Clinical Features
Colonoscopic studies have reported the following incidence of endoscopically removed polyps: HPs (23.8% to 29%), SSA/P (2.2% to 9.0%), TSA (0.7% to 1.9%), and traditional adenoma (59% to 71%) (3,4).
HPs are located in the distal colon/rectum (70% to 84%). SSA/Ps tend to be located in the proximal colon (40% to 75%), and TSAs tend to be located in the distal colon (33% to 77%) (1,3–5).
The vast majority of HPs are 5 mm or smaller in size. The majority of TSAs are greater than 10 mm, whereas SSA/Ps are 6 to 10 mm (3,4).
The incidence of BRAF (V600E) mutations is SSA/P (82%), TSA (62%), and HPs (21% to 76%). The incidence of CIMP-high (CIMP-H) is as follows: SSA/P (76%), TSA (79%), and HPs (14% to 47%) (1). Some studies have shown that left-sided TSAs have a higher incidence of K–ras mutations and are more often CIMP-low (CIMP-L), giving rise to microsatellite-stable carcinomas (6).
The recognition of SSA/P and TSA is important because of the association of SSA/P and TSA as precursors for cancers that evolve through methylation (e.g., CIMP-H or CIMP-L; see section on CIMP-related adenocarcinomas). It is extremely important to recognize these lesions and differentiate SSA/Ps and TSAs from HPs as some experts consider the need for surveillance similar to classic adenomas and advanced adenomas (7,8). One recent longitudinal follow-up study of patients with SSA/P reported a subsequent higher incidence of colorectal cancer than in patients with classic adenomas (9). Although these entities have been characterized and described in detail, the interobserver agreement for diagnosing HPs and SSA is only moderate, whereas it is excellent for TSA (10).
The entity of SPS, previously called hyperplastic polyposis, has been better characterized over the past recent years. The World Health Organization (WHO) has defined the entity as follows: (a) at least five histologically diagnosed serrated polyps proximal to the sigmoid colon, of which two are larger than 10 mm; or (b) any number of serrated polyps occurring proximal to the sigmoid colon in someone who has a first-degree relative with SPS or (c) more than 30 serrated polyps of any size that are distributed throughout the colorectum (11). The incidence of SPS is unknown; however, in a retrospective study of 929 patients with serrated polyps, 19 (1.8%) were found to meet the criteria of SPS (12). The incidence of colorectal cancer in patients with SPS has been reported to range from 25% to 50% in those studies reported at initial diagnosis (7), whereas the incidence in SPS patients undergoing surveillance follow-up is 7% at 5 years (13). A high percentage of patients with SPS have a family history of colorectal cancer (13–16). Familial occurrence with apparently dominant inheritance has been reported but is rare. The number of polyps has varied from numerous to hundreds; some studies have reported a gender bias, whereas others have not (14–16) In a recent study of patients seen in a genetic clinic (Australia, New Zealand, Canada, and United States), the number of polyps ranged in number from 6 to 150 (mean 30), and 89% of the patients had pancolonic disease. Most of the polyps (83%) were of the serrated polyp type (38.4% MVHP, 6.1% GCHP, 27% SSA/P, 6.8% SSA/P with cytologic dysplasia, and 4.4% TSA) and 16.9% of the polyps were traditional (conventional) adenomas (13). Other previous investigators have found similar polyp types (14,16). The cancer types have been reported as not otherwise specified (NOS) (71.1%), mucinous (22.2%), and serrated (8.7%) (17). Cancers arising in these patients have been reported as MSI-high (MSI-H), MSI-low (MSI-L), and microsatellite stable (MSS) (16); however, the majority of the cancers arise through the classic APC/WNT pathway rather than the serrated pathway (17). One study has reported a significant association (p = .003) between the presence of “conventional adenomas” and the presence of colorectal carcinoma (17).
Hyperplastic Polyps: Pathologic Features
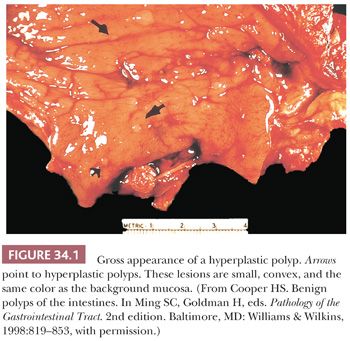
Grossly, HPs tend to be small, dome-shaped (convex) structures that are the same color as, or paler than, the surrounding mucosa. They often sit on the crest of the mucosal folds (Fig. 34.1). Occasionally, HPs may be pedunculated and may grossly mimic adenomas.
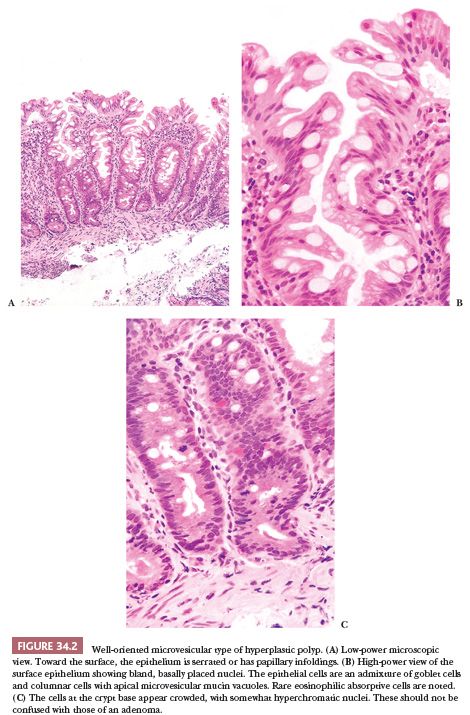
Microscopically, HPs have been divided into three types: MVHP, goblet cell hyperplastic polyps (GCHPs), and mucin-poor hyperplastic polyps (MPHPs). MVHP is the entity that correlates with what was previously designated HP of the rectosigmoid colon. The vast majority of HPs are of the MVHP type. GCHP type is probably underestimated in diagnosis, because it has limited serration but more resembles mucosal changes adjacent to mass lesions. The MPHP subtype is uncommon, histologically showing uniform and prominent serrations with almost no evidence of mucin and “cytologic atypia,” which suggests regenerative change (5). There are molecular differences between the subtypes of HPs. The incidence of K-ras mutations, BRAF (V600E) mutations, and CIMP-H is 43%, 21%, and 14%, respectively, in GCHP and 13%, 76%, and 47%, respectively, in MVHP (1).
Although the different subtypes of HPs are interesting from a research perspective, there is currently no clinical utility in diagnosing these subtypes during routine sign-out. The only important clinical distinction is between an HP and an SSA/P or TSA.
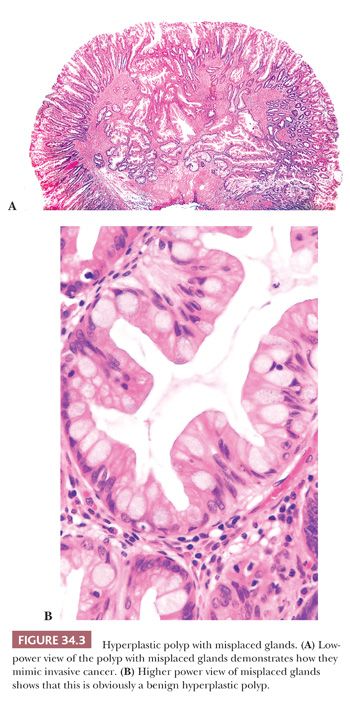
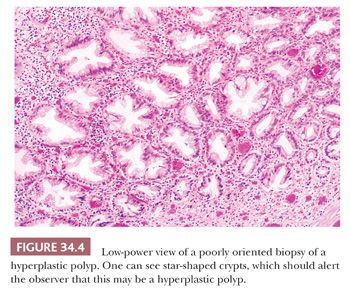
Microvesicular Hyperplastic Polyp. This lesion is characterized by columnar cells with apical vesicular mucin that alternate with eosinophilic-like absorptive and goblet cells (Fig. 34.2). The goblet cells are decreased in numbers compared with normal mucosa. Serration is seen in the upper one-third to one-half of the crypt, and often there is thickening of the subepithelial basement membrane. MVHP has a large proliferative zone that occupies much of the basal half of the mucosa. The cells at the base of the crypt show depletion of mucin and often have scattered Paneth cells. In well-oriented material, the cells at the basal portion of the crypt tend to show nuclear elongation, crowding, and increased mitotic rate compared with the normal surrounding mucosa. These findings can be impressive and should be not be interpreted as adenomatous change in an HP. The muscularis mucosae often show a characteristic splaying, with muscle fibers extending into the mucosa and surrounding individual crypts. An extensive splaying of the muscularis mucosae or misplacement of the glands into the submucosa may be observed, giving the false impression of invasion of glands into the submucosa (Fig. 34.3). One should be cognizant of this phenomenon and should not diagnose it as cancer (18). The presence of misplaced crypts (inverted crypts) beneath the muscularis propria is significantly associated with SSA, and if present, the pathologist’s suspicion for SSA should be raised (19). Often the tissue sections are cut tangentially. When this occurs, the glands take on a starlike shape, allowing the diagnosis of HP (Fig. 34.4).
Goblet Cell Hyperplastic Polyp. This type of polyp (Fig. 34.5) is seen predominantly in the distal colon and rectum and is often unrecognized and therefore underdiagnosed. At low power, it has features that are associated with mucosal changes adjacent to “mass lesions.” To my eye, it bears great similarity to aberrant crypt foci seen in both the human and in experimental animal models of colorectal cancer. Serrations are not prominent and, when present, are usually limited to the surface/luminal aspects of the mucosa. The mucosa is thickened, as is the subepithelial basement membrane, and goblet cells are quite prominent (5).
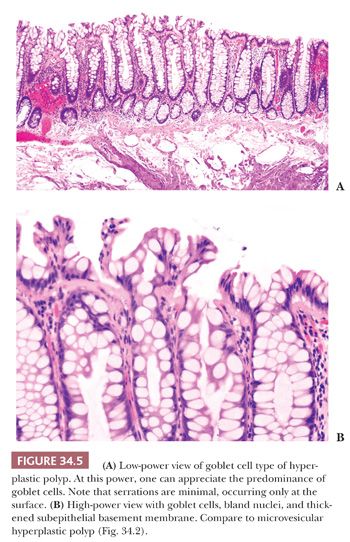
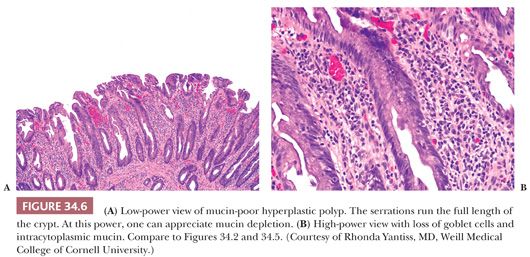
Mucin-Poor Hyperplastic Polyp. This variant (Fig. 34.6) is very uncommon. At low power, one is struck by the prominent serration that often runs the entire length of the crypt. Goblet cells are uncommon, and the columnar cells are mucin depleted. The changes mimic those of epithelial regeneration or are similar to those seen in prolapse.
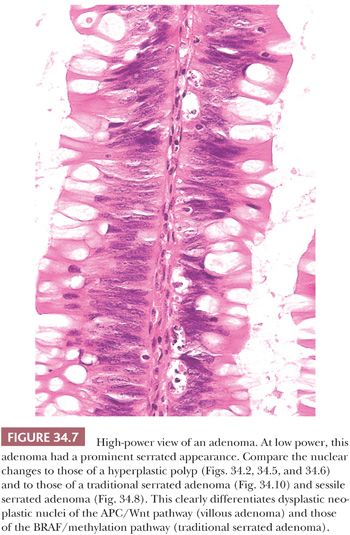
HP-type epithelium can be seen in juvenile polyps, Peutz-Jeghers polyps, and inflammatory polyps, especially inflammatory cloacogenic polyps of the anal region. At low-power microscopy, some villous adenomas have a serrated appearance and eosinophilic cytoplasm that can mimic HPs and TSA. However, on closer inspection at high power, the finding of nuclear changes of classic adenoma differentiates HP and TSA from villous adenoma (Fig. 34.7).
