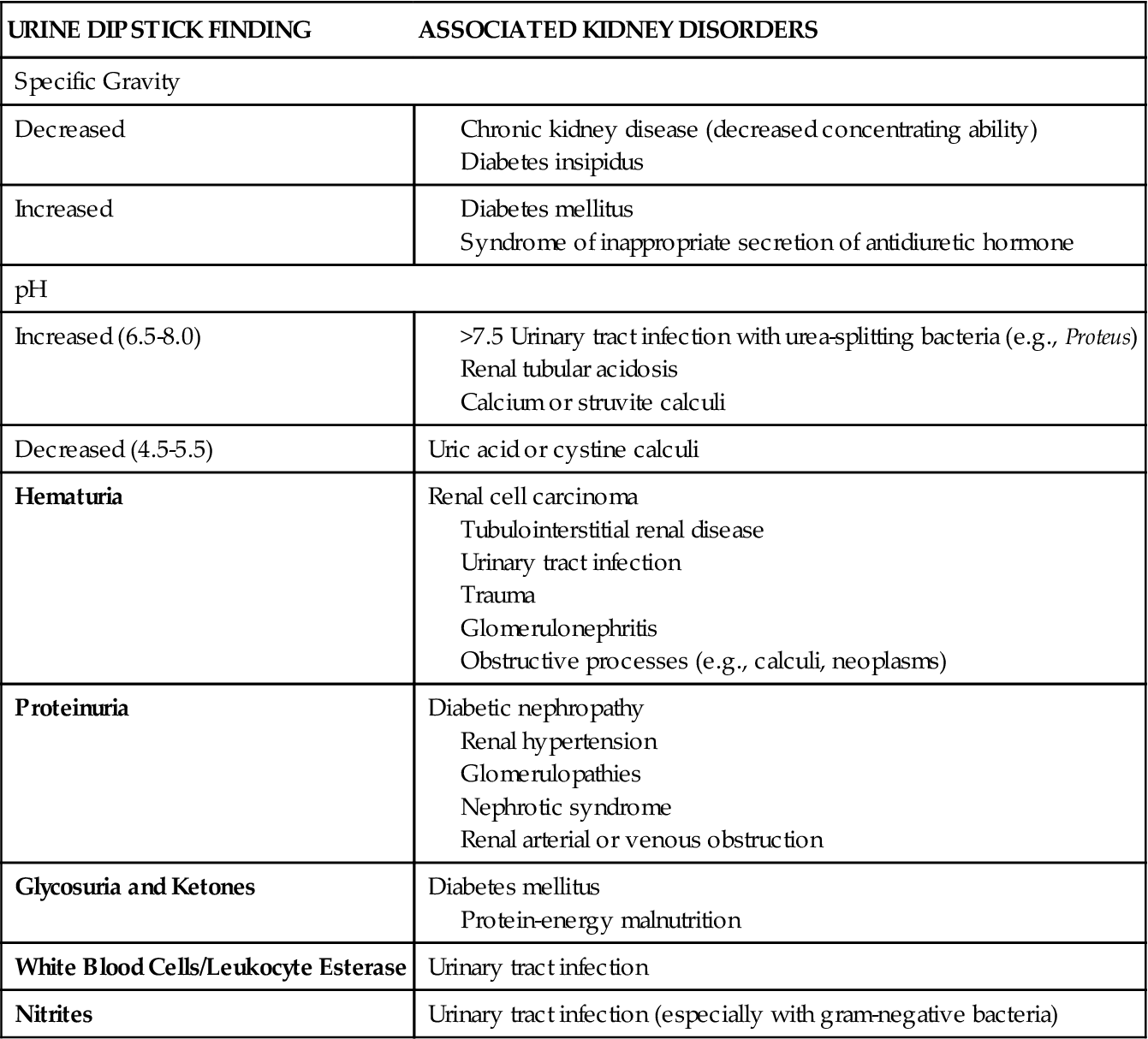
Data derived from Gerber GS, Brendler CB: Evaluation of the urologic patient: history, physical examination, and urinalysis. In Wein JA et al, editors: Campbell-Walsh urology, ed 9, Philadelphia, 2007, Saunders, pp 97-105.
