Chapter 10
Intermediate metabolism, diabetes, and atherosclerosis
10.1 Overview
Genetic enzyme defects rare; comparatively little effort spent on targeted drug development; only a few defects can be treated with drugs
Gout more common; multiple causes, similar manifestation and treatment
Diabetes mellitus very common; treatment with insulin injections (types 1 and 2) and oral antidiabetics (type 2)
Atherosclerosis exceedingly common; drug therapy targets underlying metabolic conditions, other risk factors, and consequences of advanced disease
Notes: Intermediate metabolism involves numerous enzymes, and inheritable defects for many of them may cause disease. Only in a few cases can these genetic diseases be treated with drugs, and therefore, it does not make sense here to treat the entirety of metabolism in a systematic manner. Instead, we will look at a few selected defects for which some form of pharmacological treatment is available.
Diabetes is caused by endocrine dysregulation rather than primarily by enzyme deficiencies, so it could as well have been covered in chapter 7. It was included here because its manifestations are mostly metabolic. Also somewhat arbitrarily, atherosclerosis is covered here because of its important connections to cholesterol metabolism.
10.2 Enzyme defects in amino acid metabolism
The two hereditary enzyme defects that we will discuss here, phenylketonuria and tyrosinemia, both affect the same pathway, namely the degradation of phenylalanine and tyrosine. Their clinical manifestations are quite different, however, and so are their treatments.
10.2.1 Degradation of phenylalanine and tyrosine
Notes: This slide shows the degradation pathway for phenylalanine and of tyrosine, and the structure of NTBC (2-[2-nitro-4-(trifluoromethyl)benzoyl]cyclohexane-1,3-dione), an inhibitor of p-phenylpyruvate dioxygenase (3) that is used in tyrosinemia type I. The enzyme defect in phenylketonuria concerns phenylalanine hydrolase (1), and the one in tyrosinemia affects fumarylacetoacetate hydrolase (6).
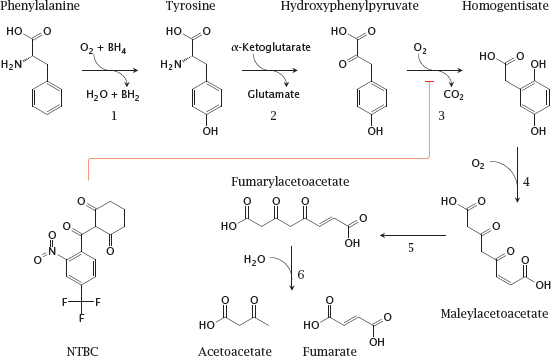
BH2 and BH4, Di- and tetrahydrobiopterin, respectively. Enzymes: 1, phenylalanine hydroxylase; 2, tyrosine transaminase; 3, p-phenylpyruvate dioxygenase; 4, homogentisate dioxygenase; 5, maleylacetoacetate isomerase; 6, fumarylacetoacetate hydrolase.
10.2.2 Phenylketonuria (PKU)
- Homozygous defect of phenylalanine hydroxylase
- Frequency: 1 newborn among 10,000 in Caucasians; frequency differs with race
- Symptomatic excess of phenylalanine manifest only after birth; intrauterine development normal
- Cognitive and neurological deficits, probably due to cerebral serotonin deficit
- Treated with phenylalanine-restricted diet
- Some cases due to reduced affinity of enzyme for cofactor THB, can be treated with high dosages of THB
Notes: As with most genetic enzyme defects, the clinical disease is manifest only in homozygous individuals. Dietary phenylalanine that is not used for protein synthesis accumulates and causes toxicity. It appears that the excess phenylalanine crowds out tryptophan at the L-aromatic amino acid transporter in brain capillaries, which is the same transporter that also transports tyrosine and L-DOPA to the brain (see slide 3.5.5). Since tryptophan is the precursor of serotonin (see slide 6.12.1), this results in a lack of cerebral serotonin [61], which is believed to cause the observed cerebral deficits.
In addition to phenylalanine itself, some aberrant metabolites derived from it also occur at increased levels, and the appearance of ketone derivatives such as phenylpyruvic acid in the urine has given the disease its name. These metabolites have no proven connection to the pathogenesis of the disease.
10.2.3 Ochratoxin A inhibits phenylalanyl-tRNA synthetase
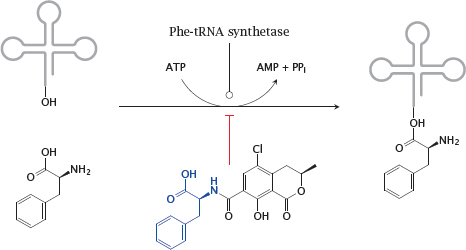
Notes: The variation of the gene frequency for PKU between races and geographical areas suggests that some regional environmental conditions may confer a selective advantage of the heterozygous state, as is the case with sickle cell anemia and glucose-6-phosphate dehydrogenase deficiency and malaria. It has been proposed that the heterozygote advantage in PKU consists in protection from the fungal toxin ochratoxin A, which is produced by some Aspergillus molds that cause food to rot [62].