Sessile Serrated Adenoma/Polyp
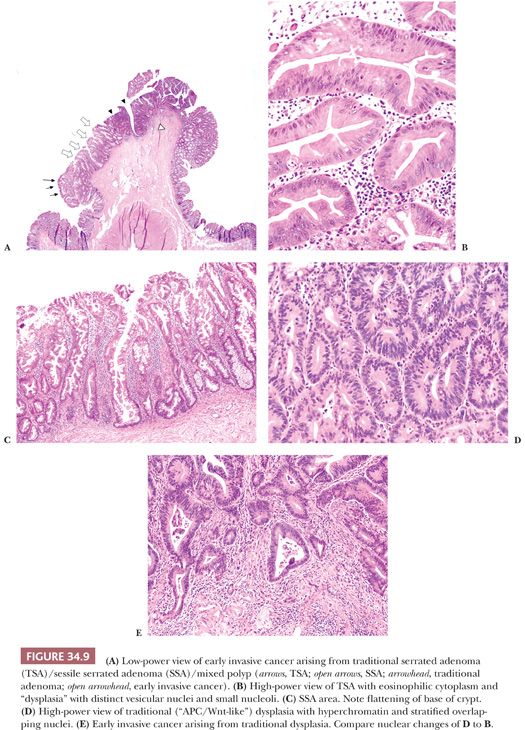
SSA/Ps (Fig. 34.8) are flat sessile lesions. The term adenoma conjures up morphologic changes of cytologic dysplasia (adenomatous changes) rather than “architectural dysplasia.” The diagnosis of SSA is based mainly on architectural features that emanate from abnormal proliferation or decreased apoptosis. The architectural changes are branching and dilation of the base of the crypt with a peculiar growth pattern where the crypts seem to grow parallel to the muscularis mucosae, often creating an inverted T shape or L shape. One may see the presence of mature cells with a goblet cell or gastric phenotype at the base of the crypt. The prominent serrations are often seen at the base or throughout the crypt. Asymmetry of cell maturation or proliferation may be seen within a single crypt or adjacent crypts. One can see pseudostratification of epithelium at the crypt surface. The nuclei are bland, but one can see nuclei with prominent nucleoli, open chromatin, irregular contour, and mitoses in the upper one-third of the crypt (5,20). SSA/Ps have a characteristic Ki-67 pattern that shows irregular, asymmetric, and highly variable expression of Ki-67. This contrasts to the regular, symmetric, and increased Ki-67 expression of the crypt base in HPs (21). SSA/P with areas of traditional adenoma (see the following texts) are now classified as SSA/P with dysplasia (11). Previously, they were termed mixed polyp, which is no longer in use. In every day clinical practice, one often receives multiple fragments of tissue in a single container. In this instance, it is difficult to determine if this represents a piecemeal polypectomy of a single lesion or multiple polyps from different sites. This makes it difficult to differentiate SSA/P with dysplasia from separate TSA and SSA/P. Close communication with the endoscopist is essential to reconcile this issue. Cancers evolving from an SSA/P do so through the entity of SSA/P with dysplasia (Fig. 34.9). The presence of traditional adenoma (“APC/Wnt-like”) mixed with SSA/P presages a rapid progression to cancer, and the pathologist should communicate the significance of this finding to the clinician (20,22).
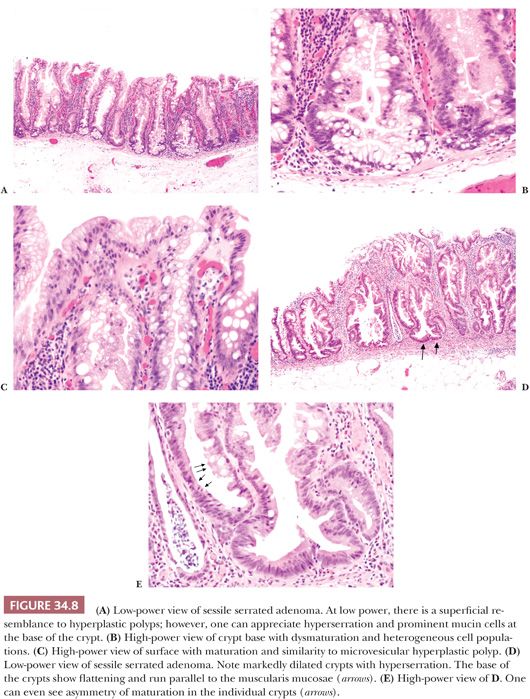
The minimal criteria for the diagnosis of an SSA/P is not clear. A consensus paper (7) reports that a single unequivocal architectural crypt is sufficient to diagnose a polyp as an SSS/P. The WHO (11) requires at least two to three abnormal contiguous crypts to make the diagnosis of SSA/P. Because of overlapping histologies, poor orientation, poor staining, cautery artifact, or scanty tissue, there is an acceptable category of unclassified serrated polyp. In my personal experience, obtaining serial and deeper sections often helps one resolve this matter.
Traditional Serrated Adenoma
TSA (Fig. 34.10) was one of the serrated adenomas originally described by Longacre and Fenoglio-Preiser (23). In contrast to SSA, TSA tends to be pedunculated/polypoid in nature. At low power, TSA has a very pronounced complex serrated architecture that is often misdiagnosed as villous adenoma. The cytologic appearance of the cells characteristically shows a uniform population of pencillate cells with bright eosinophilic cytoplasm. The nuclei are pseudostratified and elongated, have an increased nuclear-to-cytoplasmic ratio, and are vesicular with small nucleoli, but they are not markedly hyperchromatic, and mitoses are rare. In TSA, the nuclei show a wide range of change from vesicular nuclei with prominent nucleoli to bland mildly hyperchromatic nuclei and small indistinct nucleoli. These nuclei, although “atypical,” do not have the dysplastic features of nuclei seen in traditional adenomas, reflecting a different molecular pathway than traditional adenomas (“APC/Wnt-like”) (Figs. 34.7, 34.9, and 34.10). I have personally seen TSA or TSA with associated cancer in which the nuclei are distinct and clear with large nucleoli appearing to be high grade or “malignant” (see Fig. 34.49). This represents the distinctive nuclear morphology seen in CIMP (see section on CIMP cancer). These atypical features have been called serrated dysplasia and maybe a marker for neoplastic progression. TSA can also contain distinct areas of classic tubular adenoma, HP, or SSA/P (5,24,25). Cancers can arise within TSAs with traditional adenoma.
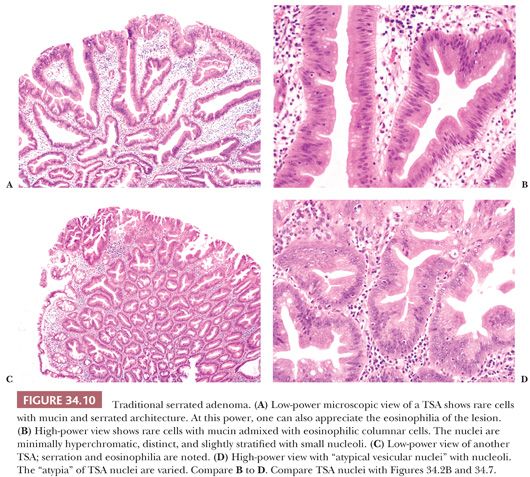
TSAs uniformly have ectopic crypt foci (ECF), which are defined as abnormal development of crypts with loss of orientation to the muscularis mucosae with the luminal end of the crypt oriented in a normal relationship to the lumen but not abutting on the muscularis mucosae. These ECFs are not seen in SSA/P or HPs. In TSA, the Ki-67 pattern is limited to the ECF in contrast to SSA/P and HP (21).
Sessile Serrated Adenoma/Polyp with Dysplasia
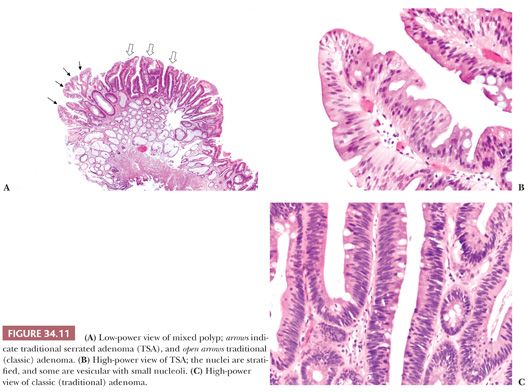
These polyps have been defined as containing combinations of areas of classic adenoma, HP, SSA/P, and TSA (Fig. 34.11). Previously, they were termed mixed polyps (5,24,25).
PEUTZ-JEGHERS POLYP
Clinical Features
The Peutz-Jeghers polyp is a hamartomatous lesion that can occur in the stomach, small intestine, and colon. These polyps are most often identified in patients with Peutz-Jeghers syndrome. Solitary Peutz-Jeghers polyps do occur in patients without obvious manifestations of the syndrome; however, the incidence of solitary Peutz-Jeghers polyps is unknown (26,27). Peutz-Jeghers syndrome (28,29) is an autosomal dominant disorder consisting of gastrointestinal (GI) hamartomatous polyps associated with mucocutaneous pigmentation. However, up to 25% of patients with the syndrome do not have a family history but represent a spontaneous mutation. The diagnostic criteria for the syndrome are as follows: (a) two or more histologically confirmed Peutz-Jeghers polyps; (b) any number of Peutz-Jeghers polyps with a family history; (c) characteristic, prominent mucocutaneous pigmentation with a family history; or (d) any number of Peutz-Jeghers polyps and characteristic prominent mucocutaneous pigment (28). Uncommon tumors such as ovarian sex cord tumors with annular tubules and adenocarcinoma (adenoma malignum type) of the uterine cervix in female patients and testicular Sertoli cell tumors in male patients are seen with the syndrome. Most patients are diagnosed in their 20s. The presenting clinical symptoms depend on the location of the polyps. Patients with small intestinal polyps present with symptoms of obstruction and abdominal pain, whereas those with large intestinal and rectal lesions present with bloody stools and protrusion or prolapse of the polyps.
Studies have shown a marked increased prevalence of cancer in patients with Peutz-Jeghers syndrome compared with the control population. The cumulative risk for all cancers was 37% to 93% between the ages of 15 and 65 years (30,31). The cancers have been divided between GI and non-GI types, with the highest cumulative rates for breast (54%), colon (39%), pancreas (36%), stomach (29%), and ovary (21%) (30). In some instances, GI cancers arose from the Peutz-Jeghers polyp (32). A recent study has concluded that individuals with solitary polyps with unequivocal Peutz-Jeghers polyp histology may have a cumulative risk of cancer similar to those with the syndrome (33); however, this is in contrast to another report (34).
The genetic basis for the Peutz-Jeghers syndrome is a germline mutation in the STK11/LKB1 gene located on chromosome 19. STK11 is a serine-threonine kinase which is important in cell proliferation in the mammalian target of rapamycin (mTOR) pathway (35). Clinical trials using the mTOR pathway inhibitor rapamycin are currently under way.
Pathologic Features
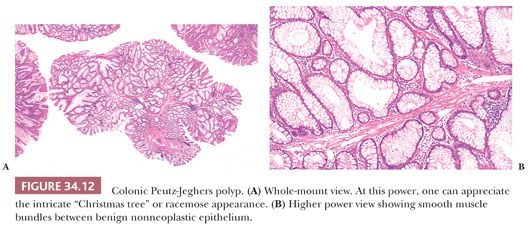
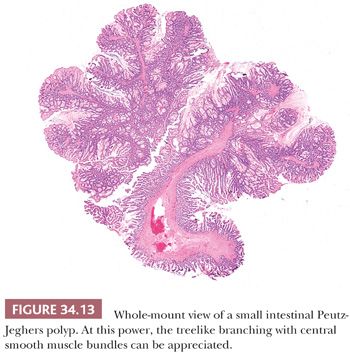
The Peutz-Jeghers polyp varies in size from less than 1 cm to more than 3.5 cm in diameter, and it may be sessile or pedunculated. The basic architecture is that of glandular epithelium resting on a branching smooth muscle framework that arises from the muscularis mucosae. This framework, which is essential in making the proper diagnosis, is easily recognized at low power; the polyp has a “Christmas tree” or arborescent-like appearance.
The epithelial component resembles the normal epithelium of the intestinal area from which the polyp arises. In the small intestine, the epithelium consists of goblet and absorptive cells with a normal component of endocrine and Paneth cells. In the large intestine, the epithelium is predominantly of the goblet cell type, often with branching crypts lined by hypertrophic goblet cells (Figs. 34.12 and 34.13). Areas of HP-type epithelium may be noted. Diagnostic difficulties can occur when ischemic necrosis that is often secondary to intussusception is present. Because of necrosis and distortion, the polyps may lack diagnostic features. In the colon and rectum, prolapse may cause histopathologic changes that can mimic the histology of Peutz-Jeghers polyp. This can be a diagnostic problem especially in the instance of solitary polyps.