TABLE 27-2
MICROSCOPIC URINALYSIS FINDINGS AND ASSOCIATED KIDNEY DISORDERS
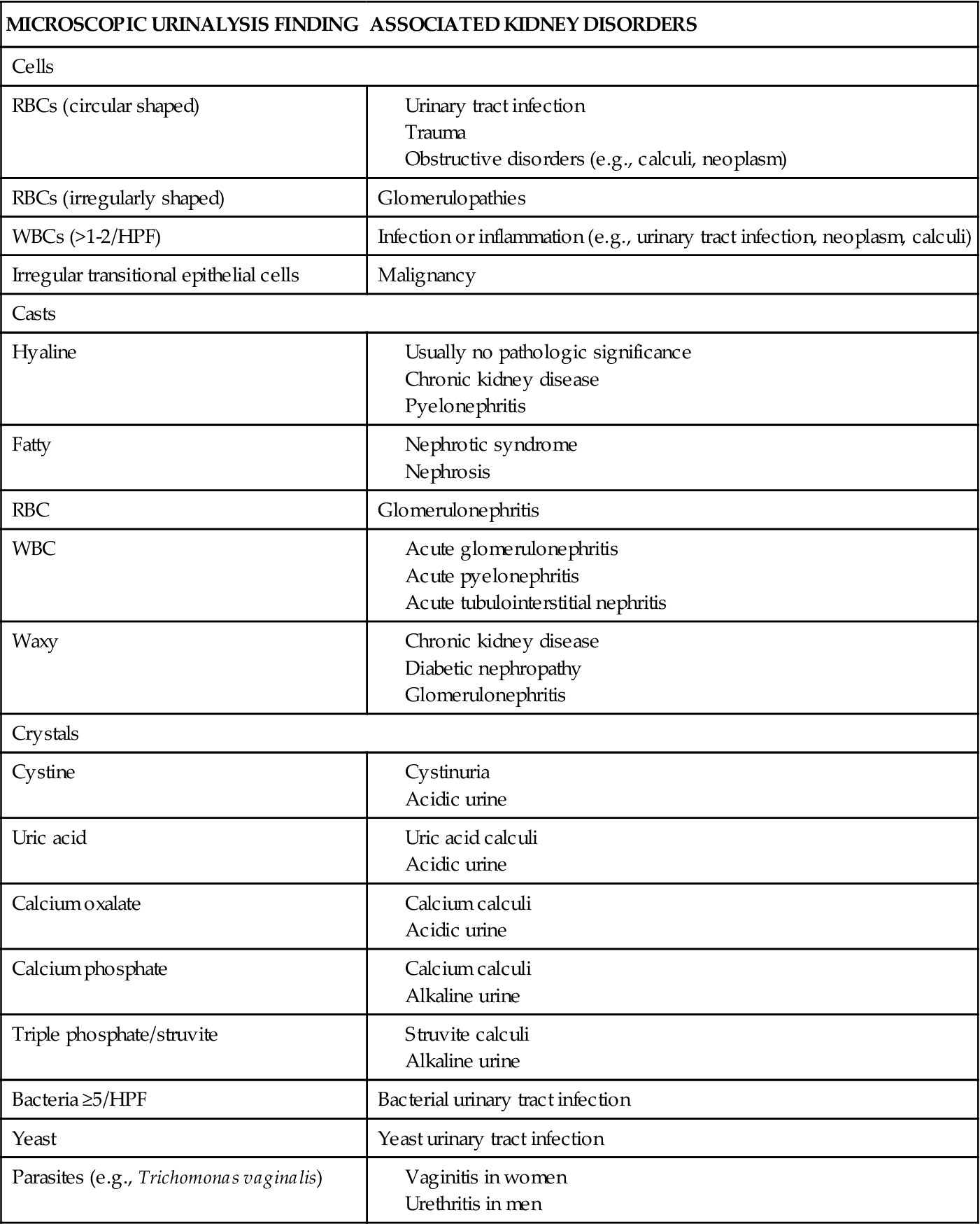
Data derived from Gerber GS, Brendler CB: Evaluation of the urologic patient: history, physical examination, and urinalysis. In Wein JA et al, editors: Campbell-Walsh urology, ed 9, Philadelphia, 2007, Saunders, pp 105-109; Pagana KD, Pagana TJ: Mosby’s manual of diagnostic and laboratory tests, ed 3, St Louis, 2006, Mosby, pp 1007-1014. HPF, High-power field; RBC, red blood cell; WBC, white blood cell.