Ochratoxin A competitively inhibits the coupling of phenylalanine to its cognate tRNA and thereby disrupts protein synthesis. It is more toxic to fetuses than to adults, most likely because fetuses are short of enzymes required for detoxifying xenobiotics. Mothers who are heterozygous for PKU will have a somewhat higher level of phenylalanine, which will be shared with the fetus via the placenta. This will counter the inhibition of tRNA aminoacylation in the fetus and thereby afford it a degree of protection.
One of the places with the highest abundance of PKU is Ireland. This country is also known for its repeated historic episodes of famine. Starving people will likely eat rotten food rather than discard it. Indeed, reference [62] reports that lowered rates of abortion were found in Irish women who were heterozygous for PKU; I have not ascertained whether these quoted statistics applied to periods of actual famine.
10.2.4 Tyrosinemia
- Homozygous defect of fumarylacetoacetate hydrolase
- Fumarylacetoacetate and preceding metabolites back up
- Fumaryl- and maleylacetoacetate react with glutathione and other nucleophiles, causing liver toxicity
- Drug NTCB inhibits p-hydroxyphenylpyruvate dioxygenase, intercepting the degradative pathway upstream of the toxic metabolites
Notes: Tyrosinemia is comparatively common in Quebec. In this case, there seems to be no heterozygote advantage; instead, the high incidence is due to the so-called “founder effect”, that is, the common descent of the afflicted population from a small group of founding settlers that happened to contain one or several carriers of the gene. (Refer to slide 10.2.1 for the relevant pathway and enzyme reactions.)
10.2.5 The urea cycle and related pathways
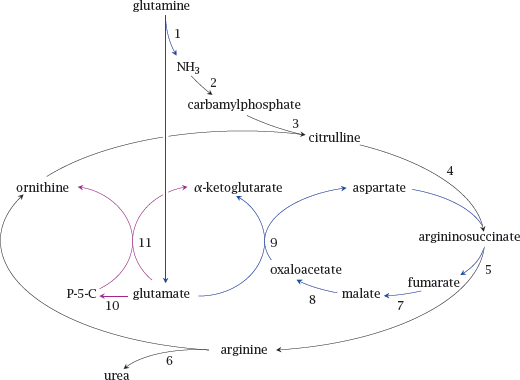
Enzymes: 1, glutaminase; 2, carbamylphosphate synthetase; 3, ornithine transcarbamylase; 4, argininosuccinate synthase; 5, argininosuccinase; 6, arginase; 7, fumarase; 8, malate dehydrogenase; 9, aspartate aminotransferase; 10, Δ1-pyrroline-5-carboxylate synthase; 11, ornithine amino-transferase.
Notes: The urea cycle runs in the liver and disposes of ammonia that accrues in the degradation of amino acids. It incorporates two molecules of ammonia into one molecule of urea, which is excreted in the urine.
While amino acid degradation occurs to a large part in the liver, other organs also participate; for example, the branched-chain amino acids leucine, isoleucine and valine are mostly degraded in skeletal muscle. Surplus ammonia is transported from these organs to the liver in the form of alanine or glutamine.
In the slide, reactions 2–6, shown in black, form the urea cycle. Reactions 1 and 7–9, shown in blue, supply the urea cycle with nitrogen from glutamine. Reaction 10 yields Δ1-pyrroline-5-carboxylate (P-5-C), and together with reaction 11 replenishes ornithine.
10.2.6 Alternate pathway therapy in urea cycle enzyme defects
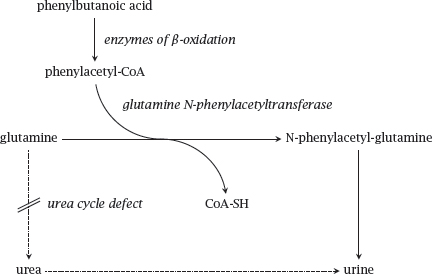
Notes: Alternate pathway takes advantage of the amino acid conjugation reactions that occur as part of phase II drug metabolism (see slide 4.4.9). In this application, the usual physiological role of amino acid conjugation is turned on its head: While in normal metabolism amino acids are expended in order to inactivate and eliminate unwanted organic acids, we here supply innocuous organic acids in order to induce their conjugation with amino acids. The elimination of these conjugates then becomes an alternate means for the disposal of surplus nitrogen. Brilliant!
The substrate acid covered in this scheme is phenylbutanoate, which undergoes β-oxidation before conjugation to glutamine and excretion in the urine. Another useful substrate acid is benzoate, which undergoes conjugation with glycine and further drains the liver’s pool of nitrogen.