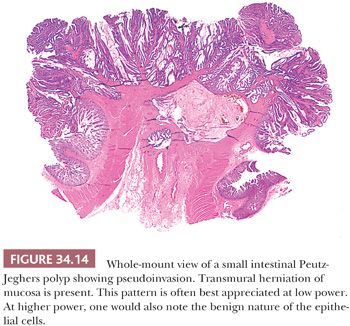
Areas of adenomatous and carcinomatous transformation within Peutz-Jeghers polyps have been reported (32,36,37), with a prevalence ranging from 6% to 12% (32,37). Importantly, one should not be misled into making a diagnosis of cancer based simply on the finding of epithelial structures admixed with smooth muscle fibers because this is the basic histologic feature of this lesion. Occasionally, there is herniation of cystically dilated epithelial structures into the bowel wall, even extending into the serosa and forming true tumor masses, analogous to colitis cystica or jejunitis cystica profunda (Fig. 34.14). In these instances, the herniated epithelium is nonneoplastic, and it may be accompanied by smooth muscle fibers, lamina propria, and mucin pools, which are important findings that help to distinguish epithelial misplacement from neoplasia. This pseudoinvasive pattern has been reported in approximately 10% of small intestinal Peutz-Jeghers polyps (38). A report of a leiomyosarcoma arising in a small intestinal Peutz-Jeghers polyp has also been published (39).
HAMARTOMATOUS POLYP: COWDEN DISEASE (PTEN-HAMARTOMA SYNDROME)
Cowden disease is an autosomal dominant disorder caused by PTEN mutations which is associated with colonic and small intestinal polyps, breast and thyroid cancer, facial trichilemmomas, acral keratoses, and oral mucosal papillomatosis (40). GI polyps are seen in 35% of cases (29).
The pathologic features of the GI polyps vary, but these lesions have been grouped as hamartomas. The most common lesion is a polyp showing a mildly fibrotic and disorganized mucosa with extensive splaying of the muscularis mucosae. These lesions are morphologically similar to the nonulcerated mucosa in the solitary rectal ulcer syndrome. Other polyps have been described as inflammatory, juvenile, lipoma, ganglioneuroma, adenomas, or nodular lymphoid hyperplasia. A recent large prospective study of 127 patients with confirmed PTEN pathogenic mutations has provided new insight into the types of GI polyps and the association of colorectal adenocarcinoma. Of the total 127 patients, 64 (50.4%) had GI polyps, whereas those patients undergoing endoscopies (93%) had GI polyps. Of the 62 patients with colorectal polyps, 18 had hamartomatous polyps; 27, HPs; 16, ganglioneuromas; 16, adenomas; and 11, inflammatory polyps. At least 9 patients had polyps of three different histologic types and 16 patients met the operational diagnosis of SPS. Nine colorectal cancers were diagnosed, with an average of 44.4 years at diagnosis, and 18% of individuals with adenomas developed colorectal carcinoma. These authors consider PTEN-associated Cowden syndrome to be a mixed polyp syndrome, with HPs most prevalent and a risk of early-onset colorectal carcinoma (41).
GANGLIONEUROMA
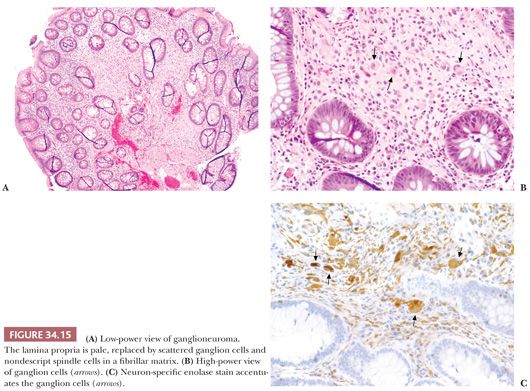
Ganglioneuromas may occur in the small and large intestine as solitary lesions, ganglioneuromatous polyposis, or diffuse ganglioneuromatosis. The former two are uncommon, whereas diffuse ganglioneuromatosis may be associated with von Recklinghausen disease and multiple endocrine neoplasia (MEN) type 2B (42). In the past, ganglionueromas have been reported in some patients with juvenile polyposis, but we now know that those cases actually represented cases of PTEN-hamartoma syndrome. The solitary lesions can be sessile or pedunculated, grossly resembling juvenile polyps, tubular adenomas, or HPs. Microscopically, the most common pattern is a superficial resemblance to juvenile polyps, with distorted crypts and cystic glands seen at low magnification. At higher magnification, one sees collections of spindle cells in a fibrillar matrix and irregular groups and nests of ganglion cells within the lamina propria. Another pattern is that of a proliferation of ganglion cells in a neurofibroma-like background (Fig. 34.15). The polyps in ganglioneuromatous polyposis are often indistinguishable from those of solitary ganglioneuroma, but they may show greater variability in supportive cells and may consist of villiform mucosal projections containing clusters of ganglion cells with no apparent neural component. In diffuse ganglioneuromatosis, one sees a diffuse intramural or transmural proliferation of ganglioneuromatous tissue involving the enteric plexus (42). The pathologist can, at times, suspect the diagnosis from biopsy material simply by identifying ganglion and Schwann cells in the mucosa. Immunohistochemistry (IHC) (e.g., use of antibodies for neuron-specific enolase for detection of neurons and Schwann cells and of S-100 protein for the detection of Schwann cells) can be helpful.
JUVENILE POLYP
Clinical Features
Although juvenile polyps occur mainly in the first two decades of life, they are not uncommonly seen in adults (43). They are localized chiefly in the rectum, and they are usually solitary. Bleeding is the most common sign, with anal protrusion of polyps seen in 20% of cases and autoamputation of the polyp seen in 10% of cases. A polyposis syndrome exists; it may be limited to the colon (juvenile polyposis coli), or it may be associated with polyps in the small intestine and stomach (29,44). The criteria for juvenile polyposis are (a) more than five juvenile polyps of the colon or rectum, (b) juvenile polyps throughout the entire GI tract, and (c) any number of polyps in a patient with a family history of juvenile polyposis (44,45). A rare and lethal form occurring in infancy consists of juvenile polyps throughout the entire GI tract, and this is associated with diarrhea, anemia, and hypoalbuminemia. In polyposis cases, the polyps are counted in the dozens to hundreds; however, they are not as numerous as in familial adenomatous polyposis (FAP). Approximately 40% of these cases may have a genetic, probably autosomal dominant, origin. Genetic heterogeneity appears to be present because subsets of families have mutations in BMPR1A (10q22) and SMAD4/DPC4 (18q21). Associated intestinal and extraintestinal congenital defects (e.g., malrotation of the gut, mesenteric lymphangioma, hypertelorism, hydrocephalus, tetralogy of Fallot, and coarctation of the aorta) have been reported.
An increased risk of colon cancer is seen in patients with juvenile polyposis. Colorectal cancer has been reported in 30% to 40% of patients with juvenile polyposis, and upper GI cancers have been reported in 10% to 15% of cases (44,45). In some familial cases, intestinal and gastric cancers have been seen in those family members without juvenile polyps (46). However, solitary juvenile polyps are not markers for subsequent colorectal cancer, and they do not require further follow-up investigation (47).
Pathologic Features
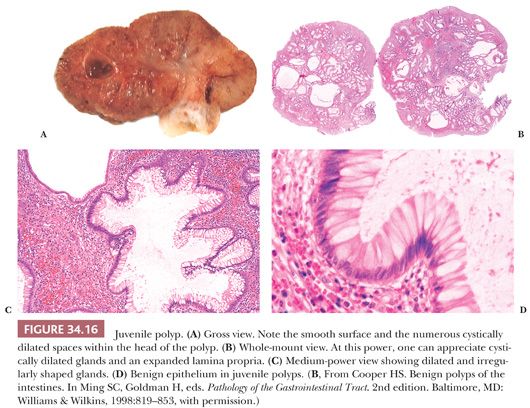
Grossly, juvenile polyps are mainly pedunculated, rarely sessile, and usually smaller than 3 cm in diameter. They have a smooth glistening surface, and they are reddish tan to red. On cut section, one can often appreciate the cystic fluid–filled spaces (Fig. 34.16). Microscopically, fully developed juvenile polyps consist of cystically dilated and tortuous glands in an inflamed stroma (Fig. 34.16). This classic pattern is readily appreciated at low power, and it should alert the pathologist to the diagnosis. The glands are made up of well-formed mucus-secreting cells that may become flattened and attenuated. In approximately 45% of cases, one can note pink regenerative epithelial cells similar to those seen in HPs. The stroma usually contains acute and chronic inflammatory cells and granulation tissue. When dilated glands rupture into the stroma, a foreign body giant cell reaction might occur. Occasionally, one can identify areas of osseous metaplasia.
The juvenile polyp is nonneoplastic. Nevertheless, in patients with solitary juvenile polyps (nonsyndromic), rare cases of polyps with areas of adenomatous transformation and even carcinoma are observed (48,49). Adenomatous change (dysplasia) has been reported in 15% to 30% of juvenile polyps from patients with juvenile polyposis (45,50). However, interobserver variation in diagnosing dysplasia in juvenile polyps is seen (50). Some studies have reported separate adenomas and HPs (45,48). A large study of juvenile polyposis reported that 9% of typical juvenile polyps (also known as type A–classic juvenile polyp) and 47% of atypical juvenile polyps (also known as type B–epithelial phenotype) showed foci of dysplasia. These atypical juvenile polyps grossly formed lobular or multilobular masses (appearance of closely packed juvenile polyps attached to a single stalk) rather than the typical smooth and spheric polyp. The atypical juvenile polyps revealed relatively less lamina propria and more epithelium than the typical juvenile polyp and often had a villous or papillary configuration (44). In these cases, the glands show more budding and branching but less cystic change than classic solitary juvenile polyps. Type A polyps predominately have BMPR1A germline mutations, whereas type B polyps predominately have SMAD4 germline mutations. Dysplasia is equally associated with SMAD4 and BMPR1A mutations; however, focal dysplasia in those with SMAD4 mutations was seen only in type B polyps. In those with BMPR1A mutations, dysplasia was seen in both type A and type B polyps (51).
Small or “early” juvenile polyps may be histologically indistinguishable from inflammatory polyps because both have a cap of granulation tissue above cystically dilated glands. In this instance, the judicious course is to comment that the lesion in question may be either a juvenile polyp or an inflammatory polyp.
INFLAMMATORY POLYP
Inflammatory polyps may be secondary to inflammatory disorders of the intestine. They are seen in 10% to 20% of cases of ulcerative colitis, and they may also be noted in Crohn disease, ischemic disease, amebiasis, and schistosomiasis; in addition, they are seen adjacent to ulcers and at surgical anastomotic sites.
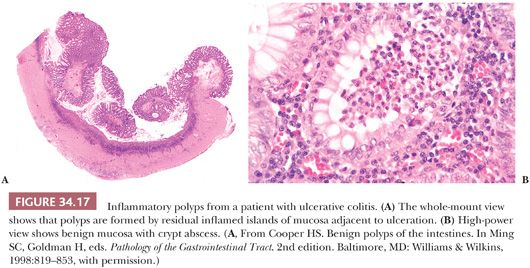
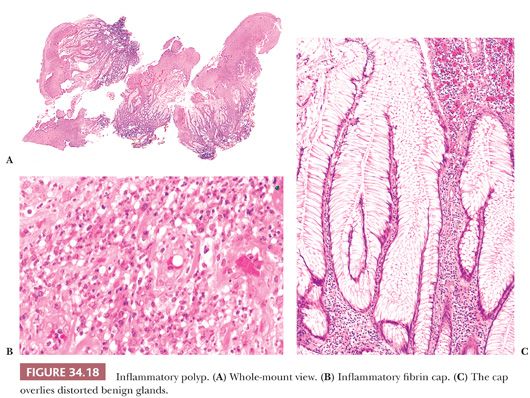
In ulcerative colitis and Crohn disease, the polyps are simply raised tags of mucosa and/or submucosa. The mucosa may show changes of inactive inflammatory bowel disease, or the epithelial crypts may show abscesses and an inflamed stroma (Fig. 34.17). Inflammatory polyps may also be rounded nodules or caps of granulation tissue overlying epithelial structures (Fig. 34.18), or they may closely resemble early juvenile polyps. In young children with no history or pathology of inflammatory bowel disease, one must be careful not to misdiagnose an early juvenile polyp as an inflammatory polyp.