Other Diagnostic Tests
Many of the diagnostic tests presented in Chapter 26 are applicable to intrarenal disorders. The simple kidney, ureter, and bladder (KUB) radiograph identifies gross abnormalities of the kidney related to position, size, and shape as well as renal calculi that are radiopaque.1 Renal vasculature can be examined by renogram or renal scan; renal scans will also identify neoplasms in the kidney. Ultrasonography differentiates the solid mass of a neoplasm from fluid-filled cysts. Computerized tomography (CT) and magnetic resonance imaging (MRI) provide detailed information regarding multiple pathologies including thrombi or other vascular occlusions, masses, and obstructions involving the kidney. In situations in which these diagnostic tests are insufficient and actual tissue examination is necessary (e.g., neoplasm assessment), a renal biopsy may be required.1
Congenital Abnormalities
A wide variety of anomalies in the development of the kidneys have been documented in the literature. One or both kidneys may be involved. In some cases, findings are associated with other abnormalities, whereas in others the renal abnormalities are isolated. Congenital kidney abnormalities may be exceptionally rare or may occur with considerable frequency. They may be identified prenatally during ultrasound assessment of the developing fetus, noted at birth, or manifest only in later life. Discussion here will focus on the more common congenital anomalies because a complete discussion of all these pathologies is beyond the scope of this text.
Renal Agenesis and Hypoplasia
Renal agenesis means a failure of one or both kidneys to embryonically develop at all.2,3 On the other hand, renal hypoplasia describes a condition in which some fetal development of the kidneys has occurred. They both can be found as a single entity or in combination with other congenital malformations.
Bilateral renal agenesis (BRA) results from failure of the metanephros (renal buds) to develop in the fetus.2 It is incompatible with extrauterine life and results in stillbirth or death shortly thereafter.2,4 It is usually found in Potter syndrome, where severely inadequate quantities of amniotic fluid result in compression of the fetus within the uterus.2 This pathology is characterized by a collection of associated anomalies that includes wide-spaced eyes with epicanthal folds, low-set ears with insufficient cartilage, a beaked nose, and a receding chin.4
Unilateral renal agenesis (URA) is more common than bilateral renal agenesis.3 URA is often associated with concurrent urologic or nonurologic congenital anomalies. Nonurologic anomalies are usually cardiac or gastrointestinal in nature. Sometimes renal agenesis has been found to be familial and inherited as a dominant trait; screening by ultrasound of parents and siblings has been recommended when infants with agenesis or dysgenesis are involved. Poorly controlled diabetes and exposure to certain drugs (e.g., those affecting angiotensin II) and chemicals have been implicated as teratogens. In URA, the remaining kidney usually enlarges as a compensatory mechanism. Lifelong monitoring of renal function is recommended.3
Congenital renal hypoplasia is estimated to cause 40% to 60% of the pediatric cases of ESRD. Gene mutation is likely responsible for the incomplete development of the kidney. Hypoplasia may be insufficient for extrauterine life if both kidneys are involved or may not impact renal functioning until later in life.2,4 When renal hypoplasia has been identified, lifelong monitoring is recommended.2
Cystic Kidney Diseases
Cystic disease of the kidneys incorporates a wide range of hereditary, developmental, and acquired conditions.5–10 Depending on the classification, these fluid-filled craters may be present at birth or only visible later in life. They may involve one or both kidneys and be accompanied by other anomalies, or they may be the only pathology present.8 More commonly found in men, and increasing in prevalence with aging, renal cysts have been reportedly identified in more than half of patients over the age of 50.6 Cysts may be found in other organs, or limited to the kidneys, depending on the disorder. Within the kidney, cysts may be diffuse or confined to one anatomic area.8 Renal cysts of significant size produce flank pain and hemorrhage.6
The two most common forms of cystic kidney disease are the autosomal recessive and autosomal dominant polycystic diseases.5,8,9 Autosomal recessive polycystic kidney disease (ARPKD) is usually diagnosed in infants and young children, whereas autosomal dominant polycystic kidney disease (ADPKD) is generally diagnosed in adulthood.8,9 Although the pathogenesis is similar, the ARPKD and ADPKD forms of the disease are genetically different, and their clinical courses are usually distinct (Table 27-3).