Alternate pathway therapy is combined with the application of arginine or citrulline. If the urea cycle is functional, arginine can be diverted to protein synthesis as required, and so is not an essential amino acid; however, disruption of the urea cycle may cause a shortage of arginine. This may induce protein catabolism, thereby exacerbating the symptoms. If the enzyme defect is not between citrulline and arginine, it is possible to supply citrulline instead, which has the advantage of picking up another surplus nitrogen en route to arginine.
10.3 Metabolic diseases related to purine nucleotide metabolism
In this section, we will consider adenosine deaminase deficiency and gout. Adenosine deaminase deficiency is a hereditary enzyme defect and, as such, a rare condition. In contrast, gout can arise from multiple causes and thus is much more common.
10.3.1 Overview of purine degradation
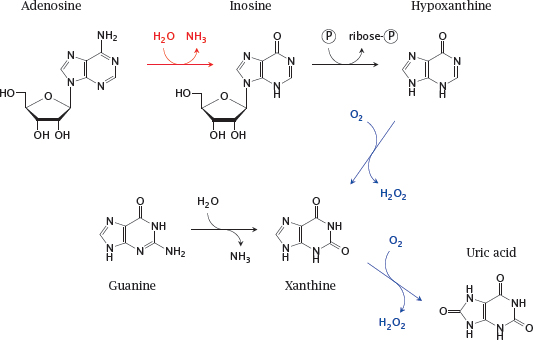
Notes: This slide shows some of the reactions involved in purine nucleotide degradation. The reaction catalyzed by adenosine deaminase is shown in red; those catalyzed by xanthine oxidase, which is a drug target in gout, are shown in blue. Uric acid is the final degradation product that is excreted in the urine.
Note that adenosine deaminase degrades both adenosine (shown here) and deoxyadenosine. Accumulation of deoxyadenosine is crucial for pathogenesis in this disease (see next slide).
10.3.2 Adenosine deaminase deficiency causes dysregulation of DNA synthesis
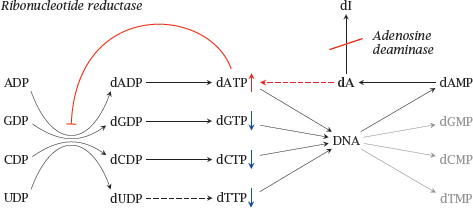
Notes: Adenosine deaminase (ADA) degrades not only adenosine but also deoxyadenosine (dA), which it converts to deoxyinosine (dI). If ADA is lacking, dA accumulates and is converted back to dATP by salvage pathway nucleoside and nucleotide kinases. Excess dATP inhibits ribonucleotide reductase and thereby throttles the supply of deoxyribonucloside triphosphates other than dATP. This interferes with DNA synthesis and promotes apoptosis.
The apoptotic stimulus created by ADA deficiency remains below the threshold at which it would trigger manifest apoptosis in most cell types. However, it is sufficiently strong in lymphocytes, which are very susceptible to apoptotic stimuli in general. Apoptosis depletes both T- and B-lymphocytes, which gives rise to severe combined immunodeficiency (SCID).1
10.3.3 Therapy of adenosine deaminase deficiency
Notes: The current standard treatment of ADA is bone marrow transplant. If the bone marrow of the patient is replaced with that of a healthy donor, the lymphocytes derived from the new bone marrow will express an intact ADA gene, and therefore be able to protect themselves from dATP accumulation and apoptosis. Gene therapy is based on the same principle, but uses replacement of the defective gene within the patients’ own bone marrow stem cells. It does work in principle but is currently still experimental.
Enzyme replacement therapy with ADA enzyme isolated from cattle is effective but inferior to bone marrow transplant. It is used mostly to bridge the time interval between diagnosis and the identification of a suitable bone marrow donor. Modification of the enzyme with polyethyleneglycol (PEG) serves to reduce its immunogenicity and extend its lifetime in the circulation.
A plausible pharmacological approach to prevent the pathology caused by deoxyadenosine accumulation would be to inhibit the salvage kinases that convert deoxyadenosine to dATP, the actual inhibitor of ribonucleotide reductase. While this approach has been demonstrated in vitro [63], I have not seen any reports describing its further development in vivo.
10.3.4 Gout
- Genetic or dietary factors promote increased production or retention of uric acid
- Uric acid has low solubility and easily forms crystalline deposits, preferentially in joints and soft tissue
- Urate crystals promote inflammation and lead to arthritis that is painful and destructive
Notes: Gout results from excess levels of uric acid, the end product of adenine and guanine degradation (see slide 10.3.1). The disease is not restricted to patients with mutations in a specific gene and can arise from different causes; it thus is far more common than any of the enzyme defects covered so far. Nevertheless, the clinical symptoms remain the same and are addressed by similar therapeutic measures.
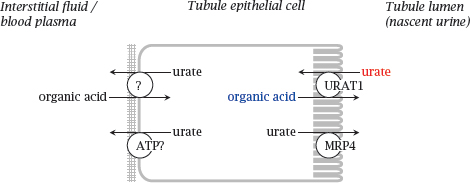
10.3.5 Transport of uric acid in the kidneys