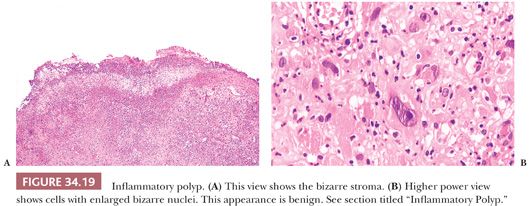
Occasionally, inflammatory polyps can have bizarre stromal changes that mimic sarcoma (46,52). Most of these patients have inflammatory bowel disease. These polyps with bizarre stroma are usually solitary and are smaller than 2 cm. Histologically, one sees bizarre spindle, epithelioid, and large ganglion-like or multinucleated cells in a fibroblastic or granulation tissue stroma. Mitoses are rare, and atypical mitoses are absent. Often, a zonation effect separates the bizarre cells from the more characteristic spindle cells; however, this appearance is not always evident in small biopsies (Fig. 34.19). In distinguishing this reaction from a malignant process, one should take into consideration the pattern of zonation of bizarre cells, scant mitoses, lack of atypical mitoses, solitary occurrence, small size, and association with inflammatory bowel disease.
Inflammatory Polyp Secondary to Mucosal Prolapse
The entities of inflammatory cap polyposis, inflammatory myoglandular polyp, and diverticular polyp are thought to be different spectra or stages in mucosal prolapse (53).
Inflammatory cap polyps are limited to the rectosigmoid and distal colon. They are sessile and can be solitary or numerous. Histologically, one sees an eroded surface with a fibrinopurulent inflammatory cap. The body of the polyp consists of acute and chronic inflammation, proliferation of the muscularis mucosae, and fibromuscular obliteration of the lamina propria. The epithelial component shows hyperplastic changes, goblet cell hyperplasia, and tortuosity of glands (54).
Inflammatory myoglandular polyps, which are mainly located in the distal colon, can rarely be found in the ileum, where they can cause intussusception. They are solitary and usually pedunculated. Histologically, one sees branching and dilated glands within a background of radially proliferating smooth muscle, an appearance not unlike that of the Peutz-Jeghers polyp. The surface is often eroded, with a fibrinopurulent cap (55).
Diverticular polyps are those that arise in the background of diverticular disease. Grossly, these lesions may be confused with an adenoma. Early lesions have mucosal hemorrhage and congestion, whereas advanced lesions show changes resembling those of mucosal prolapse as follows: mucosal edema; fibromuscular obliteration of the lamina propria; epithelial hyperplasia and branching, dilated crypts; and occasional misplaced glands in the submucosa. Pseudosarcomatous changes in the stroma have been noted (56).
Any of the foregoing lesions may require several endoscopies with multiple biopsies or polypectomies before a diagnosis of prolapsed induced inflammatory polyp can be established because the microscopic findings may not be representative of the diagnostic histopathology or may be nonspecific. Occasionally, patients may need to undergo surgical resection to establish the correct diagnosis of this benign disorder.
ANGIOGENIC (ANGIOMATOUS) POLYP
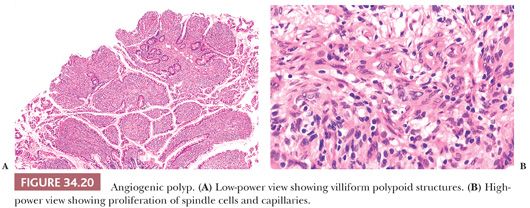
This entity, also called granulation tissue polyposis, florid angiogenesis, and angiogenic polypoid proliferation, was originally reported in ileums associated with carcinoid tumors, and these angiogenic polyps were thought to arise secondary to angiogenic growth factors secreted by the carcinoid tumors (57,58). However, they have also been reported in association with noncarcinoid tumors, and they are proposed to arise secondary to prolapse. Grossly, angiogenic polypoid proliferations are present as exuberant mucosal polyposis that may simulate cobblestoning, but they can form well-defined polyps approximately 3 mm in diameter. Microscopically, the polyps consist of various degrees of expanded villi with intramucosal capillary proliferation, ectasia, and fibromuscular proliferation; the last feature appears in mucosal prolapse as well (Fig. 34.20).
INFLAMMATORY FIBROID POLYP
Inflammatory fibroid polyps are benign tumor masses occurring in the small intestine, stomach, and, less commonly, large intestine (59). They occur at any age and are not associated with any medical conditions or syndromes. Other terms used to describe this entity are eosinophilic granuloma, submucosal fibroma, hemangiopericytoma, inflammatory pseudotumor, and fibroma.
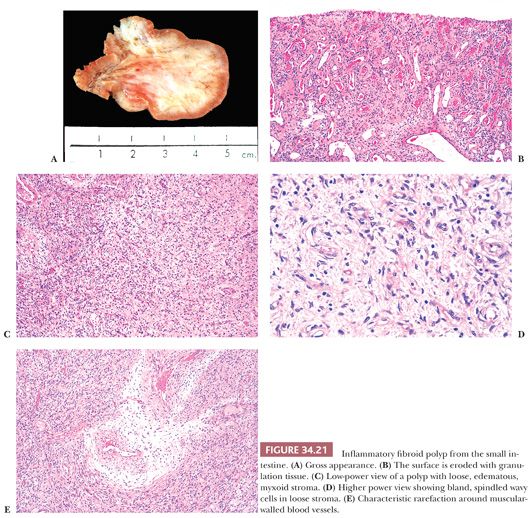
These polyps range from 1.5 to 13 cm in size (mean size of 3 to 4 cm), and they are polypoid with a broad base. Most often, they are limited to the submucosa, but they can infiltrate the mucosa or the muscularis propria and extend to the serosa. Grossly, they are tan, gray, or yellow; the overlying mucosa may be ulcerated (Fig. 34.21). Microscopically, they are mesenchymal lesions with an inflammatory infiltrate and a variable vascular component. The fibroblasts may be spindle shaped or stellate with indistinct basophilic cytoplasm. In some areas, the lesions may be sparsely cellular with a prominent myxoid component (Fig. 34.21). Cellular fields may show cells with mitotic figures, with some lesions showing up to two mitoses per high-power field (hpf). The inflammatory infiltrate consists of eosinophils, lymphocytes, plasma cells, macrophages, and mast cells. The eosinophils may vary in number from few to many. Those with small numbers of eosinophils may show areas of prominent stromal hyalinization. The vascular component may be prominent; many lesions contain a distinctive zone of loose connective tissue around the larger blood vessels (Fig. 34.21). The differential diagnoses are GIST, leiomyoma, schwannoma, perineurioma, and other benign nerve sheath lesions (60). Diagnosing these lesions from endoscopic biopsies is very difficult, if not impossible, because biopsies may show only granulation tissue from the eroded surface. Inflammatory fibroid polyp is a benign lesion. The spindle cell component expresses vimentin, muscle-specific actin, smooth muscle actin, CD34, and desmin, but it is negative for S-100 protein, CD117, and desmin (Table 34.2). One should be careful interpreting CD117 stains in these lesions because mast cells, which are often numerous, will stain positively. Activating PDGFRA mutations are seen in inflammatory fibroid polyps. Mutations in exon 12 predominate in small intestinal lesions, whereas mutations in exon 18 predominate in gastric lesions (61).
CRONKHITE-CANADA SYNDROME
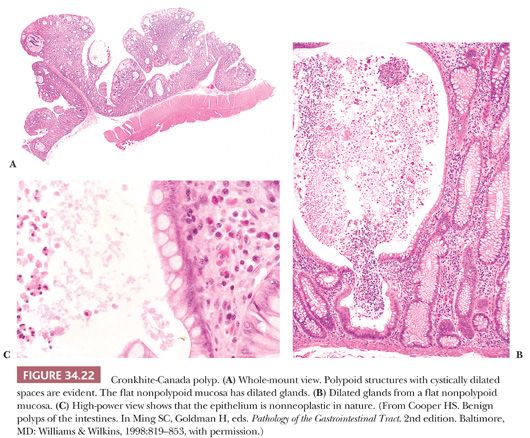
Cronkhite-Canada syndrome is a nonhereditary GI polyposis associated with alopecia, nail atrophy, and hyperpigmentation of the skin (62). Unlike most polyposis disorders in which the onset is noted earlier in life, in this disorder, 80% of cases initially present at or after 50 years of age. The clinical features are diarrhea, weight loss, abdominal pain, anorexia, and weakness. Physical findings include nail changes (dystrophy, thinning, and splitting), hair loss, and hyperpigmentation of the skin. Fifty percent of these patients die secondary to cachexia. The polyps occur in the stomach, small intestine, colon, and rectum.
Histologically, these polyps are identical to juvenile polyps, with tortuous and cystically dilated glands. However, in the Cronkhite-Canada syndrome, the nonpolypoid intervening mucosa contains cystically dilated glands, whereas in patients with juvenile polyps, the intervening mucosa is histologically normal (Fig. 34.22). Similarly, the clinical setting and symptoms of juvenile polyps and Cronkhite-Canada syndrome are entirely different. Adenomatous change in Cronkhite-Canada polyps has been reported, and occasional cases of colorectal adenocarcinoma have been reported in patients with this syndrome (63).
LYMPHOID POLYP
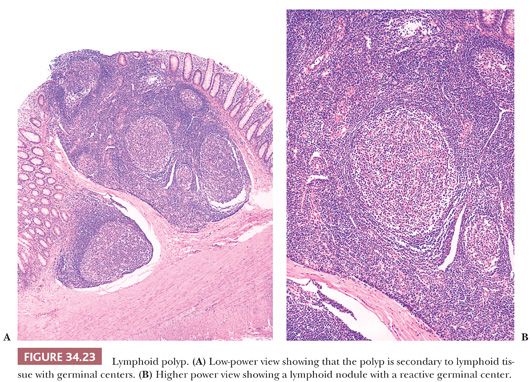
Lymphoid polyps are benign lesions that occur mainly in the rectum. They occur in all age groups; they may be found incidentally, or they may cause symptoms such as bleeding, discomfort, or prolapse. Eighty percent are sessile and solitary; the remainder are pedunculated and/or multiple (two to six in number). Their pathologic features are quite characteristic, consisting of prominent lymphoid follicles with active germinal centers that are located in the mucosa and submucosa (Fig. 34.23). Local excision is curative; occasionally, spontaneous remission of these lesions has been noted (64). Cases of multiple lymphoid polyposis have been reported. These lesions occur mainly in children; some have been associated with a family history, whereas other cases represent lymphoid polyposis of the terminal ileum in patients with FAP coli and Gardner syndrome (65).
NEOPLASTIC POLYPS (ADENOMA)
Adenomas are traditionally divided into the following three types: tubular, tubulovillous, and villous. However, classification may also be based on gross appearance (polypoid and nonpolypoid) (66). Adenomas are important clinically because they are premalignant lesions. Adenomas are quite common in the large intestine, but they are rare in the small intestine.
Small Intestine
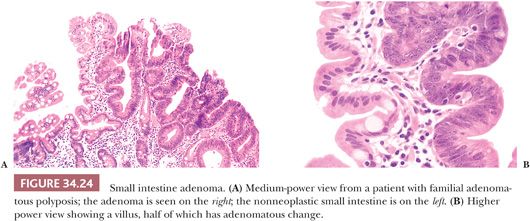
Small intestinal adenomas are tubular or villous and are usually sessile. Microscopically, they resemble adenomas of the colon. All degrees of dysplasia can occur, as can in situ and invasive cancer (Fig. 34.24). Approximately 90% of patients with FAP coli often have small intestinal adenomas, predominantly in the duodenum, many of which are only evident microscopically (67,68).
Large Intestine
The prevalence of large intestinal adenomas varies in different parts of the world. Adenomas are common in Westernized cultures (high prevalence of colorectal cancer) and uncommon in developing countries (low prevalence of colorectal cancer); autopsy studies show a prevalence as high as 60% in the former and as low as 5.5% in the latter. Most adenomas smaller than 1 cm tend to show an even distribution throughout the large intestine, whereas those larger than 1 cm tend to be localized in the distal colon. The prevalence of tubular adenomas, tubulovillous adenomas, and villous adenomas is 95%, 2% to 4%, and 1% to 3%, respectively, in autopsy studies, whereas the prevalence is 65% to 85%, 16% to 27%, and 3% to 9%, respectively, in colonoscopic studies.
Polyp (Adenoma) to Cancer Sequence
Adenomas are of clinical importance because they may develop into cancer. Invasive adenocarcinoma is found in approximately 5% of all adenomas; however, this relationship is size dependent. Invasive cancer is found in 0.5% of adenomas smaller than 1 cm, in 5.0% of adenomas 1 to 1.9 cm, and in 10% of adenomas larger than 2 cm. Adenoma type also is related to cancer, with invasive cancer found in 2% to 3%, 6% to 8%, and 10% to 18% of tubular, tubulovillous, and villous adenomas, respectively.