TABLE 27-3
COMPARISON OF AUTOSOMAL RECESSIVE AND AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE
| FEATURE | AUTOSOMAL RECESSIVE | AUTOSOMAL DOMINANT |
| Gene defect | Chromosome 6p | Gene PKD1 on chromosome 16, or gene PKD2 on chromosome 4, or gene PDK3 location unknown |
| Incidence | 1:20,000 | 1-2:1000 |
| Age at diagnosis | Usually neonate to childhood | Usually fourth to fifth decade |
| Imaging findings | Symmetrically enlarged kidneys | Enlarged kidneys, often asymmetric |
| Histologic findings | Cysts derived from epithelial cells of collecting ducts | Entire nephron involved |
| Liver involvement | Abnormal portal ducts progressing to fibrosis | Multiple cysts |
| Other systemic findings | Usually none | Cysts in other abdominal organs, aneurysms, abnormal cardiac valves, hernias, and diverticuli |
Autosomal Recessive Polycystic Kidney Disease
ARPKD is often identified in the neonatal period, and when accompanied by pulmonary hypoplasia, it may result in death.9 In ARPKD, the kidneys retain their shape but are uniformly enlarged, and collecting ducts are dilated from the medulla to the cortex. Abnormal portal ducts are found in neonates, and portal fibrosis is noted in older patients.9 The most common clinical signs in the neonatal period are respiratory distress or palpable kidneys on physical examination. Systemic hypertension is frequently most severe in infancy.9 Those who live to reach adulthood typically retain some renal function, but experience a progressive decline in liver function.9 There has been no documented evidence of ARPKD being linked with renal neoplasia.11 A recessive pattern of inheritance within the family and pathology on liver biopsy are the best diagnostic data because both the clinical presentation and the results of ultrasounds, CT, or MRI can be similar to those of the autosomal dominant form of polycystic kidney disease.10
Autosomal Dominant Polycystic Kidney Disease
ADPKD is the most common of all the hereditary cystic kidney diseases.5,7,10 Typically, both kidneys are involved, but in 17% of the cases, kidney disease is unilateral and these are most likely to be the pediatric cases. The other kidney is often affected by the time the child reaches adulthood.5 For 5% of the end-stage kidney disease patients receiving hemodialysis in the United States, the etiology of their disease is ADPKD.10 The disease affects both genders and has no racial preference.7,11 Although prenatal and neonatal cases have been reported and ADPKD can present at any age, it usually manifests in patients who are 40 to 59 years old.10,11 In less than 1% of cases, ADPKD is identified in the neonate or in utero.7 ADPKD is marked by a great deal of variability in the rate of decline in renal function.9,11 The disease process appears to advance more rapidly in men than in women.7 The incidence of renal cell carcinoma is no greater in ADPKD than in the general population, but it does occur at an earlier age.7
There are two genetically distinct but phenotypically similar forms of ADPKD: PKD1 and PKD2. Distribution of these genotypes is such that 85% of the cases are associated with PDK1 and just less than 15% with PDK2.5,7,11 Mutations of these genes add further complexity to ADPKD.7Approximately 10% of the cases of ADPKD are not familial in origin and appear sporadically.5 Because of significant variability in clinical presentation within families, environmental modifying factors have been suggested, though not yet documented.7 It does seem that the specific gene is responsible for the initiation of cystic development, but not necessarily for cyst expansion.7
While the cysts multiply and expand, the overall size of the kidneys increases and there is a progressive decline in glomerular filtration rate (GFR) as the amount of normally functioning tissue is slowly reduced.7 Figure 27-2 provides a comparison of normal and polycystic kidneys. Renal perfusion decreases, and significant remodeling occurs in the renal circulation.7 Expansion of the cysts compacts and distorts the vascular system, and the resulting local ischemia activates the intrarenal renin-angiotensin system. The progressive reduction in renal function appears to be primarily associated with an increase in the size of the kidney and the overall volume of the cysts.7