Pathologic Features
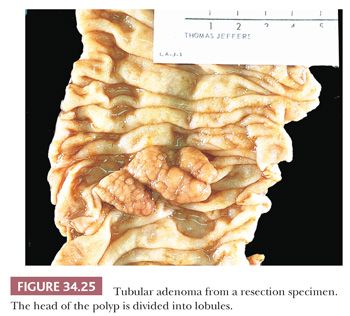
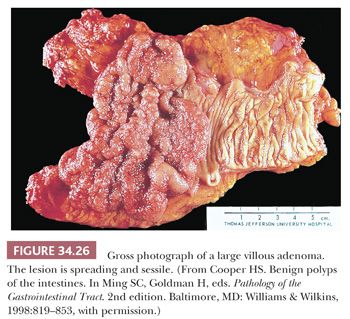
Grossly, adenomas may be polypoid (pedunculated or sessile) or nonpolypoid (see section titled “Nonpolypoid Adenomas”) (66). Classically, tubular adenomas have been described as pedunculated. In general, this is probably true; however, tubular adenomas may be sessile, and villous adenomas may be pedunculated. Tubular adenomas tend to be spheric, with a relatively smooth surface that is often divided into lobules secondary to intercommunicating clefts in the head of the adenoma. The adenoma is reddish or darker than the surrounding mucosa (Fig. 34.25). Villous adenomas tend to have a shaggy surface with obvious papillary fronds (Fig. 34.26). In general, the size of the adenoma correlates with the histologic type. Ninety-one percent of adenomas smaller than 1 cm are tubular adenomas, 7% are tubulovillous adenomas, and 2% are villous adenomas. When adenomas are larger than 2 cm, 50%, 38%, and 12% are tubular, tubulovillous, and villous adenomas, respectively.
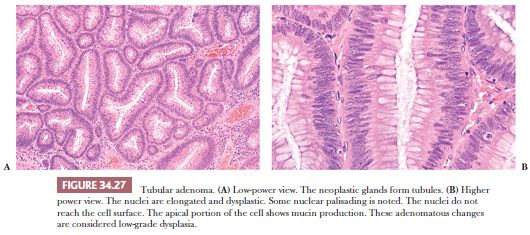
Adenomas, irrespective of type, consist of similar adenomatous epithelium. The overall growth pattern determines whether an adenoma is tubular, tubulovillous, or villous. Tubular adenomas have a proliferation of adenomatous epithelium showing gland or tubule formation. These tubules are separated from each other by normal lamina propria (Fig. 34.27).
In villous adenomas, one sees similar epithelium; however, the overall growth pattern or configuration is fine fingerlets or villi that project perpendicularly from the muscularis mucosae to the outer tip of the adenoma (Fig. 34.28). Because many lesions show mixed tubular and villous features, the question arises of how each specific type is defined. If 75% to 80% of the lesion is tubular or villous, then it is classified as tubular adenoma or villous adenoma, respectively, with all the lesions that fall in between being classified as tubulovillous adenomas (69). In adenomas of all types, the epithelial cells show enlarged, elongated, hyperchromatic nuclei that are stratified within the cell. Mucin may be absent or, when it is present, may vary in amount from well-formed goblets to small apical vacuoles.
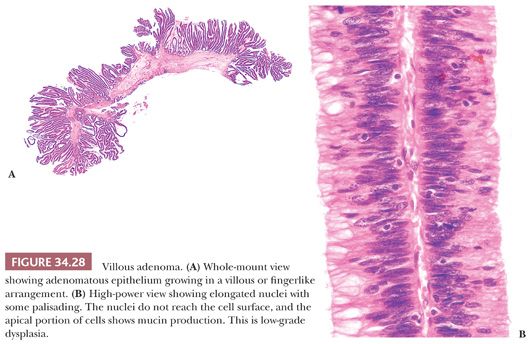
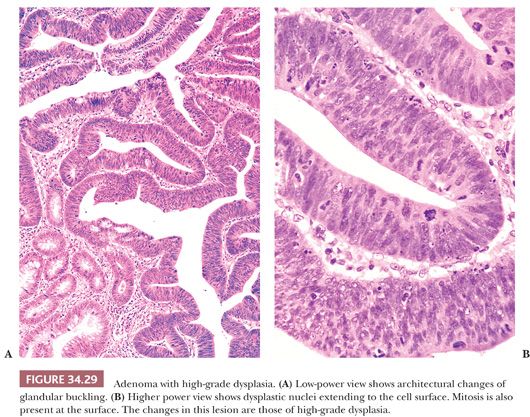
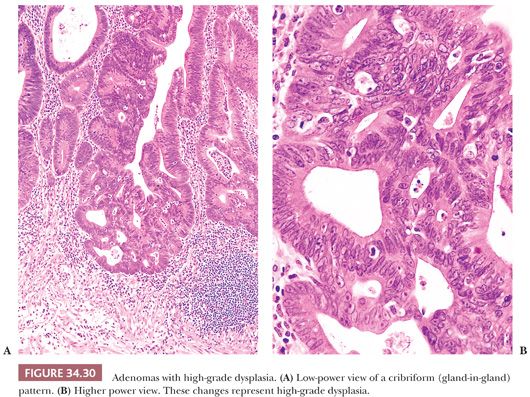
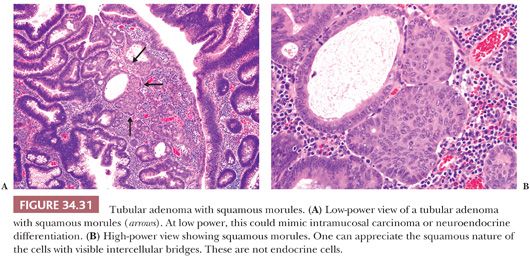
By definition, adenomas consist of dysplastic epithelium. The WHO (69) and most pathologists use the system of high-grade and low-grade dysplasia (intraepithelial neoplasia). The usual or common adenoma is considered to show low-grade dysplasia (Figs. 34.27 and 34.28). High-grade dysplasia encompasses changes that were considered in situ cancer in the past, and it is replacing that term in practice, although in situ cancer is still used in the tumor-node-metastasis (TNM) staging system of the American Joint Committee on Cancer (AJCC) (70). In high-grade dysplasia, one sees both architectural and cytologic changes. Architecturally, the crypts show irregular branching, budding, or cribriform configurations. Cytologically, the nuclei are enlarged, hyperchromatic, or vesicular with prominent nucleoli. The nuclei are stratified within the cell, and they reach the luminal border of the cell. Mitotic figures are prominent. Areas of necrosis may be present, and cytoplasmic mucin production may be reduced or absent (Figs. 34.29 and 34.30). Adenomas may contain foci of Paneth cell metaplasia, melanocytic metaplasia, and squamous metaplasia (Fig. 34.31).
NONPOLYPOID ADENOMAS
Nonpolypoid adenomas may be slightly elevated, flat, depressed, and lateral spreading. Nonpolypoid colonic adenomas have a propensity for high-grade dysplasia when the lesions are small, and the depressed variant may be a precursor of small malignant lesions that rapidly grow into deeply penetrating cancers (71). The initial reported studies regarding flat (nonpolypoid) adenomas were from Japan (71,72), a finding that led some investigators to believe that these flat aggressive adenomas were rare in Western societies (73). However, colonoscopic screening studies from Western populations that used chromoendoscopy with magnification reported a prevalence of 22% to 23% for flat adenomas (74,75). A colonoscopic screening study of 1000 persons from England reported that 36% of all adenomas were flat, whereas 63% were polypoid and 0.6% were depressed (76). A large colonoscopy study of colorectal adenomas and early carcinoma from Japan reported an incidence of 44.5% for nonpolypoid lesions (71,77).
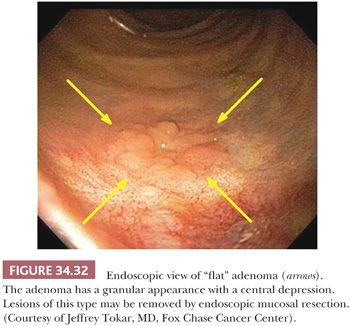
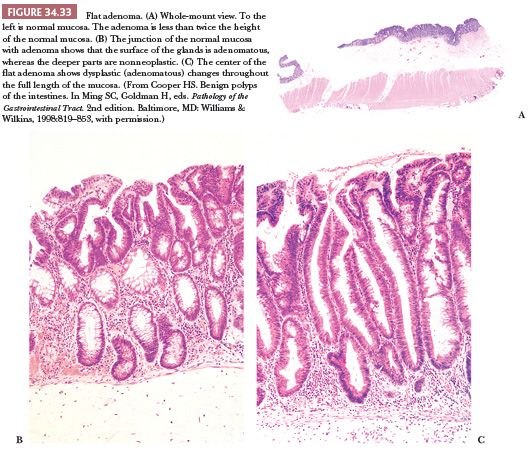
Grossly, flat lesions are flat or slightly raised plaques that often have a central depression, which can easily be missed on colonoscopy; they are usually small (Fig. 34.32). The endoscopic criteria for a flat adenoma are as follows: a mucosal elevation with a flat or rounded surface and a height less than half the diameter of the lesion, which does not have the typical appearance of a hyperplastic polyp. Most flat adenomas are less than 2 mm in height or have a height that is less than the closed cups of the biopsy forceps (2.5 mm). Lesions protruding above this level are classified as sessile (66). Large (>1 cm) nonpolypoid colorectal adenomas are often labeled as lateral spreading tumors (78). These have been divided into the granular and flat type. The granular type has a granulonodular uneven surface, whereas the flat type has a flat smooth surface. Histologically, the lesions lack a polypoid or exophytic growth pattern, and they appear plaque-like. In vertical extent, these lesions are never greater than twice the thickness of the adjacent nonneoplastic epithelium. Characteristically, they are tubular adenomas, with the adenomatous or dysplastic epithelium limited to the luminal surface of the crypt that is associated with underlying nonneoplastic epithelium. Adenomatous transformation can develop throughout the full thickness of the crypt; however, this feature is usually limited to the center of the lesion, with the radial portion showing the characteristic histologic features (Fig. 34.33). Nevertheless, this histologic finding of dysplastic glands limited to the luminal surface is not diagnostically pathognomonic for flat adenoma (nonpolypoid). One study reported that, in a review of consecutive adenomas, 25% showed the foregoing histologic features, none were flat endoscopically, and none had high-grade dysplasia (79). The diagnosis of flat adenoma should be made only when the characteristic endoscopic appearance is present. These nonpolypoid adenomas tend to have a high incidence of high-grade dysplasia (carcinoma in situ), ranging from 10% to 41% (75,80,81), which are much higher percentages than the 4% incidence seen in comparably sized classic adenomas. The incidence of submucosal cancer is high in the depressed subtype of nonpolypoid lesions (74). The incidence of submucosal invasion in adenomas less than 1 cm in size is 40.9% for depressed nonpolypoid, 1.3% for polypoid, and 0.2% for flat nonpolypoid (74). The discrepancies in the reported incidence of high-grade dysplasia and small early cancers have been said to be related to differences in diagnostic criteria among different Japanese pathologists (82) or between Japanese and Western pathologists (83). However, two studies from the West have reported a significantly greater incidence of early cancers associated with flat adenoma than with polypoid adenomas of similar size (74,76). Small flat adenomas tend to evolve into polypoid adenomas, whereas nonpolypoid depressed adenomas of the right colon become endophytically growing advanced cancers. Granular laterally spreading adenomas of the right colon are considered precursor lesions for CIMP-H/MSS and possibly a proportion of CIMP-H/MSI-H cancers (84). The flat laterally spreading lesions have a greater malignant potential than the granular spreading type (77). Granular type of laterally spreading adenoma has a significantly greater incidence (61%) of CIMP-H compared with polypoid adenomas (25%) and flat laterally spreading adenoma (8%). The incidence of K-ras mutations is significantly higher in granular laterally spreading tumors (78%) compared with polypoid adenomas (25%) and flat laterally spreading adenoma (16%) (84).