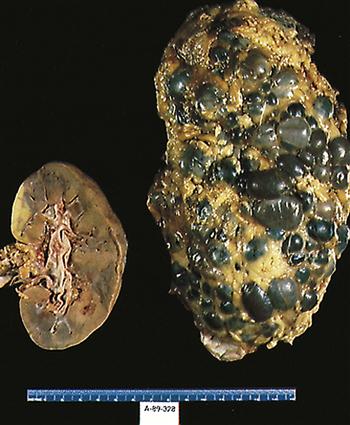
In contrast to its recessive form, ADPKD is associated with pathologies in other body systems.5,9,11 The most common extrarenal presentation is in the liver. Additional extrarenal sites include the spleen, pancreas, lung, seminal vesicles, circle of Willis, skin, and heart.5,7,11
Involvement of these other organs and tissues results in additional clinical manifestations, each requiring attention by health care providers.7 In the early phases of the disease, the ability to concentrate urine is decreased.7 Hypertension is often diagnosed late in the disease process and increases the likelihood of escalated loss of renal function, proteinuria, and hematuria.7 In 60% of adult patients with ADPKD, pain is the most frequent complaint.7 Pain may be due to bleeding within the kidney, movement of kidney stones, or the development of urinary tract infections. Kidney stones develop in 20% of the cases and are usually uric acid–based or calcium oxalate–based. Urinary tract infections are most often attributable to Enterobacter organisms. Infection of the cysts themselves may also develop; these infections are more difficult to treat, and patients with this condition may require months of antimicrobial therapy. When it occurs, hemorrhage of cysts is usually self-limited.7
Diagnosis is based on genetic history and imaging techniques, most often ultrasonography.7,9,11 When there is no family history of ADPKD, a presumption of the disease is made if imaging either identifies cysts and bilaterally enlarged kidneys or shows cysts in the liver and both kidneys. Genetic testing is then performed to substantiate the diagnosis.7,10
Treatment of ADPKD is primarily supportive, emphasizing the control of blood pressure and the management of any associated pathologic conditions.7 Once ESRD is reached, dialysis or kidney transplantation is required.7
Neoplasms
Neoplasms found in the kidney may be benign or malignant primary tumors, or result from metastases from extrarenal sites. Because the kidney is encased in a tough, fibrous capsule, growing renal neoplasms will distort the architecture of the kidney and ultimately hinder kidney function. Malignant renal neoplasms also carry the threat of metastasis to distant sites.
Benign Renal Neoplasms
Several benign neoplasms may be found in the kidney, developing from the renal cortex, medulla, or capsule, but they are less common than malignant neoplasms.6 Contrary to most benign neoplasms, those in the kidney are not truly encapsulated.6 Table 27-4 summarizes the etiology and pathogenesis of some of these benign neoplasms.
TABLE 27-4
SUMMARY OF BENIGN RENAL NEOPLASMS
| BENIGN NEOPLASM | ETIOLOGY AND PATHOGENESIS |
| Renal cortical adenomas | Small, solid growths that develop from cortical tissue; incidences of less than 1% up to 23%; typically <1 cm; patient is asymptomatic |
| Metanephric adenoma | Histologically related to Wilms tumor |
| Oncocytoma | Most difficult to differentiate from renal cell carcinoma; light brown to tan in color with well-defined borders |
| Angiomyolipoma | Composed of adipose, smooth muscle tissue, and blood vessels; believed to be hormone dependent because it is primarily found in postpubescent women |
| Nephroma | Cystic neoplasm not reliably differentiated from renal cell carcinoma in adults or nephroblastoma in children |
| Mixed epithelial stromal neoplasm | Most often found in perimenopausal women, most of whom are taking estrogen replacement therapy |
| Leiomyomas | Evolve from renal capsule, pelvis, or renal vein |
| Hemangiomas, fibromas, lipomas, lymphangiomas, and reninomas | Rare benign neoplasms |
From Campbell SC, Novick AC, Bukowski RM: Neoplasms of the upper urinary tract. In Wein JA et al, editors: Campbell-Walsh urology, ed 9, Philadelphia, 2007, Saunders, pp 1575-1582.