PROCESSING AND REPORTING OF ENDOSCOPICALLY REMOVED MATERIAL
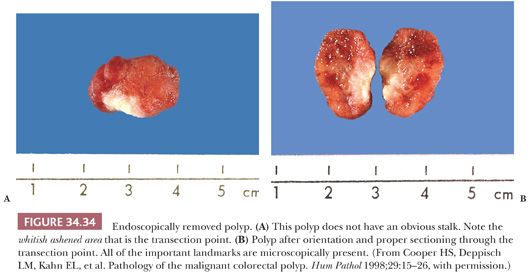
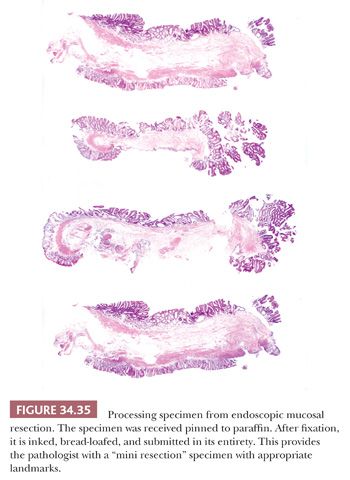
The endoscopically removed material that the pathologist receives is usually excised by either snare cautery, including polypectomy, or forceps biopsy. Recently, specimens are being received as endoscopic mucosal resections (EMRs). EMRs are often used for nonpolypoid lesions. Forceps biopsy material should be submitted in its entirety, and levels should be cut. In our laboratory, we cut three levels. Ideally, the tissue should be blocked so multiple ribbons can be placed on a slide. When a polypectomy specimen is received, the pathologist must carefully inspect the gross specimen and look for the stalk or transection point. The polyp should be cut so that all of the proper landmarks become evident on the glass slide (Fig. 34.34). The entire specimen should be submitted. For the polyp to be cut properly, it must be well fixed. Polyps less than 1.5 cm in diameter may require 2 to 3 hours of fixation of the entire lesion before gross sectioning, whereas larger polyps may require longer or possibly overnight fixation. Although presently there are no guidelines for processing colorectal EMR specimens, one might follow the guidelines for gastric EMR specimens (85). In our laboratory, we receive the specimen pinned on cork/paraffin. The specimen is inked, serially sectioned, and submitted in its entirety (Fig. 34.35).
The report that the pathologist renders from forceps biopsy material usually can provide only limited information (e.g., type of adenoma present or cancer present and degree of dysplasia or grade). Because the diagnosis of the type of adenoma rendered by the pathologist will play an important role in subsequent patient screening and predicting for subsequent advanced adenoma (86,87), one should classify the lesion as tubular adenoma, tubulovillous adenoma, or villous adenoma. One should indicate whether high-grade dysplasia is present. Because by definition all adenomas show dysplasia, I do not comment when only low-grade dysplasia is present. A diagnosis of cancer can often be rendered. If no submucosa is present, the pathologist cannot absolutely make a diagnosis of invasive cancer. However, in the presence of marked desmoplasia, an infiltrative pattern, and ulceration, one can be confident that it is invasive cancer, especially if the sample is taken from a mass lesion. At times, forceps biopsy material will provide nondiagnostic material (e.g., the eroded surface of an inflammatory fibroid polyp), and resection or polypectomy is required for diagnosis. Obviously, polypectomy material enables the pathologist to categorize lesions exactly and, if cancer is present, to determine (a) whether or not it is invasive; (b) its grade; (c) its relation to the margin of transection; and (d) whether lymphatic or venous invasion is present (see section titled “Examples of Reporting Malignant Polyps”). Because EMR specimens are a “mini” local excision, we try to report the same parameters as in local excision (e.g., type of lesion and grade of cancer if present, status of margins, and presence or absence of lymphatic invasion if cancer is present) (Fig. 34.35).
ADENOMAS WITH CANCER: DEFINITIONS
The incidence of high-grade dysplasia (in situ cancer) and invasive cancer arising in adenomas is approximately 12.3% and 5%, respectively. Differentiating between invasive cancer and high-grade dysplasia is important because the former has the biologic potential to metastasize, whereas the latter does not. The term invasive adenocarcinoma should be restricted to the situation in which cancer has invaded through the muscularis mucosae and into the submucosa.
High-grade dysplasia is a pathologic process that is limited to the mucosa and that, more precisely, is confined within its crypt or gland basement membrane. The diagnosis of high-grade dysplasia is made on architectural and cytologic changes. At low and medium power, the glands show an irregular complex branching, budding, or cribriform pattern. Cytologically, the individual cells usually show loss of mucin, severe nuclear atypia (hyperchromasia or vesicular pattern with enlarged nucleoli), and stratification reaching the luminal surface of the cell; increased mitoses; and, occasionally, necrosis (Figs. 34.29 and 34.30). The term intramucosal cancer is often used by pathologists. It is defined as cancer that shows invasion into the lamina propria and/or muscularis mucosae, but not into the submucosa (Fig. 34.36). Intramucosal carcinoma has no biologic potential for metastases. Lewin et al. (88) have reported that intramucosal high-grade poorly differentiated carcinoma can be managed endoscopically.
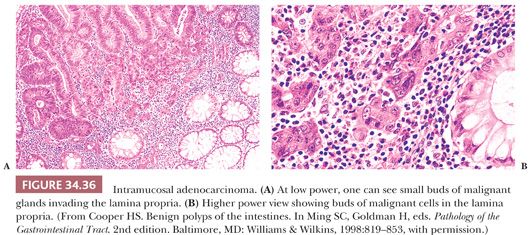
ENDOSCOPICALLY REMOVED MALIGNANT POLYP
A malignant polyp is one in which cancer has invaded into the submucosa. Cancer limited to the mucosa or only to the muscularis mucosae is not a malignant polyp and has no metastatic potential.
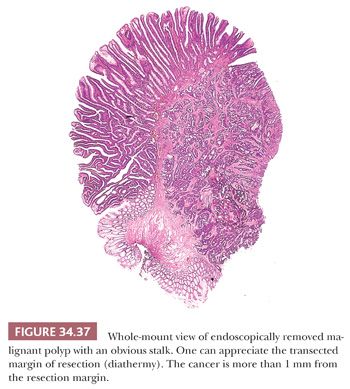
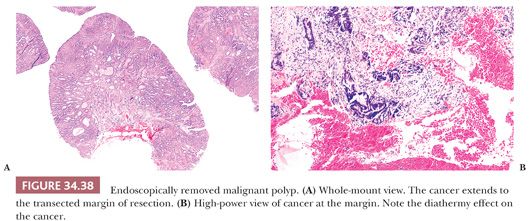
The evaluation of an endoscopically removed malignant polyp is a stepwise process that involves (a) adequate fixation, (b) sectioning, (c) knowledge of the type of removal, (d) examination of slides, and (e) pathologist–clinician interaction. Fixation and sectioning are discussed earlier in the section titled “Processing and Reporting of Endoscopically Removed Material.” If the polyp is not properly fixed, sectioning may be difficult, and it may result in a less than optimum specimen, which may preclude the pathologist from properly evaluating the lesion. The pathologist should be informed whether the polyp was removed piecemeal or in one piece. Piecemeal polypectomy results in multiple fragments of tissue showing diathermy and may preclude one from properly evaluating the true margin of resection. The pathologist should be informed about this so that he or she can make a true and accurate statement about the margin of resection.
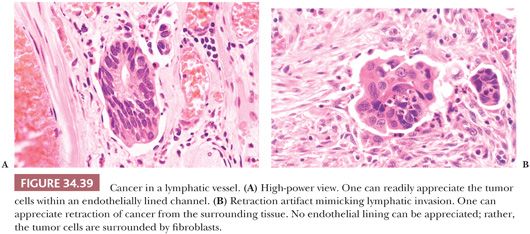
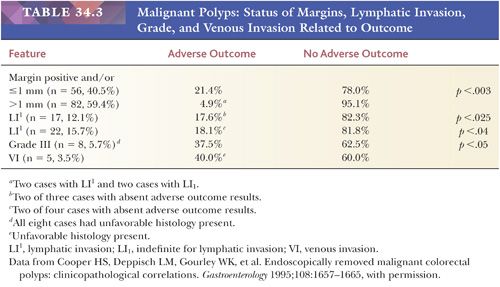
When examining the slides, one should evaluate and report on the following: (a) status of resection margin, (b) grade of cancer, and (c) presence or absence of lymphatic or venous invasion (Figs. 34.37 to 34.40). Most investigators believe that if cancer is at or near the resection margin and/or is grade III and/or is in lymphatics or veins, then polypectomy should be followed by definitive surgical resection. However, if the margin is negative, the cancer is grade I or II, and no lymphatic or venous invasion is noted, then polypectomy should be curative (89,90). The definition of cancer at the margin is the presence of tumor cells in the actual free edge of the submucosal transection point that contains diathermy. Tumor close to the margin has been defined as cancer within the actual diathermy, one hpf from the diathermy, less than 1 mm from the transected margin, or less than 2 mm from the transected margin. The detection of lymphatic invasion requires tumor emboli in true endothelially lined channels; this should be differentiated from retraction artifact, which is commonly present. Retraction artifact is most often present within the tumor itself, whereas true lymphatic invasion is more confidently diagnosed in the peritumoral submucosa (away from the tumor). Great interobserver variation exists in diagnosing lymphatic invasion (91); because of this, some investigators do not even bother evaluating such invasion (92,93).
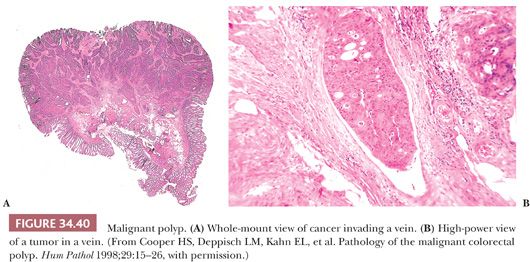
In a large interinstitutional study (91), the adverse outcome rate (i.e., recurrent disease in those treated by polypectomy only or lymph node metastases or residual local disease in those treated by resection after polypectomy) was 19.7% in patients with cancer at or near the margin, and/or grade III cancer, and/or lymphatic or venous invasion. Analyzed further, the adverse outcome rate was 21.4% for cancer at or near the margin, 16.6% for lymphatic invasion, and 37.5% for grade III cancer (Table 34.3). Polypoid carcinomas (cancers with no residual adenoma) are treated in a fashion similar to that of cancers with background adenoma.
There is controversy concerning whether malignant colorectal polyps with a sessile configuration can be treated by endoscopic removal. Overall, the literature seems to indicate that endoscopically removed sessile malignant polyps have a significantly greater adverse outcome than do polypoid malignant polyps. However, close review of the data indicates that configuration itself is not a significant variable for adverse outcome, and endoscopically removed sessile malignant polyps with a negative margin, no lymphovascular invasion, and grade I or II can be successfully treated by endoscopic polypectomy (94).
New potentially significant histopathologic parameters that may help predict treatment of endoscopically removed malignant polyps are depth of invasion, tumor budding, lymphatic vessel density, and cribriform histology. From a cohort of patients whose initial treatment was either endoscopy or primary surgery, the incidence of lymph node metastasis was 0% in cancers that had invaded to a depth less than 2000 µm when criteria of lymphatic invasion, grade, and margin of resection were also taken into consideration (90,94). Kaneko (95) reported an incidence of lymph node metastases of 1.5% and 13.8% in cancers invading less than 1000 µm and greater than 1000 µm, respectively. In multivariate analysis, studies of T1 cancers have shown that the presence of tumor budding (see later “Tumor Budding” section) was significantly associated with lymph node metastasis and/or local regional failure (90,94,95). In T1 cancers, those with high lymphatic density (counted on D2-40–stained slide) had a significantly greater incidence of lymph metastasis than those with low lymphatic density (23.3% vs. 8.4%, respectively) (95). T1 colorectal cancers with a cribriform histology were significantly associated with lymph node metastasis using multivariate analysis. However, the study was a cohort treated initially with either endoscopic polypectomy or primary resection and not analyzed separately (90).
Examples of Reporting Malignant Polyps
Example 1. Sigmoid colon, polypectomy: grade III adenocarcinoma arising within an adenoma. The cancer extends to the transected margin of resection. No evidence of lymphatic or venous invasion.
Example 2. Rectum, polypectomy: grade II adenocarcinoma arising within an adenoma. Cancer is 2.5 mm from the transected margin (the margin is negative for cancer). No evidence of lymphatic or venous invasion.
ADENOMA WITH PSEUDOINVASION OR MISPLACED EPITHELIUM
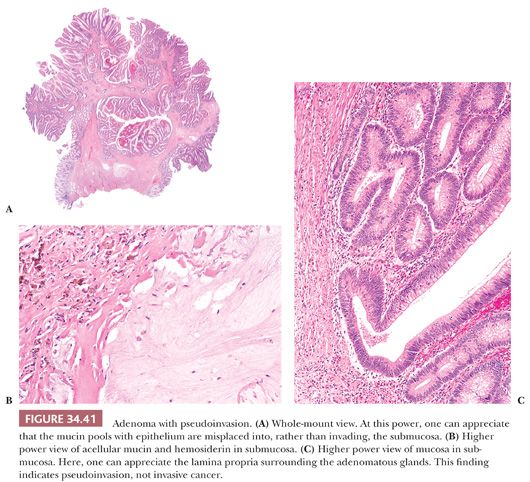
Pseudoinvasion is defined as the situation in which the mucosa (or epithelium) of the polyp has been “misplaced” into the submucosa, thus mimicking invasive carcinoma. Other terms to describe this entity are epithelial misplacement and hamartomatous inverted polyp. Differentiating pseudoinvasion from invasive cancer is one of the most difficult problems in GI surgical pathology. It is important for the pathologist to be able to recognize this entity so that a mistaken diagnosis of cancer is not rendered and unnecessary surgery is not performed. The prevalence of pseudoinvasion varies from 2.5% to 3.5% of all adenomas. The male-to-female ratio is 3:1. Most of these lesions are located in the sigmoid colon, and they occur in adenomas with well-defined stalks. The diagnosis of pseudoinvasion is best appreciated at low-power magnification, which reinforces the impression of misplacement rather than invasion.
The pathology is characterized by the presence of epithelial structures misplaced into the submucosa. In invasive cancer, malignant epithelial glands infiltrate into the submucosa and cause a desmoplastic reaction. In pseudoinvasion, the misplaced structures consist of both epithelium and surrounding lamina propria. The finding of lamina propria surrounding these misplaced glands is extremely helpful in differentiating pseudoinvasion from invasive cancer (Fig. 34.41). This situation is somewhat analogous to adenomyosis of the uterus. The misplaced glands can show histologically normal epithelium, adenomatous changes, or even high-grade dysplasia (96). The submucosal glands can show cystic dilation with rupture and secondary inflammation. The submucosa often contains fresh hemorrhage and/or hemosiderin deposition. In cases with possibly misplaced glands, the determination of whether cancer is present in the mucosa is important. Without the presence of cancer in the mucosa, one should consider pseudoinvasion as the correct diagnosis rather than invasive carcinoma.
Epithelial misplacement can also occur in colorectal adenomas secondary to biopsy before polypectomy, and it is important for the pathologist to be cognizant of this possibility. Within the first few days after forceps biopsy, ulceration develops at the biopsy site. As a result, an exudate with admixed mucus and small groups of dysplastic (adenomatous) epithelial cells in the lamina propria is seen. By day 7, one can see small pools of mucus-containing proliferating dysplastic cells embedded in abundant granulation tissue within the submucosa. These changes are different from the type of epithelial misplacement described earlier. However, the history of a recent biopsy and the presence of granulation tissue, ulceration, reepithelialization, and small groups of dysplastic epithelium in mucin pools are distinctive enough to diagnose epithelial misplacement rather than invasive cancer (97).
FAMILIAL ADENOMATOUS POLYPOSIS
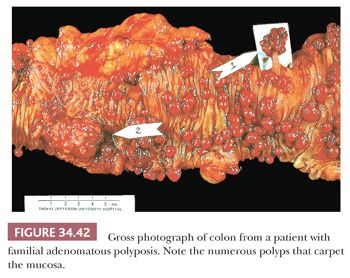
This condition is an autosomal dominant disorder in which the large intestine is carpeted with adenomas (ranging from hundreds to thousands) (Fig. 34.42) (29,67). The diagnostic criteria for FAP are (a) 100 or more colorectal polyps; (b) germline mutation in adenomatous polyposis coli (APC) gene; (c) family history of APC; and (d) at least one of the following: epidermoid cyst, osteoma, or desmoid tumor (67). Extraintestinal manifestations such as thyroid cancer, osteoma, epidermoid cysts, fibromas, supernumerary teeth, congenital hypertrophy of the retinal pigment epithelium, and desmoid tumors are part of this syndrome. Desmoid tumors occur in 10% of cases. They often occur in areas of previous surgery, and they can be quite aggressive, often causing death (98).
All patients with classic FAP develop colonic adenocarcinoma if the disorder is left untreated. The incidence of this disorder has been estimated at 1 in 8000 births, with 20% of cases representing a new mutation. The average age of detection in patients with symptoms is 36.5 years; however, the average age of detection is 23.5 years in patients screened because of a family history. Adenomas rarely appear before the age of 10 years, and no extraintestinal manifestations are observed. Approximately 90% of patients with FAP develop small-bowel adenomas, primarily in the duodenum, and gastric polyps, mainly fundic gland polyps and adenomas (67,68). Approximately 10% of patients will develop duodenal adenocarcinoma by the age of 60 years (98). The genetic defect consists of a germline mutation in the APC gene on chromosome 5q21 (29,68,98). In classic FAP, most mutations occur between codons 169 and 1393, with mutations between codons 1250 and 1464 being associated with particularly severe polyposis (98).
Attenuated Familial Adenomatous Polyposis
This condition is a less severe form of polyposis with a low number of polyps (adenomas), usually numbering less than 100; however, these patients sustain a high risk of colorectal cancer. The cancers usually develop 15 years later than in patients with classic FAP but 10 years earlier than in patients with sporadic cancer. Compared with classic FAP, there are no appreciable differences in upper GI tract lesions (e.g., fundic gland polyps or duodenal adenoma/adenocarcinoma). In attenuated FAP, the mutations tend to occur in the extreme 5′ and 3′ ends of the APC gene. The previously described entity of hereditary flat adenoma syndrome is currently thought to be a variant of attenuated FAP (98).
MYH POLYPOSIS SYNDROME
MYH-associated polyposis (MAP) is a recently described autosomal recessive disorder. Clinically, patients show features of attenuated FAP (<100 adenomas); however, 7.5% of patients with full-blown classic FAP without detectable APC mutations were found to carry biallelic inactivation of MYH. These patients often resemble cases of FAP with de novo APC mutations. In some cases, extracolonic features such as congenital hypertrophy of retinal pigment epithelium and duodenal polyps have been reported (99). In MAP, the precise colon cancer risk has not yet been ascertained; however, it is probably similar to FAP. The mean age of adenoma and cancer diagnosis is 46 and 49.7 years, respectively (98). Patients with MAP have microadenoma similar to that seen in FAP and attenuated FAP (99). MYH participates in base excision repair, and dysfunction of MYH results in the accumulation of somatic G:C → T:A mutations in specific growth regulatory genes, one of them being APC (99). The MYH protein is normally expressed in the cell nucleus. A recent publication has reported that loss of nuclear MYH protein (via IHC) may be a sensitive test for patients with MAP (100). HPs and SSA/P have also been shown to be a common finding in MAP, with 47% of MAP patients having SSA/Ps and/or HPs. The detection of almost exclusively G:C → T:A transversions in the K-ras gene of SSA/P and HP suggests that these polyps are causally related to MYH deficiency (101).
MUIR-TORRE SYNDROME
The Muir-Torre syndrome is an autosomal dominant hereditary disorder with a variant phenotype that consists of skin tumors of sebaceous differentiation and visceral malignancies. Fifty percent of patients have colorectal cancer, and 15% of female patients develop endometrial cancer (102,103). The median age of onset of colorectal neoplasms is 50 years of age, and the colon cancers are more often located in the proximal colon (103). Kindreds of Muir-Torre patients show mutations similar to patients with Lynch syndrome (hereditary nonpolyposis colon cancer [HNPCC]). However, 35% of the tumors are MSS, and these have a later onset and a less pronounced family history (103).
TURCOT SYNDROME
Turcot syndrome was originally described in two siblings with polyposis coli who developed malignant brain tumors. This rare disorder is probably autosomal dominant. The true frequency of Turcot syndrome is difficult to assess because the brain tumors are associated with a high mortality and may precede the detection of colonic polyps. Studies indicate that two forms of Turcot syndrome may be seen; one group consists of patients with gliomas and colorectal adenomas (nonpolyposis patients), and the other group consists of patients with brain tumors (predominantly medulloblastomas) associated with adenomatous polyposis. Molecular studies have shown that the former group has germline mutations of mismatch repair genes (hMLH1 and hMSH2) and is part of the hereditary nonpolyposis syndrome, whereas the latter group is associated with germline mutations of the APC gene (29,98) and is part of the FAP syndrome.
HEREDITARY MIXED POLYPOSIS SYNDROME
Hereditary mixed polyposis syndrome is an autosomal dominant disorder involving a single Ashkenazi Jewish family that was initially mapped to chromosome 15q13q14, which overlaps the region containing the CRAC1 gene (colorectal adenomas and carcinoma gene) located at 15q14-q22 (22). More recent data on this kindred and other Askenazi kindreds has found a mutation just upstream of the GREM1 gene that appears to be the culprit (104). The following types of polyps have been described in individuals with this disorder: (a) classic adenomas, (b) HPs, (c) SSA/Ps, and (d) atypical juvenile polyps. The number of polyps is usually less than 15 per patient. Colorectal cancer is seen in this disorder. The characteristic lesion appears to be an atypical juvenile polyp. This disorder may be a variant of juvenile polyposis; however, in juvenile polyposis, adenomas are uncommon, whereas in hereditary mixed polyposis, most of the polyps are adenomas. In hereditary mixed polyposis, the number of polyps is lower than that seen in juvenile polyposis. Juvenile polyposis usually presents a decade earlier than hereditary mixed polyposis (105). Recently, data has suggested that Cowden syndrome may also be a form of a mixed polyposis syndrome (41).
CARCINOMA
SMALL INTESTINE
Small intestinal adenocarcinomas account for approximately 40% of small intestinal malignant tumors; however, compared with colonic adenocarcinoma, they are uncommon. The distribution along the small bowel varies depending on whether the series includes ampullary cancers. In one series, 48.4%, 32.5%, and 19.2% of tumors were located in the duodenum, jejunum, and ileum, respectively (106). Grossly, the cancers may be flat, stenosing, ulcerative, infiltrative, or polypoid. They are morphologically similar to adenocarcinomas elsewhere in the GI tract, but more often, they are papillary. These cancers are often associated with adenomas; their diagnosis by endoscopic biopsy may be difficult because they are frequently interpreted as adenomas with dysplasia.
LARGE INTESTINE AND RECTUM
Carcinoma of the colon and rectum is a disease of Western lifestyle. Among both male and female patients, it is the second most common visceral malignancy in the United States, with an expected 185,000 new cases each year.
Pathologic Features
Macroscopically, most colorectal cancers are of either the polypoid or the ulcerative-infiltrating type. Generally, polypoid cancers have a better prognosis than ulcerative lesions; however, this association most probably is directly related to the finding that polypoid cancers are usually of a lower clinicopathologic stage than are ulcerative lesions at the time of diagnosis. Rarely (0.3% of cases), colorectal cancer may grossly resemble linitis plastica of the stomach. However, many cases with a gross linitis plastica appearance may not be primary colorectal lesions but may, instead, be metastases from other sites. Table 34.4 lists the histopathologic classification of colorectal cancer according to the WHO.

Adenocarcinoma
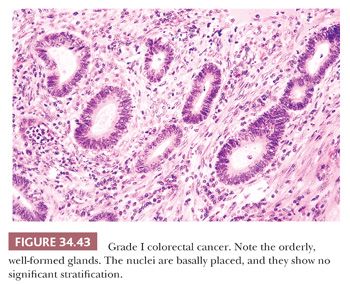
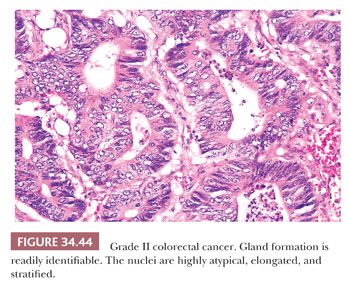
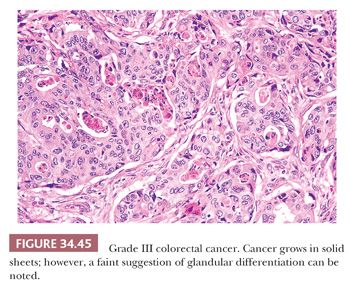
Adenocarcinoma can be divided into three grades based primarily on an overview of the arrangement of cells with regard to the degree of tubular (acinar) formation. Fifteen percent to 20% of colorectal adenocarcinomas are grade I (low-grade or well-differentiated tumors), 60% to 70% are grade II (average-grade or moderately differentiated tumors), and 15% to 20% are grade III (poorly differentiated tumors). Grade I cancers are composed mainly of simple tubules, in which the nuclear polarity is easily discerned and the nuclei are of uniform size. A distinct resemblance to adenomatous epithelium is present (Fig. 34.43). Grade II cancers are composed of tubules that may be simple, complex, or slightly irregular, in which the nuclear polarity is barely discernible or is lost (Fig. 34.44). Grade III tumors are characterized by a predominance of the absence of glandular differentiation (solid-like pattern) as well as by loss of nuclear polarity (Fig. 34.45). The terms low grade and high grade are now favored for clinical usage. Grades I and II have been collapsed into low grade and grade III and undifferentiated carcinoma (see the following discussion) have been combined into high grade. Partially, this is based on the similar behavior of grades I and II carcinomas and greater reproducibility among pathologists (107–109). Grade III cancers have a significantly poorer prognosis than grade I and II lesions (110,111). To define a colorectal carcinoma as a grade III lesion, the poorly differentiated component should form more than 50% of the tumor rather than representing only small rare foci of the neoplasm. This grading system is based on the predominant pattern; however, some recommend that grading be based on the worst area even if it does not predominate (112). The disorganized glands commonly seen at the advancing edge of the cancer should not be construed as high-grade malignancy, and these should not be assessed in grading (69,111). These disordered glands at the advancing edge of cancer have been called tumor budding, and they have been associated with a significantly poorer prognosis within tumors of similar stage (see later section titled “Tumor Budding”) (113,114).