Diagnosis and treatment
Renal neoplasms are typically detected incidentally during abdominal imaging for other reasons.35 The neoplasm may attain sufficient size to be detected with abdominal palpation, produce flank pain, and cause hematuria. Generally, benign renal neoplasms are treated by removal of the kidney (nephrectomy) because they are space-occupying lesions and because of their propensity to undergo malignant changes.6 More recently, laparoscopic and tissue-sparing approaches have been developed, allowing the retention of more functioning renal tissue.14
Renal Cell Carcinoma
The vast majority of renal cancer is due to renal cell carcinoma (RCC) (85% to 90%).13,15,18 The remaining 10% to 15% of cases are primarily transitional cell cancers of the pelvis. Figure 27-3 illustrates a cross-sectional view of RCC. In the United States in 2011, the number of new cases of renal cell and renal pelvis cancers combined was estimated to be 60,920, with 13,120 fatalities.17 Approximately 57,760 new cases of RCC were diagnosed in 2009,16 with about 14,000 deaths.15 This represents only 2% of all adult malignancies.15 Since the early 1970s, the incidence has progressively risen in the United States and most of Europe,13,15 occurring two to three times more frequently in men than in women.6,13,15 Incident rates among African Americans are 20% higher6 and continue to increase more rapidly than the incidence in Caucasians.18 Rates among Hispanics are also rising.18 Taken as a whole, these trends are gradually altering the overall distribution of the disease. Age at diagnosis averages the early sixties.13,15
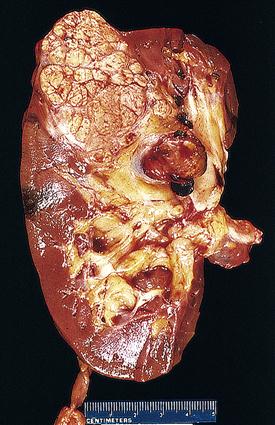
Etiology and pathogenesis
Although only a small portion of the cases of RCC are attributed to genetic factors, the risk of its development is greater than twofold when there is a first-degree relative with the disease. These hereditary kidney cancers have been the source of most of the knowledge regarding RCC. Several specific genes have been identified in these cases and are associated with specific subtypes.13,15,16,19
The identification of risk factors has been derived from pooled data from a large number of studies worldwide18 and these are listed in Box 27-1. The primary risk factors are obesity, cigarette smoking, and hypertension with risk reduction documented with weight loss, smoking cessation, and decreased blood pressure. Evidence linking trichloroethylene to renal cell cancer risk is accumulating.18 Low fruit/vegetable intake has not been adequately substantiated.18
Renal cell carcinoma encompasses several subtypes, histologically differentiated from one another. This variability in histology complicates diagnosis. The World Health Organization (WHO) has established a system of terminology and differentiation of these subtypes.15 The most common of these (approximately 85%) is clear cell RCC.12,13 These tumors originate in the renal cortex from cells of the proximal tubule and are usually unilateral and random in occurrence with rare cases of familial patterns of inheritance.12 Clear cell RCC is most frequently associated with metastatic disease.15 Next in frequency is papillary RCC, representing 10% to 15% of all renal cell carcinomas.12,16 Papillary RCC evolves from cells of the distal tubule. This type of RCC may also be sporadic or hereditary, and two subtypes of familial papillary RCC have been identified.12,16 When compared to clear cell RCC, papillary RCC is less likely to metastasize and therefore has a better prognosis; however, if it does metastasize, the prognosis is poorer than that of clear cell RCC.12 Chromophobe RCC represents 4% to 6% of all RCC cases.12,16 This subtype originates from the renal parenchyma.12 Because of slow growth and infrequent metastasis, 5-year survival rates exceed 90%.12 Cell membranes are prominent. Finally, medullary and collecting duct carcinomas are the least common, representing less than 1% of all RCC cases.12 Unfortunately, they occur in younger patients, develop rapidly, and carry only a 30% 5-year survival rate.12 Medullary RCC has been found to be associated with the sickle cell trait.