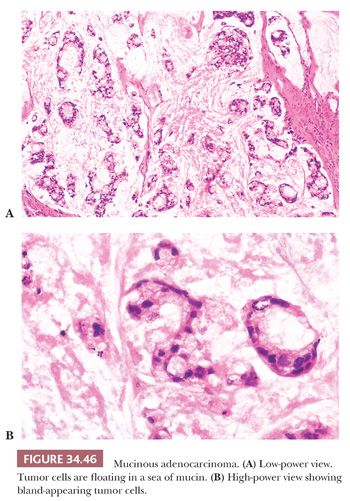
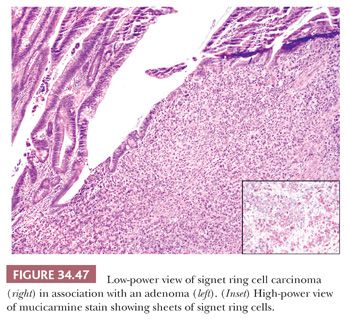
Mucinous Adenocarcinoma. Mucinous adenocarcinomas, including signet cell cancers, account for approximately 10% of colorectal cancers. The definition of mucinous carcinoma is that at least 50% of the lesion must be mucinous (69,111). Mucinous cancers are associated with tumors with MSI (sporadic and hereditary nonpolyposis syndrome), CIMP, young adults and children, villous adenomas, cancers arising secondary to therapeutic irradiation, ulcerative colitis, and colorectal cancers in low-incidence developing countries. Compared with nonmucinous colorectal cancers, mucinous carcinomas usually present at a more advanced stage, they have more extensive perirectal spread, they show a greater incidence of lymph node involvement, and they tend to have an overall poorer prognosis (115). However, multivariate analysis has shown that stage is important in the prognosis, and mucinous histology itself is not an independent prognostic factor in colorectal cancer (115,116). Among the mucinous tumors, the extracellular type is much more common than the intracellular, or signet cell, type. The extracellular type is characterized by tumor cells floating freely in large pools of mucin. The cells that are floating in the mucin often have a bland histologic appearance (Fig. 34.46). The intracellular type is morphologically identical to the signet cell cancer seen in the stomach. The signet cell variant behaves very aggressively, and it has a poor prognosis (Fig. 34.47).
In a univariate analysis, patients with mucinous tumors with MSI (sporadic), expanding growth pattern, and lack of venous invasion had a significantly improved survival compared with patients with mucinous tumors that were MSS, had an infiltrative pattern, and had venous invasion; however, in multivariate analysis, only the expanding growth pattern and Dukes stage were shown to be independent prognostic factors (117).
Cribriform Comedo-Type Adenocarcinoma. This type of adenocarcinoma has extensive areas of cribriform glands with central necrosis similar as to what one would see in cribriform and comedo in situ duct carcinoma of the breast. This phenotype has been correlated with microsatellite stable CIMP (118).
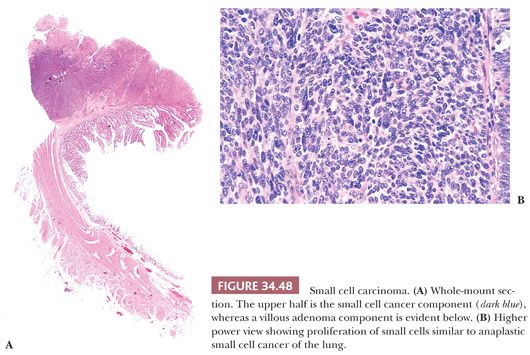
Small Cell Cancer
A rare variant is small cell cancer, which comprises less than 1% of colorectal cancers (119). Histologically, these cancers are identical to small cell carcinoma of the lung (oat cell type and intermediate type) (Fig. 34.48). They have an extremely poor prognosis, and almost all cases have lymph node and liver metastases. A third of these cancers have arisen within typical adenomas. These small cell cancers may also show areas of squamous differentiation and intimate association with the usual type of adenocarcinoma. Immunopathology may be helpful in making a diagnosis of small cell cancer. These neoplasms express neuron-specific enolase (84% of cases), Leu-7 (18%), synaptophysin (50%), and chromogranin (37%). However, the histopathology is so characteristic that, in most instances, the diagnosis can readily be made by the examination of routine hematoxylin and eosin–stained slides.
Undifferentiated Cancer
Undifferentiated colorectal cancers are uncommon, representing less than 1% of colorectal cancers. They are malignant epithelial tumors that have no glandular structures or other features to indicate definite differentiation. The absence of intracytoplasmic mucin helps to differentiate these tumors from poorly differentiated adenocarcinoma (120). Many of the cases originally described as undifferentiated carcinoma represented what is now defined as medullary carcinoma (69,120). According to Jass (121), undifferentiated carcinomas lack circumscription, can be pleomorphic, and lack tumor-infiltrating lymphocytes (TILs) or Crohn-like reaction, whereas medullary carcinomas have a uniform cell type (lack pleomorphism), are circumscribed, and have TILs and a Crohn-like reaction.
Medullary Carcinoma
Medullary carcinomas are located in the proximal colon and have a female predominance. Grossly, these tumors are large bulky masses with an expansile growth pattern. The tumor cells have eosinophilic or amphophilic cytoplasm with rounded nuclei with prominent nucleoli. The tumor cells consist of undifferentiated small to medium rounded tumor cells arranged in closely packed, trabecular, or solid patterns. Medullary carcinomas are well circumscribed and have a prominent Crohn-like reaction, focal gland formation, and mucinous features (122,123). At low power, this pattern often mimics endocrine neoplasms (but is negative for endocrine markers), a finding that should arouse one’s suspicion that this could be a medullary carcinoma. Loss of expression of CDX-2 is strongly associated with medullary carcinoma (Fig. 34.49) (124).
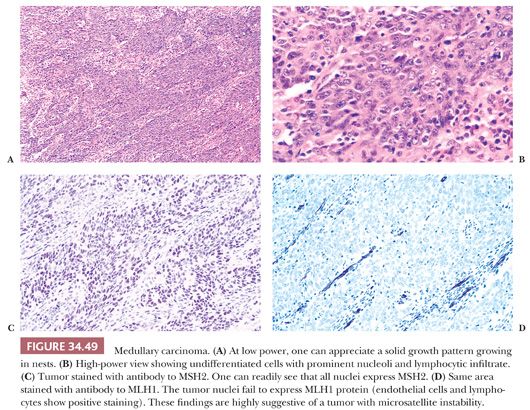
Surprisingly, these carcinomas have a good prognosis and show less frequent lymph node metastasis than other variants (120,122,123). Tumors with a medullary pattern are often diploid (85%), lack expression of p53, and are MSI-H in both sporadic colorectal cancer and HNPCC (123,125,126) (see “Microsatellite Unstable Cancers and Lynch Syndrome [Hereditary Nonpolyposis Colon Cancer]”). The pathologists should be aware that unlike most colorectal cancers, these tumors may lack expression of CK20 (127).
Squamous and Adenosquamous Cancers
Squamous cancers or adenoacanthomas of the colon are extremely rare. They have been associated with ulcerative colitis, schistosomiasis, and pelvic irradiation. To make a diagnosis of primary colorectal squamous cancer or adenoacanthoma, the following criteria must be met: there must be no other sites of squamous cancer in the body and no involvement of cloacogenic or squamous-lined mucosa. Survival correlates with Dukes staging, similar to common adenocarcinoma (128).
Serrated Adenocarcinoma
Serrated adenocarcinoma (SACA) is a newly described entity that encompasses 7.5% of all colorectal carcinomas and 17.5% of proximal carcinomas (129). It is more common in females (9.3%) than males (5.8%). SACAs belong to the category of CIMP-H tumors, the vast majority being MSI-L (and/or MSS) with TSAs as their precursor lesion. A smaller percentage are MSI-H with SSA and mixed polyps being the precursor lesions. SACAs that are MSI-L or MSS have a significantly worse prognosis than those that are MSI-H. SACAs are recognized by a constellation of histopathologic features (Fig. 34.50).
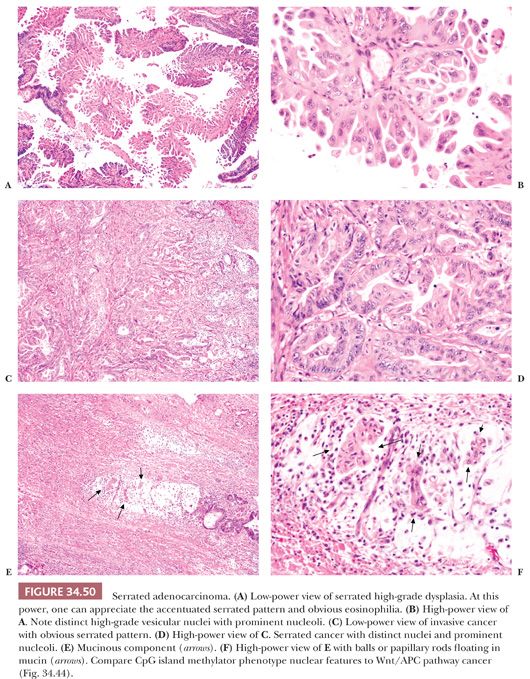
1. A serrated pattern of epithelium with eosinophilic cytoplasm with well-preserved nuclear polarity is seen. The nuclei can be bland or vesicular with prominent nuclei; however, the nuclei are not hyperchromatic, overlapping, or stratified as seen in adenocarcinomas that arise via the Wnt pathway.
2. Forty-three percent of SACAs show mucinous differentiation, and 17.6% are mucinous adenocarcinomas with a well-preserved serrated pattern. Histologically, they characteristically have eosinophilic papillary rods and eosinophilic cell balls floating in mucin. This histology is different from that seen in mucinous carcinomas that arise via the Wnt and/or CIMP-L pathways.
3. Poorly differentiated SACA cells grow in a trabecular pattern. This often bears morphologic resemblance to invasive micropapillary carcinoma and often shows lymphovascular invasion. The tumor cells show abundant cytoplasmic eosinophilia with vesicular nuclei and prominent nucleoli.
This constellation of histopathologic findings should alert the pathologist to the diagnosis of SACA. Glandular serration in isolation is a nonspecific finding that may be seen in all types of colorectal cancer.
Micropapillary Adenocarcinoma
Micropapillary carcinoma is an uncommon histologic variant with an aggressive behavior described in breast and urinary bladder. Micropapillary carcinoma of the colon and rectum has been recently reported in separate studies representing 9.0% and 19.1% of carcinomas from Houston, Texas, and Seoul, South Korea, respectively (130,131). Histologically, micropapillary carcinoma consists of balls/clusters of neoplastic cells (with eosinophilic cytoplasm and pleomorphic nuclei) surrounded by cleftlike spaces. The diagnosis is easily appreciated at low magnification (Fig. 34.51). A minimum of 5% of tumor with micropapillary features is required for the diagnosis. The micropapillary component ranges from 5% to 80% of the tumor, with the majority of tumors showing only a focal micropapillary component (<10% to 30%). Compared with “conventional” colorectal adenocarcinoma, the micropapillary variant has significantly greater incidence of lymph node metastasis, greater number of positive lymph nodes, more advanced stage, and greater incidence of distant metastasis, regardless of the percentage of micropapillary component. In multivariate analysis, micropapillary features are an independent predictor of lymph node metastasis. Micropapillary carcinoma, although superficially similar to what has been designated tumor budding, should not be confused with the latter. Compared with tumor budding, micropapillary carcinoma is present at both the advancing edge of the tumor and within the body of the actual tumor. The tumor nests/clusters/balls of micropapillary carcinoma are larger than what is defined as tumor budding (130,131).
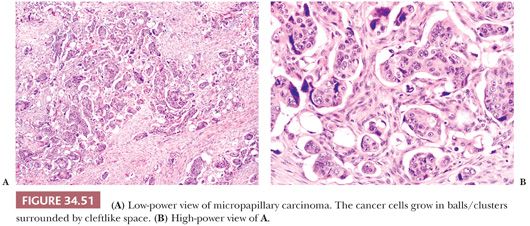
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