Clinical manifestations
RCC is often asymptomatic until it is quite advanced. When it does present with signs and symptoms, the most common are CVA tenderness, hematuria, and a palpable abdominal mass. Dyspnea, cough, and bone pain develop secondary to metastasis.
Diagnosis and treatment
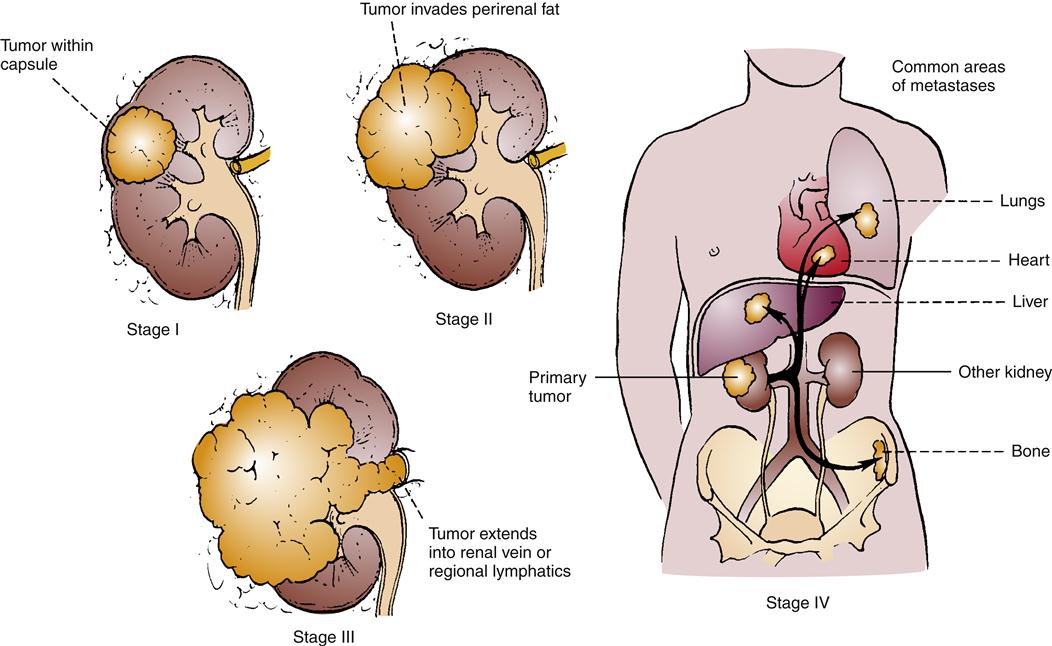
Renal ultrasound and CT of the abdomen may be of value. If cysts accompany RCC, renal ultrasound is 98% accurate in discerning them. Figure 27-4 illustrates the staging system used with RCC. When RCC is localized, it can be surgically removed in a nephrectomy, and this is the standard of treatment.12,13 Unfortunately, metastasis occurs in more than one third of cases,12,14 and metastatic RCC has been found to be unresponsive to cytotoxic chemotherapy.14,16 The standard nephrectomy is a major surgery, necessitating a large lateral incision. Other less radical approaches include nephron-sparing interventions and laparoscopic nephrectomy. In metastatic disease, spontaneous regression of metastases following nephrectomy occurs in less than 1% of cases. Conventional chemotherapeutic interventions have been found to elicit a poor response in advanced RCC. Interferon-α and interleukin-2 (IL-2) are cytokines used as tissue-specific therapies in these situations with clear cell RCC, with 6% to 27% response rates.13,14 More targeted therapies, specifically angiogenic inhibitors and tyrosine kinase inhibitors, have been studied in clinical trials or are now available for selected patient situations.13,14,20
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


